Dysfunction of Kidney Endothelium after Ischemia/Reperfusion and Its Prevention by Mitochondria-Targeted Antioxidant
S. S. Jankauskas1, N. V. Andrianova1, I. B. Alieva1, A. N. Prusov1, D. D. Matsievsky2, L. D. Zorova1,3, I. B. Pevzner1, E. S. Savchenko4, Y. A. Pirogov5, D. N. Silachev1, E. Y. Plotnikov1*, and D. B. Zorov1*
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; E-mail: zorov@genebee.msu.ru, plotnikov@genebee.msu.ru2Institute of General Pathology and Pathophysiology, Russian Academy of Medical Sciences, 125315 Moscow, Russia
3Lomonosov Moscow State University, International Laser Center, 119991 Moscow, Russia
4Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia
5Lomonosov Moscow State University, Faculty of Physics, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received July 1, 2016; Revision received August 1, 2016
One of the most important pathological consequences of renal ischemia/reperfusion (I/R) is kidney malfunctioning. I/R leads to oxidative stress, which affects not only nephron cells but also cells of the vascular wall, especially endothelium, resulting in its damage. Assessment of endothelial damage, its role in pathological changes in organ functioning, and approaches to normalization of endothelial and renal functions are vital problems that need to be resolved. The goal of this study was to examine functional and morphological impairments occurring in the endothelium of renal vessels after I/R and to explore the possibility of alleviation of the severity of these changes using mitochondria-targeted antioxidant 10-(6ʹ-plastoquinonyl)decylrhodamine 19 (SkQR1). Here we demonstrate that 40-min ischemia with 10-min reperfusion results in a profound change in the structure of endothelial cells mitochondria, accompanied by vasoconstriction of renal blood vessels, reduced renal blood flow, and increased number of endothelial cells circulating in the blood. Permeability of the kidney vascular wall increased 48 h after I/R. Injection of SkQR1 improves recovery of renal blood flow and reduces vascular resistance of the kidney in the first minutes of reperfusion; it also reduces the severity of renal insufficiency and normalizes permeability of renal endothelium 48 h after I/R. In in vitro experiments, SkQR1 provided protection of endothelial cells from death provoked by oxygen–glucose deprivation. On the other hand, an inhibitor of NO-synthases, L-nitroarginine, abolished the positive effects of SkQR1 on hemodynamics and protection from renal failure. Thus, dysfunction and death of endothelial cells play an important role in the development of reperfusion injury of renal tissues. Our results indicate that the major pathogenic factors in the endothelial damage are oxidative stress and mitochondrial damage within endothelial cells, while mitochondria-targeted antioxidants could be an effective tool for the protection of tissue from negative effects of ischemia.
KEY WORDS: ischemia, kidney, Doppler, blood vessels, oxidative stress, mitochondriaDOI: 10.1134/S0006297916120154
Abbreviations: AKI, acute kidney injury; DCF, 2,7-dichlorodihydrofluorescein; I/R, ischemia/reperfusion; IREC, isolated renal epithelial cells; OGD, oxygen–glucose deprivation; ROS, reactive oxygen species; SkQR1, 10-(6ʹ-plastoquinonyl)decylrhodamine 19.
The problem of acute kidney injury (AKI) treatment remains relevant
despite decades of intense experimental work aimed at deciphering the
mechanisms of organ damage and approaches to affect them. Many data on
AKI pathogenesis have been accumulated; however, effective ways to
treat this pathology have not been found. AKI often occurs as a
complication of various severe clinical conditions such as sepsis,
severe trauma, cardiac arrest, and dehydration; it contributes
significantly to mortality among patients in intensive care units.
Disorders in blood supply is the most common cause of AKI; it results from the drastic reduction of blood pressure in systemic vasodilatation or heart disorders or in case of excessive vasoconstriction of renal vasculature. In all cases, the renal blood flow autoregulation system is unable to maintain perfusion of renal parenchyma at the level corresponding to the energy requirement of the nephron. If such a period of ischemia is long enough, then even after removal of the factors that caused the ischemia, the restoration of blood supply does not lead to complete restoration of renal blood flow – it remains somewhat decreased [1, 2]. Such secondary hypoperfusion, which develops after the initial ischemic episode, is caused by pathological changes in the functioning of the renal vascular bed. The latter leads to the loss of renal blood flow autoregulation [3, 4], enhancement of constrictor response to stimulation of the sympathetic nervous system, disappearance of reaction to vasodilating compounds [4], attraction and adhesion of immunoinflammatory cells in microvascular renal bed [5, 6], and blood stasis in capillaries [2].
These functional changes are associated with destructive processes in cells of the vascular wall, initiated by ischemia followed by reperfusion (I/R). In both in vitro and in vivo experiments, I/R was shown to damage cytoskeletal structure and organization of tight junctions in endothelial and smooth muscle vascular cells [7-10]. Similar changes in renal vascular cells were also found in patients with AKI [11]. An increase in the concentration of endothelial activation markers in blood of experimental animals [9] and the reduction of the number of endothelial cells lining the renal capillaries (visualized by immunohistochemical methods) also indicated endothelial damage after I/R [12, 13].
Mitochondria deserve special attention among cell organelles subjected to the damaging effect of I/R [14]. Increased production of reactive oxygen species (ROS) by mitochondria is one of the main consequences of I/R [15]. ROS are known to play an important role in the regulation of renal vascular tone. For example, administration of antioxidants increases renal blood flow [16], and inhibition of intracellular antioxidant systems results in its decrease [17]. In addition, ROS regulate the tone of afferent arteriole in the tubuloglomerular feedback system [18], which ultimately regulates nephron blood supply. ROS also play the key role in signaling cascades triggered by angiotensin II [19, 20].
