REVIEW: Control of Myofibroblast Differentiation and Function by Cytoskeletal Signaling
N. Sandbo1#, L. V. Smolyaninova2, S. N. Orlov2,3#*, and N. O. Dulin3,4#*
1University of Wisconsin, Department of Medicine, Madison, WI, USA2Lomonosov Moscow State University, Faculty of Biology, Laboratory of Biomembranes, 119991 Moscow, Russia; E-mail: sergeinorlov@yandex.ru
3Siberian State Medical University, 634050 Tomsk, Russia; E-mail: ndulin@medicine.bsd.uchicago.edu
4University of Chicago, Department of Medicine, 5841 IL, USA
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received August 7, 2016; Revision received September 14, 2016
The cytoskeleton consists of three distinct types of protein polymer structures – microfilaments, intermediate filaments, and microtubules; each serves distinct roles in controlling cell shape, division, contraction, migration, and other processes. In addition to mechanical functions, the cytoskeleton accepts signals from outside the cell and triggers additional signals to extracellular matrix, thus playing a key role in signal transduction from extracellular stimuli through dynamic recruitment of diverse intermediates of the intracellular signaling machinery. This review summarizes current knowledge about the role of cytoskeleton in the signaling mechanism of fibroblast-to-myofibroblast differentiation – a process characterized by accumulation of contractile proteins and secretion of extracellular matrix proteins, and being critical for normal wound healing in response to tissue injury as well as for aberrant tissue remodeling in fibrotic disorders. Specifically, we discuss control of serum response factor and Hippo signaling pathways by actin and microtubule dynamics as well as regulation of collagen synthesis by intermediate filaments.
KEY WORDS: myofibroblast, actin stress fibers, transcription, microtubules, differentiation, intermediate filaments, collagen synthesisDOI: 10.1134/S0006297916130071
Abbreviations: CArG box, CC(A/T)6GG DNA sequence; ECM, extracellular matrix; ET1, endothelin-1; GPCR, G-protein-coupled receptors; GTPases, guanosine triphosphate hydrolases; IDPN, iminodipropionitrile; IF, intermediate filaments; LPA, lysophosphatidic acid; MRTF-A, myocardin-related transcription factor; MT, microtubules; RGD, arginine/glycine/aspartate; Rho-GEFs, Rho guanine nucleotide-exchange factors; RPEL, arginine/proline/glutamate/leucine; S1P, sphingosin-1-phosphate; SRF, serum response factor; TGF-b, transforming growth factor b; WF-A, Withaferin A.
Myofibroblasts are modified fibroblasts that play an essential role in
mediating wound repair after tissue injury, but also can play an
aberrant role in the formation of exuberant extracellular matrix (ECM)
during tissue fibrosis. The original morphologic characterization of
myofibroblasts by electron microscopy showed them to have numerous
well-formed cytoskeletal microfilaments [1-3]. These are microfilaments, which were subsequently
found to be actin filaments (stress fibers comprised of both cytosolic
actin isoforms (b and g) and smooth muscle “specific”
actin, smooth muscle a-actin). The presence of actin stress fibers was
similar to that seen in smooth muscle cells, but these cells also
retained typical fibroblast features such as a robust endoplasmic
reticulum. Thus, the term “myo”-fibroblasts was used to
describe these cells.
Myofibroblasts also display increases (compared to undifferentiated fibroblasts) in the expression of ECM genes that are involved in wound healing or fibrotic tissue formation. These are genes encoding for periostin [4, 5], fibronectin (including the splice isoform of fibronectin that contains an extra type III domain (EDA fibronectin)) [6, 7], and multiple collagen isoforms [8]. Likewise, there is an increase in the expression of components of the actin cytoskeleton and its associated contractile machinery, which enables the myofibroblast to generate increased contractile forces [9]. This, together with induction of adhesion related proteins such as integrins and proteoglycans, couple the cytoskeleton to ECM across the cell membrane, enabling transmission of intracellular forces to the surrounding matrix. Thus, in normal tissue, the myofibroblast is a contractile cell that regulates normal organ function. They have been found in alveolar interstitium, hepatic sinusoids, and adjacent to intestinal crypts [10]. In healing dermal wounds, myofibroblasts are essential for the contracting newly formed granulation tissue, closing the wound and providing ECM for restoration of epithelial monolayer. However, myofibroblasts also exhibit cellular behavior that propagates tissue fibrosis. They are major producers of collagen and other ECM components and have enhanced ability to assemble nascent fibronectin matrix [11]. They are also characterized by resistance to apoptosis, which results in persistence of the population in response to injury, thereby propagating the fibrotic response [12, 13]. Myofibroblasts are invariably found in fibrotic tissues of various organs. Therefore, targeting myofibroblast differentiation could be an attractive strategy for treatment of fibrotic diseases.
In this review, we discuss critical roles of actin cytoskeleton in myofibroblast force generation, mature focal adhesion formation, ECM remodeling, and regulation of gene transcription that is critical for myofibroblast differentiation. We also discuss emerging roles of two other cytoskeleton systems, microtubules and microfilaments, in control of myofibroblast differentiation and ECM remodeling.
ACTIN STRESS FIBER FORMATION
The actin cytoskeleton is comprised of actin monomers that consist of six isoforms [14]. The b- and g-actins are ubiquitous and are the primary components of actin filaments in most cells. In myofibroblasts, the smooth muscle a-actin isoform is upregulated, along with smooth muscle g-actin, skeletal a-actin, and cardiac a-actin. Actin filaments can be organized in several arrays that can alter cell shape, promote cell motility, and alter the transmission of force to the surrounding matrix environment.
One of the signature morphologic features of the myofibroblast is a robust actin stress fiber network, which provides an important means of influencing the ECM across the cell membrane. Actin stress fibers are bundles of polymerized actin filaments that are typically attached at one or both ends to focal adhesion sites [15, 16]. Actin stress fibers are nucleated at focal adhesion sites from actin filaments. The actin-interacting protein a-actinin binds filaments and bundles them into “stress fibers”. a-Actinin is periodically arrayed along actin stress fibers [17, 18]. Similarly, non-muscle myosin is also periodically localized on actin stress fibers in an alternating pattern with a-actinin [19]. From a signaling perspective, actin filament formation occurs in response to activation of the Rho family of guanosine triphosphate hydrolases (GTPases) [20], with activated Rho family members (Rho A, Rho B, Rho C) being sufficient to drive actin filament formation [21, 22]. Rho family GTPases mediate this effect on actin filament formation via binding to and activation of two serine/threonine kinases: rho-associated coiled-coiled kinases 1 and 2 (ROCK1 and ROCK2) (Fig. 1). ROCK1 is essential for actin filament formation by the phosphorylation (via LIM kinase) and inactivation of the actin depolymerizing factor cofilin [23]. The ROCKs also exert their effects on cell contraction via phosphorylation and inactivation of myosin light chain phosphatase, thereby increasing myosin light chain phosphorylation, myosin-mediated contractility, and association with actin filaments [20, 24, 25]. Activation of non-muscle myosin and its association with actin filaments is essential for actin stress fiber assembly via its role in bundling existing actin filaments [20, 26].