We have previously shown that mitochondria-targeted antioxidant SkQR1 (10-(6ʹ-plastoquinonyl)decylrhodamine 19) has a pronounced nephroprotective effect in a number of renal pathologies, including experimental cold and warm kidney ischemia, rhabdomyolysis, gentamicin nephrotoxicity, and acute pyelonephritis [21-26]. SkQR1 effects in kidneys are primarily associated with the reduction of oxidative stress associated with these pathologies and increased tolerance of kidney cells to damaging factors. However, the effects of this antioxidant on renal hemodynamics and the functional state of renal endothelium have not been previously studied. The aim of this study was to test the hypothesis that the protective effect of SkQR1 on renal I/R is linked to the prevention of the renal vascular dysfunction.
MATERIALS AND METHODS
Experiments on cell cultures. Cells of endothelial line EA.hy926 were studied (courtesy of Prof. C. J. Edgel, North Carolina State University, USA). Cell culture EA.hy926 was obtained by hybridization of the primary endothelial line HUVEC with A549 lung carcinoma cells; it reproduces all the main morphological, phenotypic, and functional characteristics inherent in microvessel endothelial cells. EA.hy926 line cells were cultured in complete growth medium consisting of DMEM/F-12 (1 : 1), 10% fetal calf serum (PAA, USA), 100 U/ml penicillin-streptomycin, 2 mM L-glutamine, 0.1 mM hypoxanthine, and 16 µM thymidine (all from PanEco, Russia). For the experiments, cultures were seeded in 96-well plastic plates. Oxygen–glucose deprivation (OGD) was carried out by a conventional procedure; cultured cells were washed twice with balanced salt solution containing (in mM) NaCl 116, KCl 5.4, CaCl2 1.8, MgSO4 0.8, NaH2PO4 1, pH 7.3, and then incubated for 5 h in the same solution in a humidified chamber filled with nitrogen at 37°C. Culturing conditions remained the same for control cultures. Immediately after completion of OGD, saline solution was replaced by cultivation medium. The number of surviving cells was assessed after 24 h based on their ability to reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to formazan as measured by spectral properties (standard MTT-test [27]).
Induction of renal I/R. Experiments were performed on outbred male albino rats (350-400 g) kept in a vivarium on a standard diet. Work with laboratory animals was carried out in accordance with the requirements of the Commission of Bioethics of Belozersky Institute of Physico-Chemical Biology (protocol #1, April 8, 2013).
Left kidney ischemia was performed as previously described [21]. The animal was anesthetized with chloral hydrate intraperitoneally (i.p., 300 mg/kg), abdominal incision was performed, and the vascular bundle of the left kidney was clamped with an atraumatic microvascular clamp for 40 min. In acute experiments, the right kidney stayed intact. To assess the level of damage in the kidney subjected to I/R, we conducted nephrectomy of the contralateral (intact) kidney, because no renal failure is observed when at least one kidney remains intact. Ligation of the right ureter was performed to prevent retrograde urine release into the abdominal cavity. During anesthesia, the animal’s temperature was maintained at 37°C. Control animals were subjected to similar procedures except the clamping of the vascular bundle of the left kidney (sham-operated animals). Since elevation of the creatinine concentration in blood is an indicator of the degree of deterioration of renal filtration function, creatinine concentration in blood served as an indicator of renal failure in animals. Creatinine was measured in serum taken from the tail vein using an AU480 Chemistry System (Beckman Coulter, USA).
Magnetic resonance imaging. Renal vessels were studied using magnetic resonance imaging with a BioSpec 70/30 device (Bruker, Germany) with induction of magnetic field 7 T and gradient system 105 mT/m. To obtain T2-weighted imaging, a pulse sequence based on the spin echo RARE (Rapid Acquisition with Relaxation Enhancement) was used with the following parameters: TR = 2800 ms, TE = 90 ms, slice thickness 1.5 mm, field of view 8 × 4 cm, matrix size 256 × 384. Before conducting magnetic resonance imaging, the animals were anesthetized and placed in a positioning device equipped with a system of stereotaxis and thermoregulation.
Study of blood flow using high-frequency Doppler technology. High-frequency ultrasound Doppler operating at a frequency of 27 MHz was used in the experiments on hemodynamics (developed in the Laboratory of Bioengineering, Institute of General Pathology and Pathophysiology, Russian Academy of Medical Sciences). Blood flow was measured using bandage-type transducer with internal diameter of 1.5 to 2 mm, calibrated in units of volumetric flow rate. The transducer was applied to the renal artery. Blood flow velocity was registered 10-15 min after fixation of transducers (this time was needed for blood flow stabilization).
Blood flow velocity was recorded for short time periods (up to 30 s) at regular intervals: after blood flow stabilization, immediately after the clamping of the vascular bundle (clamps were placed distal to the transducer), 40 min after ischemia, during the first 60 s of reperfusion, and then every 5 min until the 30th min. For experiments with Nω-L-nitroarginine (L-NA) administration, blood flow velocity was recorded also immediately after the administration of this compound and 5 and 10 min after its administration.
Linear (V) and volumetric (Q) blood flow velocity were recorded during the experiment. Data from the ultrasonic device was digitized using the Graph2Digit software. Renal hemodynamics was analyzed based on the following parameters: heart rate (HR, beats/min), volumetric blood flow velocity (Q, ml/min), peak systolic blood flow velocity (Vmax, cm/s), end diastolic flow velocity (Vmin, cm/s), resistivity index (Pourcelot index): (Vmax – Vmin)/Vmax.
Evaluation of permeability of the renal vascular endothelium. In vivo administration of Evans Blue dye was used to evaluate permeability of the renal vascular bed. Anesthetized animals were injected intravenously with 2 ml 0.5% Evans Blue. After 70 min, the animals were transcardially perfused with 200 ml PBS to wash Evans Blue out of blood vessels. After that, the left kidney was excised, dried, weighed, cut, and photographed. Then the kidney was homogenized, centrifuged for 10 min at 14,000g, and the supernatant was collected. For protein precipitation, the supernatant was mixed with 50% trichloroacetic acid (1 : 2 v/v) and centrifuged for 20 min at 14,000g. Then 90 µl of ethanol was added to 30 µl of the supernatant, and fluorescence intensity was measured (excitation – 535 nm, emission – 625 nm). Solution with known Evans Blue concentration was used for calibration.