Fig. 1. Signaling mechanisms controlling myofibroblast differentiation through actin and microtubule cytoskeletons. TGF-b promotes activation of Rho through Smad (SBE)-dependent expression of sphingosine kinase 1 (SphK1) producing GPCR ligand sphingosine-1-phosphate (S1P), through the expression of endothelin-1 (also acting through a specific GPCR), or through the expression of Rho guanine nucleotide exchange factor GEF-H1 that directly activates Rho. Rho can also be activated by other GPCR ligands (lysophosphatidic acid, LPA), or by stiff ECM or tension. Activation of Rho leads to filamentous F-actin formation by a dual mechanism involving: (i) ROCK/LIMK-dependent phosphorylation and inactivation of actin depolymerization factor, cofilin; and (ii) through mDia-promoted actin polymerization by profilin. Together, this results in a decrease in the amount of monomeric (globular) G-actin (that sequesters MRTF), release and translocation of MRTF to the nucleus, and activation of SRF-dependent transcription of CArG-dependent genes driving myofibroblast differentiation. Microtubules (MT) negatively regulate this pathway through multiple possible mechanisms, including sequestration of Smads, GEF-H1, or mDia2. Myofibroblast differentiation is also controlled by YAP/TAZ transcription factors acting through TEAD elements on the promoters of target genes. The Rho/F-actin pathway promotes the activity of YAP/TAZ through inhibition of the LATS-dependent phosphorylation of YAP/TAZ, leading to their release from sequestration by phosphoserine-binding protein 14-3-3, and their translocation to the nucleus to drive gene transcription. For abbreviations, see text.
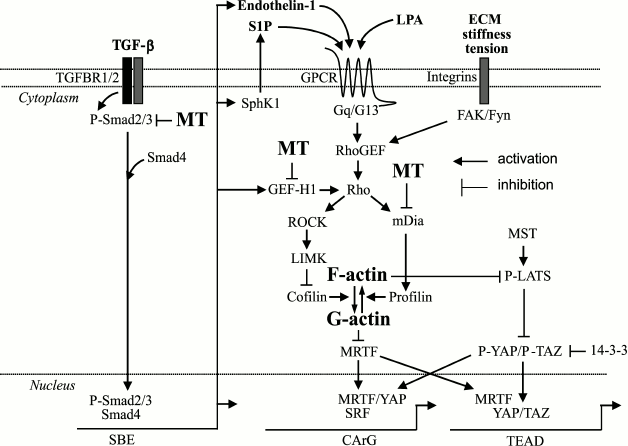
A second key set of Rho effectors for actin filament formation is the family of formin molecules (diaphanous-related formin 1, 2, and inverted formin-2 (mDIA1, mDIA2, and INF2) (Fig. 1). Diaphanous-related formins are autoinhibited by a C-terminal autoregulation domain. GTP-bound Rho displays the autoregulatory domain, exposing the formin homology FH2 domain that nucleates actin filaments [27, 28]. mDIA1 drives the formation of actin filaments from focal adhesion sites during stress fiber development [29]. These nascent filaments then can be “bundled” in a ROCK-dependent manner to form mature stress fibers [30].
Extracellular stimuli that are important in inducing actin stress fiber formation in the context of myofibroblast differentiation and tissue fibrosis include increased matrix stiffness and profibrotic molecules such as transforming growth factor-b (TGF-b), sphingosine-1 phosphate (S1P), lysophosphatidic acid (LPA), endothelin-1 (ET1), etc. (Fig. 1). Tension, which can be transmitted from the ECM to the cell, appears to play an important role in the formation of actin stress fibers. This is supported by the observation of actin microfilaments in granulation tissue, but not in many “normal” tissues [1]. Culture of fibroblasts on attached, non-contractile collagen results in the formation of actin stress fibers, which rapidly disassemble upon release of the collagen from attachment to the cell culture plate support [31, 32].
Transmission of tension occurs via focal adhesion associated integrins, such as b1-integrin [33]. On the cytoplasmic face, applying force to integrins induces RhoA activation [34]. This occurs via the tyrosine kinases Fyn and focal adhesion kinase (FAK) and subsequent downstream activation of the Rho-guanine exchange factors LARG and GEF-H1, respectively [35].
Similarly, TGF-b treatment results in the further induction of actin stress fibers in cultured fibroblasts on stiff matrices [36]. This effect is likely due to the activation of Rho GTPases by Rho-GEFs, whose expression and activation require Smad-mediated gene expression [37, 38]. Stimulation with TGF-b also results in the induction of the smooth muscle a-actin isoform, which augments force generation of actin stress fibers [39].
ROLE OF ACTIN STRESS FIBERS IN ECM REMODELING
Formation of filamentous actin and actin stress fibers plays a key role in the tissue remodeling function of myofibroblasts. It is essential for maturation of the focal adhesion, which in turn results in signal transfer to ECM and several adhesion-related kinases. As detailed above, focal adhesion can serve as nucleating sites for actin filaments and stress fibers. The actin stress fiber itself serves as a structural template for the recruitment of adhesion proteins. However, actomyosin contractility derived from the actin stress fibers also appears essential for focal adhesion maturation [20, 40]. The incorporation of smooth muscle a-actin into actin stress fibers is associated with increased actomyosin force generation and is essential for the maturation of enlarged “supermature” adhesions in myofibroblasts [41]. Although transmission of myosin-derived force to the bound molecules of the focal adhesion site appears essential for adhesion assembly, the exact mechanisms that account for this effect are not fully elucidated. One possibility is via the force-mediated unfolding of mechanosensitive elements of the adhesion complex. Possible candidates include talin, which upon direct force application unfolds to expose binding sites to the focal adhesion protein vinculin [42]. Alternatively, p130 Crk-associated substrate (p130Cas), which is a scaffolding protein at adhesion sites, contains a substrate domain that can be extended in response to force, rendering it more susceptible to phosphorylation by tyrosine kinases, such as Src, thereby promoting recruitment of adhesion molecules [43]. Formation of robust actin stress fibers and their associated mature adhesion complexes results in transmission of force from the cell into the surrounding ECM. Integrin heterodimers that nucleate adhesion complexes recognize RGD (arginine/glycine/aspartate) sites on ECM molecules such as fibronectin. Force transmission from myofibroblasts to the surrounding matrix is essential for contracting and closing healing wounds. The appearance of actin stress fibers in myofibroblasts of granulation tissue correlates with the contractile force generation by the tissue [39], and reorganization and contraction of collagen gels by fibroblasts is disrupted by treatment with inhibitors of actin filament formation [44, 45].
However, force transmission to individual ECM components can alter the remodeling function of fibroblasts. For example, fibronectin matrix is actively assembled by fibroblasts and myofibroblasts. Fibronectin fibrils are assembled by a stepwise cell-mediated process that proceeds by binding of a fibronectin dimer to the cell surface at its N-terminus, as well as a5b1 integrin-mediated binding to the fibronectin RGD domain [46, 47]. This process unfolds the fibronectin dimer, exposing fibronectin binding sites that were previously localized to the interior of the tertiary structure. Specialized a5b1-containing cell adhesions, called fibrillar adhesions, couple extracellular fibronectin with the actin cytoskeleton via tensin (TNS1), an integrin and actin binding protein [48]. Actomyosin mediated contraction results in stretching of the fibronectin molecule and exposure of additional cryptic fibronectin binding sites [49, 50]. This process can be regulated by alterations in Rho-dependent actomyosin contractility, such as via stimulation by lysophosphatidic acid [51]. As with focal adhesion maturation, fibronectin fibrillogenesis requires an intact actin cytoskeleton [52] and is regulated by ROCK 1/2 and myosin II [53]. Likewise, induction of a myofibroblast phenotype is associated with increased rates of fibronectin fibril assembly, due in part to the increased expression of smooth muscle a-actin [11]. This effect is possibly via increases in the amount of force transmission to the fibronectin molecule leading to increased unfolding.
REGULATION OF TRANSCRIPTION BY ACTIN DYNAMICS
Structural alterations in actin filament configuration play an important role in force transmission to the matrix and to facilitating increases in fibronectin matrix remodeling by myofibroblasts. In addition to these important and essential roles for tissue remodeling, actin cytoskeleton regulates gene transcription in response to profibrotic growth factors and increases in matrix stiffness.
Regulation of transcriptional activity by the actin cytoskeleton is accomplished via the localization of actin-interacting transcriptional coactivators. The first well characterized actin-regulated signaling pathway is through signaling leading to activation of serum response factor (SRF) – a transcription factor that is activated through signaling of Rho GTPases and actin filament dynamics (Fig. 1) [54, 55]. Subsequent studies identified an actin-interacting protein, myocardin-related transcription factor A (MRTF-A, also known as MAL and MKL1), as essential coactivators of SRF [56]. MRTF-A is localized in the cytosol bound to monomeric/globular (G) actin via N-terminal RPEL (arginine/proline/glutamate/leucine) motifs. Induction of Rho-dependent actin polymerization results in the dislocation of MRTF-A from monomeric actin and subsequent translocation to the nucleus, where it associates with and activates DNA-bound SRF. SRF binds to DNA at its consensus 10-bp binding sites, which have a CC(A/T)6GG consensus sequence termed the CArG-box. MRTF/SRF-target genes encode many cytoskeletal and contractile proteins [57-59].
Rho-dependent MRTF/SRF transcription can be activated by any extracellular signal that leads to actin polymerization. G-protein coupled receptor (GPCR) ligands such as lysophosphatidic acid and endothelin-1 were the first identified as extracellular stimuli of this pathway [54, 60, 61]. We have shown that the profibrotic cytokine TGF-b that is not directly coupled to GPCR also activates RhoA, MRTF/SRF, and actin polymerization and during myofibroblast differentiation [62, 63]. TGF-b canonically signals via receptor serine/threonine kinases to phosphorylate Smad proteins (Smad 2/3), leading to their heterotrimerization with Smad4, and translocation of the complex to the nucleus, where it binds to Smad-binding elements in the promoter region of target genes (Fig. 1). Through the Smad-dependent mechanism, TGF-b induces the expression of molecules that activate the Rho/actin/MRTF-A/SRF signaling pathway, such as GPCR agonists, Rho proteins, and Rho GEFs [37, 38, 64]. Furthermore, TGF-b increases the expression of MRTF-A and SRF itself [62, 63]. Thus, MRTF-A/SRF activation in response to TGF-b may augment Smad-dependent gene expression during myofibroblast differentiation by increasing the expression of SRF-dependent genes beyond levels induced by Smads alone. Furthermore, upregulation of components of this pathway may facilitate activation by other GPCR agonists. Knockout of MRTF-A abolishes expression of myofibroblast marker SM-a-actin (smooth muscle a-actin) in myofibroblasts and results in attenuated development of experimental cardiac and pulmonary fibrosis [65, 66], establishing this pathway as an important mediator of fibrotic diseases.
Rho-dependent MRTF-A activation also occurs in response to biomechanical signals, such as direct force application to integrins [67] or culturing fibroblasts on pathologically stiff ECM leading to the induction of a myofibroblast phenotype [68, 69]. The MRTF-A/SRF-mediated pathway has also been shown to promote expression of the antiapoptotic protein BCL2 during development of pulmonary fibrosis [70, 71]. Pharmacologic disruption of MRTF-A/SRF promotes myofibroblast apoptosis and ameliorates pulmonary fibrosis [72]. Together, these mechanisms suggest that the Rho/Actin/MRTF/SRF signaling axis is an important convergence point for multiple profibrotic signals that promote myofibroblast differentiation and tissue fibrosis.