Electron microscopy. The renal cortical section was cut into pieces of approximately 1 mm3 and fixed with 2.5% glutaraldehyde (Sigma, USA) in phosphate buffer (pH 7.4), then the samples were additionally fixed with 1% OsO4 solution, dehydrated (70% ethanol containing 2% uranyl acetate), and embedded in Epon 812 (Fluka, USA). After polymerization in Epon, ultrathin sample sections were made on an LKB Ultratome III microtome. Sections were contrasted with citrate and examined with a JEM-1400 electron microscope (JEOL, Japan) at accelerating voltage 100 kV.
Detection of endothelial cells circulating in blood using polymerase chain reaction with reverse transcriptase (RT-PCR). A 1.5-ml blood samples were taken from the rat jugular vein with a syringe containing EDTA as anticoagulant. RNA was isolated from the whole blood using a QIAamp RNA Blood Mini kit (Qiagen, USA) following the manufacturer’s instructions. The resulting total RNA was used as a template for reverse transcription and PCR. RT-PCR was performed in single vial format on a Bio-Rad CFX96 device using a One-Step RT-PCR kit (Alpha-enzyme, Russia) according to the manufacturer’s instructions. vcam-1 relative expression was evaluated from the curves of fluorescent signal accumulation, crossing the threshold, and normalized to the expression of housekeeping gene Gapdh. Amplification products were detected in real time using TaqMan probes according to the principle of 5′-terminal tag excision. The temperature parameters of RT-PCR reaction were: reverse transcription cycle, 50°C for 15 min; Taq-AT enzyme activation cycle, 95°C for 5 min; then 45 cycles of successive denaturation for 15 s at 95°C, annealing for 15 s at 50°C, and elongation for 10 s at 72°C with fluorescent signal registration. PCR efficiency for all oligonucleotides was tested by obtaining standard curves using RNA serial dilutions. As efficiency was similar and close to 100% (data not shown) for all the cases, we calculated normalized expression assuming efficiency to be 100%.
Primers and fluorochromes used for vcam-1 and Gapdh amplification are given in Table 1.
Table 1. Oligonucleotides used in the
study

Statistics. At least five animals were used in each group during in vivo experiments. For in vitro studies, the experiments were repeated at least three times. Statistical significance was evaluated using Student’s test. Data are presented as mean ± SEM.
RESULTS
Effects of ischemia on endothelial cell culture. To study endothelial vulnerability to the damaging effects of I/R, we used the model that most accurately reproduces I/R conditions in vitro – oxygen–glucose deprivation (OGD). Over 50% of endothelial cells in EA.hy926 culture died 24 h after 5-h OGD. Mitochondria-targeted antioxidant increased endothelial cells survival after ischemia: survival of EA.hy926 cells increased to 64% after incubation for 1 h with 25 nM SkQR1 prior to OGD (Fig. 1a).
Fig. 1. SkQR1 reduces ischemia-induced endothelial damage. a) Death of EA.hy926 cells after oxygen–glucose deprivation (OGD) was reduced after 1-h preincubation with 25 nM SkQR1; * p < 0.05. b, c) Evans Blue dye accumulation in kidneys 70 min after i.v. injection of 2 ml 0.5% Evans Blue. Visual analysis of the dye distribution in the kidney (b) and quantification of the amount of dye extracted from kidney tissue homogenates (c). SkQR1 was administered i.p. 3 h prior to the incidence of ischemia; 1, 18, 30, 42 h after the beginning of reperfusion (each injection – 100 nmol/kg). d) Changes in the level of vcam-1 mRNA in blood after 40 min of ischemia and 10 min of reperfusion; # p < 0.1.
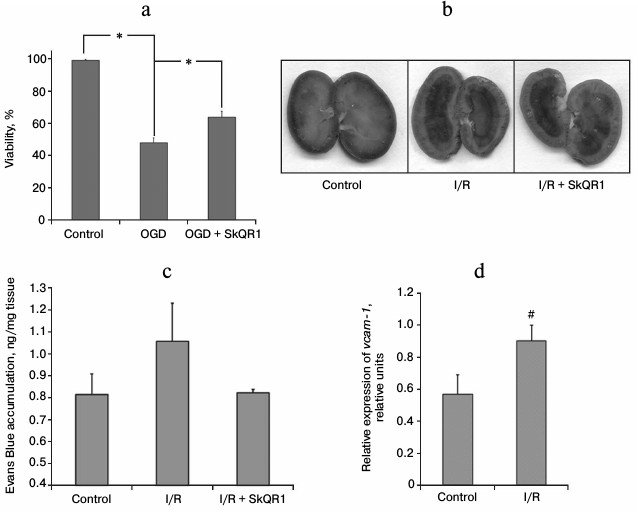
Effects of ischemia on endothelial barrier function. The degree of vascular endothelial damage after renal ischemia was also evaluated in vivo. In 48 h after 40-min renal ischemia, the permeability of the renal vascular bed was studied, which reflects the state of one of the main endothelial functions – barrier function. Comparing the accumulation of Evans Blue in the kidneys of control rats and rats subjected to I/R, we found a small increase in its concentration in postischemic kidneys (Fig. 1, b and c), suggesting increased permeability of renal vessels to albumin (99% of the dye is bound to this protein). Visual analysis of Evans Blue localization in different kidney portions showed that the main accumulation of the dye is observed in the renal pelvis and inner medulla, whereas it is hardly detectable in the cortex and outer medulla (Fig. 1b). I/R results in the increase in Evans Blue content in all kidney parts (Fig. 1b). We observed the dye accumulation in the cortex and outer medulla, i.e. in the kidney sections most sensitive to ischemic damage. We measured the amount of Evans Blue extracted from the total kidney homogenates and found the difference between the control animals and rats subjected to I/R (Fig. 1c). After the animals were treated with mitochondria-targeted antioxidant (i.p. administration of 100 nmol/kg SkQR1 3 h prior to ischemia; 1, 18, 30, and 42 h after the beginning of reperfusion), we observed a tendency toward normalization of the renal vascular bed permeability. In SkQR1-treated animals, Evans Blue concentration after I/R in the total kidney homogenate did not differ from that of intact animals (Fig. 1c). Visual analysis also showed extremely low dye content in the cortex and outer medulla (Fig. 1b).