Like MRTF/SRF, the Hippo/Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) pathway can be regulated by actin dynamics (Fig. 1). The mammalian Hippo pathway relies on regulated translocation of two related transcription factors, YAP and TAZ, from the cytosol into the nucleus [73]. YAP/TAZ are phosphorylated at serine motifs by LATS1/2 (Large Tumor Suppressor homolog) kinases, resulting in high affinity binding by cytosolic phosphoserine-binding protein 14-3-3 and sequestration in the cytosol. LATS1/2 are themselves phosphorylated by mammalian Ste20-like kinases (Mst1/2), which are orthologs of drosophila Hippo. Inhibition of LATS-mediated phosphorylation of YAP/TAZ results in loss of 14-3-3 sequestration and in nuclear translocation of YAP/TAZ. In the nucleus, YAP/TAZ bind to TEA domain (TEAD, named after TEF-1 and abaA)-containing transcription factors and regulate cell growth and proliferation [74]. YAP/TAZ can accumulate in the nucleus in response to extracellular matrix stiffness in a manner dependent on intact actin cytoskeleton [75]. YAP/TAZ localization is regulated by actin polymerization via inhibition of LATS1/2 [76, 77]. However, Rho itself may also regulate the localization of YAP/TAZ independently of LATS1/2 [75]. Translocation of YAP/TAZ is essential for expression of profibrotic genes in fibroblasts cultured on a stiff matrix, while overexpression of YAP and TAZ conveyed a profibrotic phenotype [78]. Finally, YAP/TAZ and MRTF-A may interact with each other in the nucleus to mediate transcriptional regulation [79-81].
CONTROL OF MYOFIBROBLAST DIFFERENTIATION BY MICROTUBULES
Microtubules comprise an equally important structural component of all cells, providing the motile force for chromosomal separation during mitosis, but also playing essential roles in organelle positioning, migration, the formation of cilia, and resistance to cell deformation. Microtubules are filaments formed from a and b tubulin subunits that form heterodimers. These a/b heterodimers assemble into the cylindrical structure that comprises the microtubule [82]. The microtubule filament is polarized with a faster growing “plus” end where most of the elongation of the filament occurs, governed by hydrolysis of b-tubulin-bound GTP [83]. Microtubules demonstrate dynamic instability, where periods of filament elongation alternate with sudden depolymerization events. In the context of cell–matrix interactions, microtubule polymerization state can be modified by substrate strain [84], extracellular matrix protein density [85], and matrix stiffness [86], indicating that microtubules are a mechanosensitive structure.
In addition to “sensing” extracellular biomechanical cues, microtubule polymerization state can lead to “inside-out” signals that can promote ECM remodeling by fibroblasts. Disruption of microtubule polymers induces actin filament formation, thereby increasing contractile force generation [87]. Coupling to the actin cytoskeleton occurs either mechanically (the tensegrity model) [88] or biochemically via the microtubule-associated nucleotide exchange factor, p190RhoGEF (GEF-H1), which activates Rho-mediated actin polymerization [89] or direct actin filament nucleation by mDIA1 and mDIA2 [90-93] (Fig. 1). Microtubule-mediated actin filament assembly leads to rapid assembly of fibronectin fibrils into extracellular matrix [51], along with the induction of the profibrotic genes plasminogen activator inhibitor-1 (PAI-1) and connective tissue growth factor (CTGF) [94, 95].
From a signaling standpoint, a few studies have implicated microtubules in regulation of Smad2/3 phosphorylation and cytosolic localization via binding to b-tubulin in mink lung epithelial cells, cardiac myocytes, and C2C12 myoblasts [96, 97]. These reports showed that disruption of microtubules by nocodazole or colchicine resulted in the induction of both basal and TGF-b-induced Smad-phosphorylation and Smad-dependent gene transcription. Mechanistically, these studies propose a model wherein Smad2/3/4 bind microtubules; and TGF-b triggers dissociation of Smads from microtubules, allowing phosphorylation and nuclear translocation of Smads, with consequent activation of Smad-dependent gene transcription. Thus, these studies suggest that microtubules may serve as a “cytoplasmic sequestering network for Smads, controlling Smad2 association with and phosphorylation by activated TGF beta receptor I” [97]. However, it is not clear from this model if and how TGF-b disrupts microtubules, and how the disruption of microtubules by nocodazole drives Smad2 phosphorylation and Smad-dependent gene transcription on its own without TGF-b treatment [97].
The model proposed above may suggest that microtubule dynamics would regulate myofibroblast differentiation. Indeed, we showed that in human lung fibroblasts, stabilization of microtubules by taxol abolished TGF-b-induced myofibroblast differentiation [93]. However, according to our data, the mechanism is different from what was initially proposed in studies using lung epithelial cells, cardiac myocytes, and C2C12 myoblasts [96, 97]. In our studies on human lung fibroblasts, TGF-b had no effect on a gross microtubule polymerization state; disruption of microtubules by colchicine drove the expression of myofibroblast marker SM-a-actin on its own without affecting phosphorylation or nuclear translocation of Smad2/3; stabilization of microtubules by taxol blocked the effect of TGF-b on these processes without affecting TGF-b-induced Smad phosphorylation, Smad nuclear translocation, or Smad-dependent gene transcription [93].
Our data on human lung fibroblasts suggest that microtubules may control TGF-b-induced myofibroblast differentiation at the level of actin stress fiber formation [93]. One proposed mechanism could be through the Rho guanine exchange factor GEF-H1 (activator of Rho) that binds to and is sequestered by microtubules [89]. It was shown that TGF-b promotes the expression of GEF-H1 in retinal pigment epithelium cells, where GEF-H1 may mediate TGF-b-induced expression of SM-a-actin in these cells [95]. In our experiments, TGF-b also promoted the expression of GEF-H1 in human lung fibroblasts; however, the knockdown of GEF-H1 in these cells by the siRNA (small interfering RNA) approach had no effect on TGF-b-induced myofibroblast differentiation (our unpublished data). Alternatively, we found that microtubules may control actin stress fiber formation, SRF activation, and myofibroblast differentiation through sequestration of mDIA2 [93], which is critical for actin stress fiber formation [29].
CONTROL OF COLLAGEN SYNTHESIS AND DEPOSITION BY INTERMEDIATE
FILAMENTS
Intermediate filaments are fibers with a diameter of ~10 nm made of various proteins encoded by at least 65 genes [98, 99]. The major cellular function of intermediate filaments is to provide mechanical strength and resistance to share stress. Mammalian cells contain two principally different intermediate filament systems: nuclear and cytoplasmic. The nuclear intermediate filaments are built from lamins, which together with multiple other proteins constitute nuclear lamina that supports nuclear structure and assembly of various nuclear protein complexes [100]. The cytoplasmic intermediate filaments are mainly responsible for stabilizing the cell shape as demonstrated by selective disruption of individual intermediate filaments [101]. Based on sequence homology, intermediate filament proteins are grouped into six types including (i) acidic keratins, (ii) basic keratins, (iii) vimentin, desmin, glial fibrillary acidic protein (GFAP), and peripherin, (iv) the neurofilament triplet proteins, a-internexin and nestin, (v) the lamins, and (vi) the lens-specific proteins, beaded filament structural proteins (Bfsp1 and Bfsp2) [102].