Assessment of vascular endothelium damage by levels of endothelial cells markers in circulating blood. Appearance in the bloodstream of freely circulating endothelial cells is one of the conventional markers of vascular endothelium damage. In this study, we evaluated the content of mRNA of VCAM-1 in peripheral rat blood (this protein is a specific marker of endothelial cells). Ten minutes after the beginning of reperfusion, the amount of VCAM-1 mRNA was significantly increased compared to sham-operated animals, indicating a significant increase in the number of severely damaged endothelial cells desquamated into the vessel lumen and/or the level of endothelium activation, as VCAM-1 expression increases in the inflammatory response of endothelial cells (Fig. 1d).
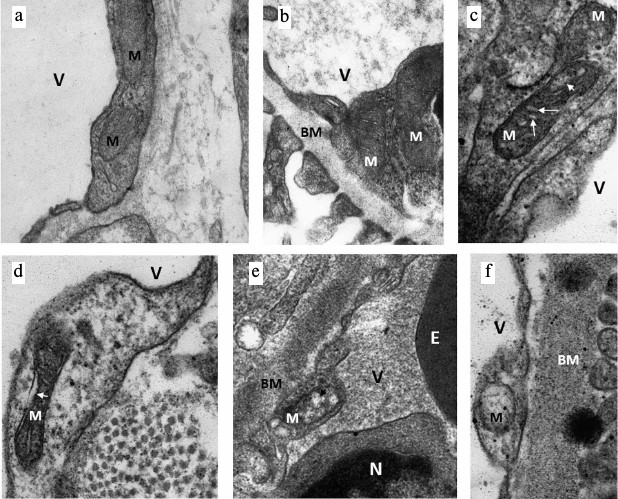
Ultrastructural changes in mitochondria of endotheliocytes. Since mitochondria play the key role in the fate of cells subjected to I/R, and mitochondria-targeted antioxidant SkQR1 had a positive effect on the survival and maintenance of the normal functioning of endothelium after I/R, we evaluated the effect of I/R on the ultrastructure of the mitochondrial reticulum of endothelial cells. Electron microscopic study of the renal cortical zone showed significant damage in endothelial mitochondria already in the early stages of reperfusion after 40 min of ischemia. Endothelial cells in the cortex of an intact kidney with mitochondria in orthodox configuration were considered as normal (Fig. 2, a and b). In a similar zone of the kidney fixed 10 min after the beginning of reperfusion we detected both endotheliocytes with mitochondria maintaining orthodox structure and endotheliocytes with altered mitochondrial morphology. In particular, we observed a significant increase in intercristae and intermembrane space accompanied by a local enlightenment of the mitochondrial matrix and their swelling (Fig. 2, c and d). In some mitochondria, the number of visually determined cristae was decreased up to their complete disappearance (Fig. 2, e and f); cells with such mitochondria showed signs of degradation. This data indicated that mitochondrial damage in renal endothelial cells is an early event observed in the first minutes after the beginning of reperfusion.
Fig. 2. Ultrastructural changes in renal endothelial mitochondria after ischemia. a, b) Intact kidney, endothelial cells with mitochondria (M) in the orthodox state; BM, basal membrane, V, vessel lumen. c-f) Kidney after 40 min of ischemia and 10 min of reperfusion. Alterations in mitochondrial morphology (M) starting from minor (c, d: swelling of inter-cristae (large arrows in (c)) and intermembrane space (arrow in (d)) and matrix (small arrow in (c)) to significant (local, shown by arrow in (e) or spacial in (f)) mitochondrial swelling. N, nucleus; E, part of erythrocyte in vessel lumen.
Changes in renal hemodynamics parameters. Renal vascular response to I/R was studied at the same time frames, in which mitochondria of endothelial cells are damaged. We visualized segmental and partially interlobar renal arteries using magnetic resonance imaging. Ten minutes after the beginning of reperfusion, the number of visualized segmental and interlobar renal arteries was significantly reduced, indicating a strong decrease in the diameter of their lumen. Ten minutes after the beginning of reperfusion, the number of visualized vessels was higher in the kidney of animals injected with SkQR1 (100 nmol/kg, i.p.) 3 h before I/R, compared to animals which did not receive the treatment, but lower than in intact kidneys (Fig. 3a).
Fig. 3. Changes in renal blood flow after 40 min ischemia. a) Representative kidney images obtained using magnetic resonance imaging after 40 min ischemia and 10 min reperfusion. SkQR1 increases the number of blood-filled renal vessels after 40 min ischemia. b-e) Representative recording of the linear (b, d) and volumetric (c, e) blood flow velocity in the left renal artery using a high frequency ultrasound (27 MHz) bondage-type Doppler transducer. Rat after I/R (b, c) and rat after I/R with SkQR1 (100 nmol/kg) administered i.p. 3 h prior to I/R (d, e).
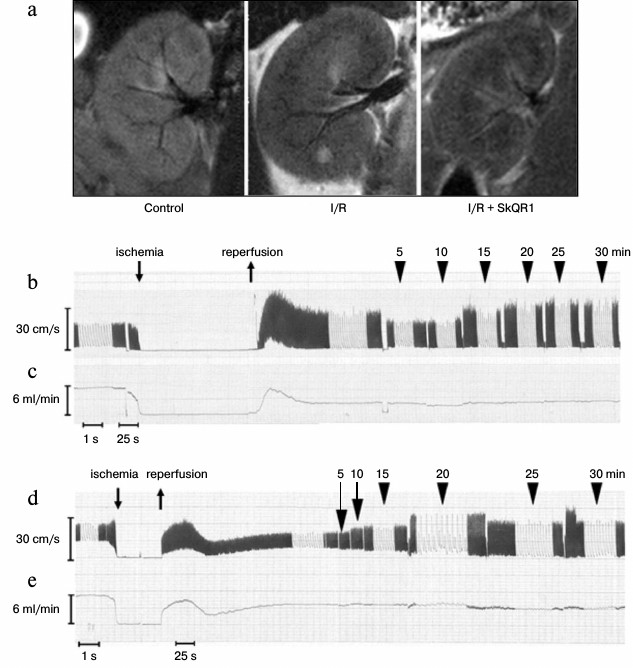
The ultrasound Doppler was used for quantitative assessment of renal blood flow after I/R. We observed severe changes in linear and volumetric blood flow velocity in the renal artery after 40 min of ischemia, which were largely prevented by preliminary SkQR1 administration (Fig. 3, b-e).
Measurement of volumetric blood flow rate in the renal artery, reflecting the total renal blood flow, showed significant renal hypoperfusion after 40 min of ischemia (Fig. 4a). One minute after the beginning of reperfusion, the volumetric blood flow rate was only about 30% of the level prior to ischemia. Measurements of this parameter at the 5th, 10th, 15th, and 20th min of reperfusion showed that renal blood supply remains low. Some recovery in blood flow was found only at the 25th min, but hypoperfusion persisted until the end of the observation period (30 min) (Fig. 4a).
Fig. 4. SkQR1 reduces the negative effect of 40 min ischemia on renal hemodynamics. Volumetric blood flow rate (a) and resistivity index (b) of the left renal artery. Administration of 100 nmol/kg SkQR1 3 h prior to I/R increased renal blood flow and reduced vasoconstriction of renal vessels as evidenced by a decrease in resistivity index. Intravenous administration of 1 mg/kg L-NA abolished the positive effect of SkQR1 on renal hemodynamics; * p < 0.05, ** p < 0.1 compared to I/R.
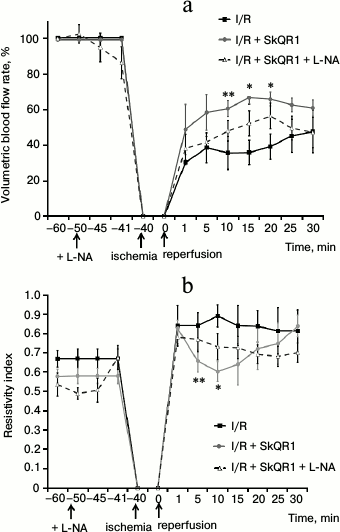
We calculated the resistivity index using the maximal peak systolic and end diastolic blood flow velocity during the cardiac cycle (the parameter indicates the degree of resistance to blood flow in the areas distal to the spot of blood flow registration). Since in our experiments the transducer was applied to the renal artery, changes in the resistivity index reflected the changes in renal vascular resistance. After 40 min of ischemia, resistivity index significantly increased, and within 1 min after the beginning of reperfusion, it was about 20% higher than before ischemia (Fig. 4b). Resistivity index reached its maximum value in the 10th min and remained increased until the 30th min of reperfusion (Fig. 4b), when the experiment was terminated. The data was consistent with the results obtained with MRI scanning of the kidney and indicated pronounced vasoconstriction of renal vessels after I/R.
Administration of SkQR1 (i.p. 100 nmol/kg) 3 h prior to the beginning of ischemia resulted in a more rapid recovery of renal blood flow. For example, at the 1st min of reperfusion, the volumetric blood flow rate in rats treated with the mitochondrial antioxidant increased to 49% (compared to the values prior to ischemia), while in rats received no treatment this parameter was only 30%. Volumetric rate continued to grow and reached 67% at the 15th min, while in animals received no treatment the blood flow was restored only to 36% of the pre-ischemic value (Fig. 4a).
Resistivity index before ischemia did not differ significantly in animals with or without administered SkQR1. After 40 min of ischemia, we observed no positive effect of mitochondria-targeted antioxidant on the increase in resistivity index caused by ischemia in the 1st min of reperfusion. However, resistivity index decreased already in the 5th and 10th min of reperfusion, returning to the values observed before ischemia. Starting from the 15th min, vascular resistance again began to increase (Fig. 4b). Thus, the mitochondria-targeted antioxidant reduced renal vascular resistance after ischemia at the very time when mitochondrial damage occurred in vascular endothelium.
We previously showed that systemic hemodynamics did not undergo significant changes after I/R [28]. In this study, heart rate measurements also showed no statistically significant differences, both with and without SkQR1 (Table 2).
Table 2. Changes in heart rate (HR) during
the experiment
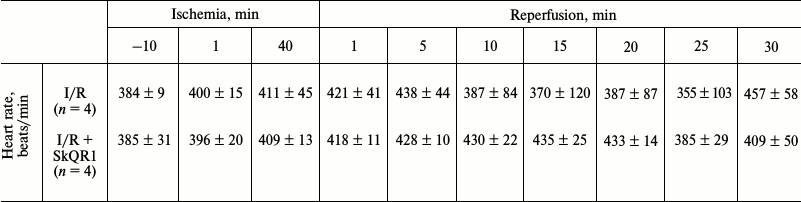
Note: Animals were subjected to 40-min renal ischemia. SkQR1 was
administered 3 h prior to the beginning of ischemia (i.p.
100 nmol/kg).
Since NO is the main vasodilatory factor, we studied the effect of SkQR1 on renal hemodynamics against the background of inhibited NO synthesis. Treatment with nonselective inhibitor of NO-synthases L-NA led to the disappearance of the positive effect of SkQR1 on renal hemodynamics. Dynamics of renal blood flow restoration under coadministration of SkQR1 (i.p. 100 nmol/kg, 3 h prior to ischemia) and L-NA (i.v. 1 mg/kg, 10 min prior to ischemia) was not significantly different from the animals that received no treatment (Fig. 4a). Decrease in the resistivity index was not observed after coadministration of L-NA and SkQR1, in contrast with animals treated only with SkQR1 (Fig. 4b).