Vimentin is the most abundant intermediate filament protein and is a major building block of cytoplasmic intermediate filaments in mesenchymal cells including fibroblasts. Surprisingly, deletion of the vimentin gene in mice resulted in no obvious defects in the development, reproduction, and structure/function of organs [103]. However, several differences between the wild type and vimentin-knockout mice were revealed under stress conditions, including impaired wound healing [104] and reduced colitis in the murine model of disease [105]. Furthermore, recent study has shown that vimentin knockout results in impaired lung injury and fibrosis in lipopolysaccharide (LPS) and bleomycin mouse models [106]. It was shown that vimentin is required for activation of the inflammasome – a multiprotein complex that mediates early inflammatory responses of innate immune cells. Chimeric mice with bone marrow lacking vimentin have attenuated lung injury and fibrosis following bleomycin exposure [106].
In cultured cells, it was shown that primary fibroblasts derived from vimentin-deficient mouse embryos displayed reduced stiffness, mechanical stability, contractility, motility, and directional migration towards fibronectin, platelet-derived growth factor, or conditioned medium collected from wild-type monolayer cultures [107]. This was associated with disturbed spatial organization of focal contact proteins and of actin microfilament organization [107]. Given the latter and the established role of actin filaments in control of SRF and Hippo pathways described above, it would be interesting to examine whether vimentin knockout affects these pathways and myofibroblast differentiation in this system.
Even though the effect of vimentin knockout on myofibroblast differentiation has not been evaluated, it was shown that vimentin deficiency leads to impaired synthesis of collagen-1 in fibroblasts, but through a unique mechanism [108] (Fig. 2). It is thought that stabilization of collagen mRNAs is an important mechanism for collagen synthesis [109-111]. The 5¢-untranslated region (UTR) of Col1A1 and Col1A2 mRNAs contain a “stem-loop” sequence (5¢SL) that controls the stability and translation of their mRNAs through LARP6 (La ribonucleoprotein domain family member 6) that binds 5¢SL with high affinity [112]. It appears that Col1A1 and Col1A2 mRNAs interact with vimentin filaments (but not with soluble vimentin) in a 5¢SL/LARP6-dependent manner, resulting in stabilization of their mRNA [108]. Knockout of vimentin or disruption of intermediate filaments by iminodipropionitrile (IDPN) results in a dramatic reduction in collagen mRNA half-life and in Col1A1 and Col1A2 protein levels [108].
Fig. 2. Control of collagen synthesis by vimentin intermediate filaments. Col1A1 and Col1A2 mRNAs interact with and are stabilized by LARP6, which also interacts with vimentin intermediate filaments (IF), and this interaction is required for the mRNA-stabilizing function of LARP6. Disruption of IF by IDPN or by WF-A promotes degradation of Col1A1 and Col1A2 mRNAs. WF-A also inhibits TGF-b-induced transcription of the Col1A1 and Col1A2 genes through inhibition of TGFBR1 phosphorylation; however, the role of vimentin IF in the latter mechanism has not been explored. For abbreviations, see text.
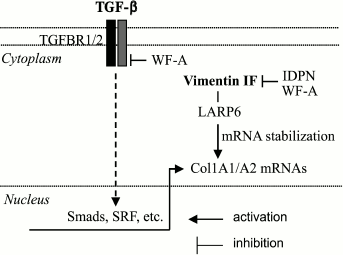
The follow-up study [113] used Withaferin-A (WF-A) from Withania somnifera – a drug that binds vimentin and disrupts vimentin filament network by covalent modification of a conserved cysteine on vimentin [114]. Disruption of vimentin filaments by WF-A in human fibroblasts was associated with accelerated decay of Col1A1 and Col1A2 mRNA and decreased levels of their mRNA and protein (Fig. 2). These effects of WF-A were also reproduced on rat hepatic stellate cells and cardiac fibroblasts. WF-A also inhibited TGF-b-induced expression of collagen-1. Interestingly, signaling studies revealed an additional mechanism of WF-A action through inhibition of TGF-b-induced phosphorylation of TGF-b-receptor-1 and of Smad3, and decreased transcription of Col1A2 at promoter level in human lung fibroblasts. Furthermore, WF-A inhibited isoproterenol-induced myocardial fibrosis in vivo [113]. However, it remains to be determined if inhibition of TGF-b signaling by WF-A is mediated by disruption of vimentin filaments.
Finally, an additional mechanism for control of collagen-1 secretion by LARP6 has been recently described. It appears that LARP6 is phosphorylated by AKT protein kinase at serine-451, and this phosphorylation is important for the rate of collagen-1 secretion, having no role in a control of collagen-1 mRNA levels [115]. Again, however, the role of vimentin filaments in LARP6 phosphorylation and/or control of collagen secretion by LARP6 has not been investigated.
CONCLUSIONS
The cell cytoskeleton, comprised of actin fibers, microtubules, and intermediate filaments, has traditionally been viewed as key to cellular structure and organization. As an example, modified pro-fibrotic (myo)fibroblasts were originally morphologically characterized by prominent actin stress fiber cytoskeleton. Later studies determined the dynamic nature of each of these structural elements, and most surprisingly, led to the discovery of their role in mediating cell–matrix interactions and the regulation of cell signaling pathways and cell fate determination. Thus, these components define the epitome of structure–function relationships for cells, especially regarding (myo)fibroblast responses to extracellular growth factors and biomechanical matrix cues, as well as the formation and remodeling of nascent matrix and granulation tissue during wound healing and fibrosis. Additional studies should be performed to develop tissue-specific approaches for therapeutic intervention in wound healing and fibrosis-mediated disorders.
Acknowledgements
This work was supported by American Thoracic Society/Pulmonary Fibrosis Foundation/Coalition for Pulmonary Fibrosis Research Award (N. S.), University of Wisconsin Graduate School Research Funding (N. S.), by the National Institutes of Health Awards HL126190 (N. S.), R56HL127395 (N. O. D), and by the National Center for Advancing Translational Sciences of the National Institutes of Health Award UL1TR000430 (N. O. D). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This publication was also supported by Dr. Ralph and Marian Falk Medical Research Catalyst Award (N. O. D.), the Russian Foundation for Fundamental Research (15-04-00101) (S. N. O.), and the Russian Scientific Foundation (14-15-0006 and 16-15-10026) (S. N. O.).