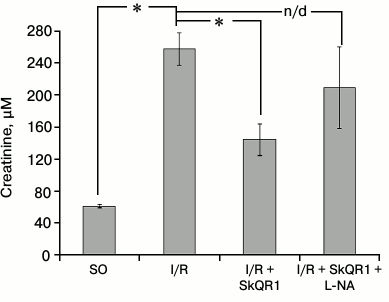
We previously showed that SkQR1 preserved the function of kidney subjected to I/R [22]. To assess the role of this effect on the renal vascular bed for the nephroprotective SkQR1 impact, we evaluated renal function 48 h after I/R based on serum creatinine concentration. SkQR1 treatment resulted in almost twofold reduction of creatinine concentration compared to animals that received no treatment, indicating a decrease in the severity of renal failure. However, when this SkQR1 treatment protocol was combined with intravenous L-NA administration 10 min before I/R, we observed no decrease in creatinine concentration (Fig. 5).
Fig. 5. Effect of L-NA on the protective effect of SkQR1 on renal function after 40-min ischemia. Renal function was assessed by creatinine concentration in blood serum 48 h after ischemia. L-NA (1 mg/kg) was administered intravenously 10 min prior to the beginning of ischemia. SkQR1 (100 nmol/kg) was injected i.p. 3 h before the beginning of ischemia and 1, 18, 30, and 42 h after the beginning of reperfusion; * p < 0.001; n/d, no significant differences; SO, sham-operated animals.
DISCUSSION
In this study, we show that damage of endothelial mitochondria plays an important role in the mechanisms of impairment of renal hemodynamics resulting from I/R. This is indicated by the fact that pronounced impairments in renal vascular bed functioning (vasoconstriction of blood vessels, increase in vascular resistance, reduction of renal blood flow) occur within the same timeframe as the impairments of mitochondrial ultrastructure. Electron microscopic study showed that already 10 min after the beginning of reperfusion, destructive processes are triggered in mitochondria of endothelial cells, leading to the impairment of their ultrastructure. In addition, 10 min after the beginning of reperfusion, we observed the reduction of the renal blood flow, which obviously leads to insufficient oxygen supply to cells. Renal blood flow does not return to the pre-ischemic values after the cessation of ischemia; it remains considerably reduced for a long time due to a persistent increase of renal vascular resistance. Administration of SkQR1, a compound selectively accumulated in mitochondria that protects endothelial cells in vitro, had a beneficial effect on the observed parameters.
It is known that I/R increases ROS production in mitochondria [14]. Since I/R results in the damage to endothelial cells of glomerular and peritubular capillaries and tubular epithelial cells, ROS hyperproduction may be within all these types of cells. It has been shown in a number of studies that ROS can regulate renal blood flow [16-18]. In these studies, the effect of ROS on blood flow was similar to the post-I/R changes that we observe; i.e. a decreased renal perfusion. We previously showed that I/R leads to considerable oxidative stress in renal parenchyma, and mitochondria are the main source of ROS [29]. It is important to note that the peak of ROS production in the kidney was observed 10 min after the beginning of reperfusion – the time when endothelial mitochondria were damaged. Administration of SkQR1 3 h before ischemia prevented the development of oxidative stress in the kidney 10 min after the beginning of reperfusion [22]. Although in that study the ROS production in renal tubules only was studied, we assumed that similar changes also take place in endothelium. This was confirmed by our observation that mitochondria-targeted antioxidant improves renal blood flow, which emphasizes the possible role of mitochondrial ROS in changes of renal blood flow. This conclusion is confirmed by other studies. For example, it has been shown that mitochondrial ROS production plays an important role in the development of experimental hypertension [20, 30].
Noteworthy that disruptions in mitochondrial functioning occurring in the first minutes of reperfusion, apparently have a long-term effect on the processes taking place in the vascular wall, because hemodynamic parameters remain abnormal for a long time after I/R – at least for 30 min. Moreover, in the preliminary experiments we observed that hemodynamic parameters reached by the 10th min of reperfusion remained basically unchanged even at the 60th min of reperfusion. We consider that the primary oxidative stress of mitochondrial nature induced in the early phase of I/R is sustained for a long time due to a positive feedback between ROS-producing systems [14, 31]. For example, it has been shown that mitochondrial ROS can activate NADPH-oxidase (a ROS-producing enzyme), and ROS generated by this enzyme can, in turn, cause oxidative stress in mitochondria [32].
Since SkQR1 is accumulated in mitochondria of all renal cells, it raises a question: Is the positive effect of SkQR1 specifically related to the protection of endothelial cells? (Earlier it has been shown that SkQR1 normalizes mitochondrial functions in tubular cells [22].) Therefore, in this study we assessed the effect of SkQR1 on the consequences of I/R in endothelial cells in the absence of any intermediates. I/R simulation in vitro showed that preincubation of EA.hy926 endothelial cells with SkQR1 decreased OGD-induced cell death, indicating the possibility of the primary protection of endothelial cells.
In our work, we demonstrate an increase in the number of endothelial cells circulating in blood 10 min after the beginning of reperfusion, indicating that I/R may cause the destruction of endothelium in vivo. The phenomenon is a marker of pronounced damage of the vascular bed [33]. Apparently, it happens due to the desquamation of the critically damaged cells into the vessel lumen. Loss of endothelial lining may be another cause of the long-term disorders in renal hemodynamics. Thus, the density of the renal capillary bed has been shown to be reduced even one week after renal ischemia [12]. In our study, we observed significant impairment of endothelial barrier function 2 days after I/R. SkQR1 treatment reduced the permeability of renal blood vessels to normal values.
Decrease in NO availability or reduction of its production by endothelial cells due to disorders in their functioning and their destruction are the most probable mechanism of vasoconstriction after ischemia. We can increase NO production in post-ischemic kidney by protecting endothelial cells with SkQR1; it allows to avoid (at least partially) the harmful effects of the secondary ischemia due to vasoconstriction. In our study, we used the nonselective inhibitor of NO-synthases L-NA to test whether the positive effect of SkQR1 on blood flow is associated with an increase in NO production. Indeed, when L-NA was administered 10 min before ischemia to rats who had previously received SkQR1, we observed disappearance of the positive effect of the mitochondria-targeted antioxidant on the renal blood flow.
Positive effect of SkQR1 on restoration of renal blood flow was limited in time (improvement of these parameters became statistically unreliable already by the 30th min of reperfusion), and L-NA inhibition of this short positive effect at the early stages of reperfusion also resulted in the reduction of the protective effect of the mitochondria-targeted antioxidant on the renal functions 2 days after I/R. Our data suggest the fundamental importance of inhibiting the very first events of the pathological cascade of vascular dysfunction, which are apparently associated also with the dysfunction of endothelial mitochondria.
Early studies have shown the positive effect of the mitochondria-targeted antioxidant SkQ1 on endothelial cell communication impaired by tumor necrosis factor. It led to general conclusions on the involvement of ROS in the inflammatory response and SkQ1 having antiinflammatory activities [34, 35].
Acknowledgements
This study was supported by the Russian Science Foundation (project No. 14-15-00147).
REFERENCES
1.Ajis, A., Bagnall, N. M., Collis, M. G., and Johns,
E. J. (2003) Effect of endothelin antagonists on the renal hemodynamic
and tubular responses to ischemia-reperfusion injury in anesthetized
rats, Exp. Physiol., 88, 483-490.
2.Olof, P., Hellberg, A., Kallskog, O., and Wolgast,
M. (1991) Red cell trapping and postischemic renal blood flow.
Differences between the cortex, outer and inner medulla, Kidney
Int., 40, 625-631.
3.Adams, P. L., Adams, F. F., Bell, P. D., and Navar,
L. G. (1980) Impaired renal blood flow autoregulation in ischemic acute
renal failure, Kidney Int., 18, 68-76.
4.Kelleher, S. P., Robinette, J. B., and Conger, J.
D. (1984) Sympathetic nervous system in the loss of autoregulation in
acute renal failure, Am. J. Physiol., 246, F379-F386.
5.De Greef, K. E., Ysebaert, D. K., Dauwe, S., Persy,
V., Vercauteren, S. R., Mey, D., and De Broe, M. E. (2001) Anti-B7-1
blocks mononuclear cell adherence in vasa recta after ischemia,
Kidney Int., 60, 1415-1427.
6.Takada, M., Nadeau, K. C., Shaw, G. D., Marquette,
K. A., and Tilney, N. L. (1997) The cytokine-adhesion molecule cascade
in ischemia/reperfusion injury of the rat kidney. Inhibition by a
soluble P-selectin ligand, J. Clin. Invest., 99,
2682-2690.
7.Kwon, O., Phillips, C. L., and Molitoris, B. A.
(2002) Ischemia induces alterations in actin filaments in renal
vascular smooth muscle cells, Am. J. Physiol. Renal Physiol.,
282, F1012-F1019.
8.Kuhne, W., Besselmann, M., Noll, T., Muhs, A.,
Watanabe, H., and Piper, H. M. (1993) Disintegration of cytoskeletal in
energy-depleted endothelial structure cells of actin filaments in
energy-depleted endothelial cells, Am. J. Physiol. Heart Circ.
Physiol., 264, H1599-1608.
9.Sutton, T. A., Mang, H. E., Campos, S. B.,
Sandoval, R. M., Yoder, M. C., and Molitoris, B. A. (2003) Injury of
the renal microvascular endothelium alters barrier function after
ischemia, Am. J. Physiol. Renal Physiol., 285,
F191-198.
10.Kevil, C. G., Oshima, T., Alexander, B., Coe, L.
L., and Alexander, J. S. (2000) H(2)O(2)-mediated permeability: role of
MAPK and occludin, Am. J. Physiol. Cell Physiol., 279,
C21-C30.
11.Kwon, O., Hong, S.-M., Sutton, T. A., and Temm,
C. J. (2008) Preservation of peritubular capillary endothelial
integrity and increasing pericytes may be critical to recovery from
postischemic acute kidney injury, Am. J. Physiol. Renal
Physiol., 295, F351-F359.
12.Cantaluppi, V., Gatti, S., Medica, D.,
Figliolini, F., Bruno, S., Deregibus, M. C., Sordi, A., Biancone, L.,
Tetta, C., and Camussi, G. (2012) Microvesicles derived from
endothelial progenitor cells protect the kidney from
ischemia-reperfusion injury by microRNA-dependent reprogramming of
resident renal cells, Kidney Int., 82, 412-427.
13.Basile, D. P., Friedrich, J. L., Spahic, J.,
Knipe, N., Mang, H., Leonard, E. C., Changizi-Ashtiyani, S., Bacallao,
R. L., Molitoris, B. A., and Sutton, T. A. (2011) Impaired endothelial
proliferation and mesenchymal transition contribute to vascular
rarefaction following acute kidney injury, Am. J. Physiol. Renal
Physiol., 300, F721-F733.
14.Zorov, D. B., Juhaszova, M., and Sollott, S. J.
(2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release, Physiol. Rev., 94, 909-950.
15.Silachev, D. N., Plotnikov, E. Y., Pevzner, I.
B., Zorova, L. D., Babenko, V. A., Zorov, S. D., Popkov, V. A.,
Jankauskas, S. S., Zinchenko, V. P., Sukhikh, G. T., and Zorov, D. B.
(2014) The mitochondrion as a key regulator of ischaemic tolerance and
injury, Heart Lung Circ., 10, 897-904.
16.Ahmeda, A. F., and Johns, E. J. (2012) The
regulation of blood perfusion in the renal cortex and medulla by
reactive oxygen species and nitric oxide in the anesthetized rat,
Acta Physiol., 204, 443-450.