REFERENCES
1.Gabbiani, G., Ryan, G. B., and Majne, G. (1971)
Presence of modified fibroblasts in granulation tissue and their
possible role in wound contraction, Experientia, 27,
549-550.
2.Majno, G., Gabbiani, G., Hirschel, B. J., Ryan, G.
B., and Statkov, P. R. (1971) Contraction of granulation tissue in
vitro: similarity to smooth muscle, Science, 173,
548-550.
3.Ryan, G. B., Cliff, W. J., Gabbiani, G., Irle, C.,
Montandon, D., Statkov, P. R., and Majno, G. (1974) Myofibroblasts in
human granulation tissue, Hum. Pathol., 5,
55-67.
4.Horiuchi, K., Amizuka, N., Takeshita, S.,
Takamatsu, H., Katsuura, M., Ozawa, H., Toyama, Y., Bonewald, L. F.,
and Kudo, A. (1999) Identification and characterization of a novel
protein, periostin, with restricted expression to periosteum and
periodontal ligament and increased expression by transforming growth
factor beta, J. Bone Miner. Res., 14, 1239-1249.
5.Elliott, C. G., Wang, J., Guo, X., Xu, S. W.,
Eastwood, M., Guan, J., Leask, A., Conway, S. J., and Hamilton, D. W.
(2012) Periostin modulates myofibroblast differentiation during
full-thickness cutaneous wound repair, J. Cell Sci., 125,
121-132.
6.Roberts, C. J., Birkenmeier, T. M., McQuillan, J.
J., Akiyama, S. K., Yamada, S. S., Chen, W. T., Yamada, K. M., and
McDonald, J. A. (1988) Transforming growth factor beta stimulates the
expression of fibronectin and of both subunits of the human fibronectin
receptor by cultured human lung fibroblasts, J. Biol. Chem.,
263, 4586-4592.
7.Serini, G., Bochaton-Piallat, M.-L., Ropraz, P.,
Geinoz, A., Borsi, L., Zardi, L., and Gabbiani, G. (1998) The
fibronectin domain ED-A is crucial for myofibroblastic phenotype
induction by transforming growth factor-b1, J. Cell Biol.,
142, 873-881.
8.Roberts, A. B., Sporn, M. B., Assoian, R. K.,
Smith, J. M., Roche, N. S., Wakefieldm, L. M., Heine, U. I., Liotta, L.
A., Falanga, V., Kehrl, J. H., and Fauci, A. S. (1986) Transforming
growth factor type beta: rapid induction of fibrosis and angiogenesis
in vivo and stimulation of collagen formation in vitro,
Proc. Natl. Acad. Sci. USA, 83, 4167-4171.
9.Malmstrom, J., Lindberg, H., Lindberg, C., Bratt,
C., Wieslander, E., Delander, E. L., Sarnstrand, B., Burns, J. S.,
Mose-Larsen, P., Fey, S., and Marko-Varga, G. (2004) Transforming
growth factor-beta 1 specifically induce proteins involved in the
myofibroblast contractile apparatus, Mol. Cell Proteomics,
3, 466-477.
10.Powell, D. W., Mifflin, R. C., Valentich, J. D.,
Crowe, S. E., Saada, J. I., and West, A. B. (1999) Myofibroblasts. I.
Paracrine cells important in health and disease, Am. J.
Physiol., 277, C1-C19.
11.Torr, E. E., Ngam, C. R., Bernau, K.,
Tomasini-Johansson, B., Acton, B., and Sandbo, N. (2015) Myofibroblasts
exhibit enhanced fibronectin assembly that is intrinsic to their
contractile phenotype, J. Biol. Chem., 290,
6951-6961.
12.Zhang, H. Y., and Phan, S. H. (1999) Inhibition
of myofibroblast apoptosis by transforming growth factor beta(1),
Am. J. Resp. Cell Mol. Biol., 21, 658-665.
13.Desmouliere, A., Chaponnier, C., and Gabbiani, G.
(2005) Tissue repair, contraction, and the myofibroblast, Wound Rep.
Regen., 13, 7-12.
14.Perrin, B. J., and Ervasti, J. M. (2010) The
actin gene family: function follows isoform, Cytoskeleton,
67, 630-634.
15.Goldman, R. D., Lazarides, E., Pollack, R., and
Weber, K. (1975) The distribution of actin in non-muscle cells. The use
of actin antibody in the localization of actin within the microfilament
bundles of mouse 3T3 cells, Exp. Cell Res., 90,
333-344.
16.Kreis, T. E., Winterhalter, K. H., and
Birchmeier, W. (1979) In vivo distribution and turnover of
fluorescently labeled actin microinjected into human fibroblasts,
Proc. Natl. Acad. Sci. USA, 76, 3814-3818.
17.Lazarides, E. (1975) Immunofluorescence studies
on the structure of actin filaments in tissue culture cells, J.
Histochem. Cytochem., 23, 507-528.
18.Lazarides, E., and Burridge, K. (1975)
Alpha-actinin: immunofluorescent localization of a muscle structural
protein in nonmuscle cells, Cell, 6, 289-298.
19.Weber, K., and Groeschel-Stewart, U. (1974)
Antibody to myosin: the specific visualization of myosin-containing
filaments in nonmuscle cells, Proc. Natl. Acad. Sci. USA,
71, 4561-4564.
20.Chrzanowska-Wodnicka, M., and Burridge, K. (1996)
Rho-stimulated contractility drives the formation of stress fibers and
focal adhesions, J. Cell Biol., 133, 1403-1415.
21.Paterson, H. F., Self, A. J., Garrett, M. D.,
Just, I., Aktories, K., and Hall, A. (1990) Microinjection of
recombinant p21rho induces rapid changes in cell morphology, J. Cell
Biol., 111, 1001-1007.
22.Giry, M., Popoff, M. R., Von Eichel-Streiber, C.,
and Boquet, P. (1995) Transient expression of RhoA, -B, and -C GTPases
in HeLa cells potentiates resistance to Clostridium difficile
toxins A and B but not to Clostridium sordellii lethal toxin,
Infect. Immun., 63, 4063-4071.
23.Ohashi, K., Nagata, K., Maekawa, M., Ishizaki,
T., Narumiya, S., and Mizuno, K. (2000) Rho-associated kinase ROCK
activates LIM-kinase 1 by phosphorylation at threonine 508 within the
activation loop, J. Biol. Chem., 275, 3577-3582.
24.Kimura, K., Ito, M., Amano, M., Chihara, K.,
Fukata, Y., Nakafuku, M., Yamamori, B., Feng, J., Nakano, T., Okawa,
K., Iwamatsu, A., and Kaibuchi, K. (1996) Regulation of myosin
phosphatase by Rho and Rho-associated kinase (Rho-kinase),
Science, 273, 245-248.
25.Yoneda, A., Multhauptm, H. A., and Couchman, J.
R. (2005) The Rho kinases I and II regulate different aspects of myosin
II activity, J. Cell Biol., 170, 443-453.
26.Lamb, N. J., Fernandez, A., Conti, M. A.,
Adelstein, R., Glass, D. B., Welch, W. J., and Feramisco, J. R. (1988)
Regulation of actin microfilament integrity in living nonmuscle cells
by the cAMP-dependent protein kinase and the myosin light chain kinase,
J. Cell Biol., 106, 1955-1971.
27.Alberts, A. S. (2001) Identification of a
carboxyl-terminal diaphanous-related formin homology protein
autoregulatory domain, J. Biol. Chem., 276,
2824-2830.
28.Zigmond, S. H. (2004) Formin-induced nucleation
of actin filaments, Curr. Opin. Cell Biol., 16,
99-105.
29.Hotulainen, P., and Lappalainen, P. (2006) Stress
fibers are generated by two distinct actin assembly mechanisms in
motile cells, J. Cell Biol., 173, 383-394.
30.Watanabe, N., Kato, T., Fujitam, A., Ishizakim,
T., and Narumiya, S. (1999) Cooperation between mDia1 and ROCK in
Rho-induced actin reorganization, Nat. Cell Biol., 1,
136-143.
31.Farsi, J. M., and Aubin, J. E. (1984)
Microfilament rearrangements during fibroblast-induced contraction of
three-dimensional hydrated collagen gels, Cell Motil., 4,
29-40.
32.Mochitate, K., Pawelek, P., and Grinnell, F.
(1991) Stress relaxation of contracted collagen gels: disruption of
actin filament bundles, release of cell surface fibronectin, and
down-regulation of DNA and protein synthesis, Exp. Cell Res.,
193, 198-207.
33.Arora, P. D., Narani, N., and McCulloch, C. A.
(1999) The compliance of collagen gels regulates transforming growth
factor-beta induction of alpha-smooth muscle actin in fibroblasts,
Am. J. Pathol., 154, 871-882.
34.Huveneers, S., and Danen, E. H. J. (2009)
Adhesion signaling – crosstalk between integrins, Src and
Rho, J. Cell Sci., 122, 1059-1069.
35.Guilluy, C., Swaminathan, V., Garcia-Mata, R.,
O’Brien, E. T., Superfine, R., and Burridge, K. (2011) The Rho
GEFs LARG and GEF-H1 regulate the mechanical response to force on
integrins, Nat. Cell Biol., 13, 722-727.
36.Desmouliere, A., Geinoz, A., Gabbiani, F., and
Gabbiani, G. (1993) Transforming growth factor-beta 1 induces
alpha-smooth muscle actin expression in granulation tissue
myofibroblasts and in quiescent and growing cultured fibroblasts, J.
Cell Biol., 122, 103-111.
37.Shen, X., Li, J., Hu, P. P., Waddell, D., Zhang,
J., and Wang, X. F. (2001) The activity of guanine exchange factor NET1
is essential for transforming growth factor-beta-mediated stress fiber
formation, J. Biol. Chem., 276, 15362-15368.
38.Tsapara, A., Luthert, P., Greenwood, J., Hill, C.
S., Matter, K., and Balda, M. S. (2010) The RhoA activator GEF-H1/Lfc
is a transforming growth factor-beta target gene and effector that
regulates alpha-smooth muscle actin expression and cell migration,
Mol. Biol. Cell, 21, 860-870.
39.Hinz, B., Mastrangelo, D., Iselin, C. E.,
Chaponnier, C., and Gabbiani, G. (2001) Mechanical tension controls
granulation tissue contractile activity and myofibroblast
differentiation, Am. J. Pathol., 159, 1009-1020.
40.Delanoe-Ayari, H., Al Kurdi, R., Vallade, M.,
Gulino-Debrac, D., and Riveline, D. (2004) Membrane and actomyosin
tension promote clustering of adhesion proteins, Proc. Natl. Acad.
Sci. USA, 101, 2229-2234.
41.Hinz, B., Dugina, V., Ballestrem, C.,
Wehrle-Haller, B., and Chaponnier, C. (2003) Alpha-smooth muscle actin
is crucial for focal adhesion maturation in myofibroblasts, Mol.
Biol. Cell, 14, 2508-2519.
42.Del Rio, A., Perez-Jimenez, R., Liu, R.,
Roca-Cusachs, P., Fernandez, J. M., and Sheetz, M. P. (2009) Stretching
single talin rod molecules activates vinculin binding, Science,
323, 638-641.
43.Sawada, Y., Tamada, M., Dubin-Thaler, B. J.,
Cherniavskaya, O., Sakai, R., Tanaka, S., and Sheetz, M. P. (2006)
Force sensing by mechanical extension of the Src family kinase
substrate p130Cas, Cell, 127, 1015-1026.
44.Bell, E., Ivarsson, B., and Merrill, C. (1979)
Production of a tissue-like structure by contraction of collagen
lattices by human fibroblasts of different proliferative potential
in vitro, Proc. Natl. Acad. Sci. USA, 76,
1274-1278.
45.Kolodney, M. S., and Wysolmerski, R. B. (1992)
Isometric contraction by fibroblasts and endothelial cells in tissue
culture: a quantitative study, J. Cell Biol., 117,
73-82.
46.Schwarzbauer, J. E., and De Simone, D. W. (2011)
Fibronectins, their fibrillogenesis, and in vivo functions,
Cold Spring Harb. Perspect. Biol., 3.
47.Tomasini-Johansson, B. R., Annis, D. S., and
Mosher, D. F. (2006) The N-terminal 70-kDa fragment of fibronectin
binds to cell surface fibronectin assembly sites in the absence of
intact fibronectin, Matrix. Biol., 25, 282-293.
48.Zamir, E., Katz, B. Z., Aota, S., Yamada, K. M.,
Geiger, B., and Kam, Z. (1999) Molecular diversity of cell–matrix
adhesions, J. Cell Sci., 112 (Pt. 11), 1655-1669.