17.Majid, D. S. A., and Nishiyama, A. (2002) Nitric
oxide blockade enhances renal responses to superoxide dismutase
inhibition in dogs, Hypertension, 39, 293-297.
18.Liu, R., Ren, Y., Garvin, J. L., and Carretero,
O. A. (2004) Superoxide enhances tubuloglomerular feedback by
constricting the afferent arteriole, Kidney Int., 66,
268-274.
19.Zhang, H., Schmeisser, A., Garlichs, C. D.,
Plotze, K., Damme, U., Mugge, A., and Daniel, W. G. (1999) Angiotensin
II-induced superoxide anion generation in human vascular endothelial
cells: role of membrane-bound NADH-/NADPH-oxidases, Cardiovasc.
Res., 44, 215-222.
20.Dikalova, A. E., Bikineyeva, A. T., Budzyn, K.,
Nazarewicz, R. R., McCann, L., Lewis, W., Harrison, D. G., and Dikalov,
S. I. (2010) Therapeutic targeting of mitochondrial superoxide in
hypertension, Circ. Res., 107, 106-116.
21.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Jankauskas, S. S., Morosanova, M. A., Stelmashook,
E. V., Vasileva, A. K., Goryacheva, E. S., Pirogov, Y. A., Isaev, N.
K., and Zorov, D. B. (2010) New-generation Skulachev ions exhibiting
nephroprotective and neuroprotective properties, Biochemistry
(Moscow), 75, 145-150.
22.Plotnikov, E. Y., Chupyrkina, A. A., Jankauskas,
S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P., and Zorov, D.
B. (2011) Mechanisms of nephroprotective effect of
mitochondria-targeted antioxidants under rhabdomyolysis and
ischemia/reperfusion, Biochim. Biophys. Acta, 1812,
77-86.
23.Plotnikov, E. Y., Silachev, D. N., Jankauskas, S.
S., Rokitskaya, T. I., Chupyrkina, A. A., Pevzner, I. B., Zorova, L.
D., Isaev, N. K., Antonenko, Y. N., Skulachev, V. P., and Zorov, D. B.
(2012) Mild uncoupling of respiration and phosphorylation as a
mechanism providing nephro- and neuroprotective effects of penetrating
cations of the SkQ family, Biochemistry (Moscow), 77,
1029-1037.
24.Jankauskas, S. S., Plotnikov, E. Y., Morosanova,
M. A., Pevzner, I. B., Zorova, L. D., Skulachev, V. P., and Zorov, D.
B. (2012) Mitochondria-targeted antioxidant SkQR1 ameliorates
gentamycin-induced renal failure and hearing loss, Biochemistry
(Moscow), 77, 666-670.
25.Plotnikov, E. Y., Morosanova, M. A., Pevzner, I.
B., Zorova, L. D., Manskikh, V. N., Pulkova, N. V., Galkina, S.
I., Skulachev, V. P., and Zorov, D. B. (2013) Protective effect of
mitochondria-targeted antioxidants in an acute bacterial infection,
Proc. Natl. Acad. Sci. USA, 110, E3100-E3108.
26.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Mitochondria-targeted plastoquinone derivatives as tools to
interrupt execution of the aging program. 2. Treatment of some ROS- and
age-related diseases (heart arrhythmia, heart infarctions, kidney
ischemia, and stroke), Biochemistry (Moscow), 73,
1288-1299.
27.Mosmann, T. (1983) Rapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assays, J. Immunol. Methods, 65, 55-63.
28.Jankauskas, S. S., Matsievsky, D. D., Plotnikov,
E. Y., and Zorov, D. B. (2014) High-frequency ultrasound Doppler system
for study of renal blood flow during ischemia/reperfusion of kidney,
Nephrol. Dial., 16, 169-175.
29.Plotnikov, E. Y., Kazachenko, A. V., Vyssokikh,
M. Y., Vasileva, A. K., Tcvirkun, D. V., Isaev, N. K.,
Kirpatovsky, V. I., and Zorov, D. B. (2007) The role of mitochondria in
oxidative and nitrosative stress during ischemia/reperfusion in the rat
kidney, Kidney Int., 72, 1493-1502.
30.Nazarewicz, R. R., Dikalova, A. E., Bikineyeva,
A., and Dikalov, S. I. (2013) Nox2 as a potential target of
mitochondrial superoxide and its role in endothelial oxidative stress,
Am. J. Physiol. Heart Circ. Physiol., 305,
H1131-1140.
31.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) Reactive oxygen species
(ROS)-induced ROS release: a new phenomenon accompanying induction of
the mitochondrial permeability transition in cardiac myocytes, J.
Exp. Med., 192, 1001-1014.
32.Dikalov, S. (2011) Cross talk between
mitochondria and NADPH oxidases, Free Radic. Biol. Med.,
51, 1289-1301.
33.Woywodt, A., Kirsch, T., and Haubitz, M. (2008)
Circulating endothelial cells in renal disease: markers and mediators
of vascular damage, Nephrol. Dial. Transplant., 23,
7-10.
34.Zinovkin, R. A., Romaschenko, V. P., Galkin, I.
I., Zakharova, V. V., Pletjushkina, O. Y., Chernyak, B. V., and Popova,
E. N. (2014) Role of mitochondrial reactive oxygen species in
age-related inflammatory activation of endothelium, Aging (Albany N.
Y.), 6, 661-674.
35.Demyanenko, I. A., Popova, E. N., Zakharova, V.
V., Ilyinskaya, O. P., Vasilieva, T. V., Romashchenko, V. P., Fedorov,
A. V., Manskikh, V. N., Skulachev, M. V., Zinovkin, R. A.,
Pletjushkina, O. Y., Skulachev, V. P., and Chernyak, B. V. (2015)
Mitochondria-targeted antioxidant SkQ1 improves impaired dermal wound
healing in old mice, Aging (Albany N. Y.), 7,
475-485.