49.Baneyx, G., Baugh, L., and Vogel, V. (2002)
Fibronectin extension and unfolding within cell matrix fibrils
controlled by cytoskeletal tension, Proc. Natl. Acad. Sci. USA,
99, 5139-5143.
50.Zhong, C., Chrzanowska-Wodnicka, M., Brown, J.,
Shaub, A., Belkin, A. M., and Burridge, K. (1998) Rho-mediated
contractility exposes a cryptic site in fibronectin and induces
fibronectin matrix assembly, J. Cell Biol., 141,
539-551.
51.Zhang, Q., Magnusson, M. K., and Mosher, D. F.
(1997) Lysophosphatidic acid and microtubule-destabilizing agents
stimulate fibronectin matrix assembly through Rho-dependent actin
stress fiber formation and cell contraction, Mol. Biol. Cell,
8, 1415-1425.
52.Wu, C., Keivens, V. M., O’Toole, T. E.,
McDonald, J. A., and Ginsberg, M. H. (1995) Integrin activation and
cytoskeletal interaction are essential for the assembly of a
fibronectin matrix, Cell, 83, 715-724.
53.Yoneda, A., Ushakov, D., Multhaupt, H. A., and
Couchman, J. R. (2007) Fibronectin matrix assembly requires distinct
contributions from Rho kinases I and -II, Mol. Biol. Cell,
18, 66-75.
54.Hill, C. S., Wynne, J., and Treisman, R. (1995)
The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional
activation by SRF, Cell, 81, 1159-1170.
55.Sotiropoulos, A., Gineitis, D., Copeland, J., and
Treisman, R. (1999) Signal-regulated activation of serum response
factor is mediated by changes in actin dynamics, Cell,
98, 159-169.
56.Miralles, F., Posern, G., Zaromytidou, A. I., and
Treisman, R. (2003) Actin dynamics control SRF activity by regulation
of its coactivator MAL, Cell, 113, 329-342.
57.Miano, J. M. (2003) Serum response factor:
toggling between disparate programs of gene expression, J. Mol.
Cell. Cardiol., 35, 577-593.
58.Sun, Q., Chen, G., Streb, J. W., Long, X., Yang,
Y., Stoeckert, C. J., Jr., and Miano, J. M. (2006) Defining the
mammalian CArGome, Genome Res., 16, 197-207.
59.Esnault, C., Stewart, A., Gualdrini, F., East,
P., Horswell, S., Matthews, N., and Treisman, R. (2014) Rho-actin
signaling to the MRTF coactivators dominates the immediate
transcriptional response to serum in fibroblasts, Genes Dev.,
28, 943-958.
60.Mao, J., Yuan, H., Xie, W., Simon, M. I., and Wu,
D. (1998) Specific involvement of G proteins in regulation of serum
response factor-mediated gene transcription by different receptors,
J. Biol. Chem., 273, 27118-27123.
61.Gohla, A., Offermanns, S., Wilkie, T. M., and
Schultz, G. (1999) Differential involvement of Galpha12 and Galpha13 in
receptor-mediated stress fiber formation, J. Biol. Chem.,
274, 17901-17907.
62.Sandbo, N., Kregel, S., Taurin, S., Bhorade, S.,
and Dulin, N. O. (2009) Critical role of serum response factor in
pulmonary myofibroblast differentiation induced by TGF-beta, Am. J.
Respir. Cell Mol. Biol., 41, 332-338.
63.Sandbo, N., Lau, A., Kach, J., Ngam, C., Yau, D.,
and Dulin, N. O. (2011) Delayed stress fiber formation mediates
pulmonary myofibroblast differentiation in response to TGF-beta, Am.
J. Physiol., 301, L656-L666.
64.Cencetti, F., Bernacchioni, C., Nincheri, P.,
Donati, C., and Bruni, P. (2010) Transforming growth factor-beta1
induces transdifferentiation of myoblasts into myofibroblasts via
up-regulation of sphingosine kinase-1/S1P3 axis, Mol. Biol.
Cell, 21, 1111-1124.
65.Small, E. M., Thatcher, J. E., Sutherland, L. B.,
Kinoshita, H., Gerard, R. D., Richardson, J. A., Dimaio, J. M., Sadek,
H., Kuwahara, K., and Olson, E. N. (2010) Myocardin-related
transcription factor-a controls myofibroblast activation and fibrosis
in response to myocardial infarction, Circ. Res., 107,
294-304.
66.Bernau, K., Ngam, C., Torr, E. E., Acton, B.,
Kach, J., Dulin, N. O., and Sandbo, N. (2015) Megakaryoblastic
leukemia-1 is required for the development of bleomycin-induced
pulmonary fibrosis, Respir. Res., 16, 45.
67.Zhao, X. H., Laschinger, C., Arora, P., Szaszi,
K., Kapus, A., and McCulloch, C. A. (2007) Force activates smooth
muscle alpha-actin promoter activity through the Rho signaling pathway,
J. Cell Sci., 120, 1801-1809.
68.Liu, F., Mih, J. D., Shea, B. S., Kho, A. T.,
Sharif, A. S., Tager, A. M., and Tschumperlin, D. J. (2010) Feedback
amplification of fibrosis through matrix stiffening and COX-2
suppression, J. Cell Biol., 190, 693-706.
69.Huang, X., Yang, N., Fiore, V. F., Barker, T. H.,
Sun, Y., Morris, S. W., Ding, Q., Thannickal, V. J., and Zhou, Y.
(2012) Matrix stiffness-induced myofibroblast differentiation is
mediated by intrinsic mechanotransduction, Am. J. Respir. Cell Mol.
Biol., 47, 340-348.
70.Schratt, G., Philippar, U., Hockemeyer, D.,
Schwarz, H., Alberti, S., and Nordheim, A. (2004) SRF regulates Bcl-2
expression and promotes cell survival during murine embryonic
development, EMBO J., 23, 1834-1844.
71.Zhou, Y., Huang, X., Hecker, L., Kurundkar, D.,
Kurundkar, A., Liu, H., Jin, T. H., Desai, L., Bernard, K., and
Thannickal, V. J. (2013) Inhibition of mechanosensitive signaling in
myofibroblasts ameliorates experimental pulmonary fibrosis, J. Clin.
Invest., 123, 1096-1108.
72.Sisson, T. H., Ajayi, I. O., Subbotina, N., Dodi,
A. E., Rodansky, E. S., Chibucos, L. N., Kim, K. K., Keshamouni, V. G.,
White, E. S., Zhou, Y., Higgins, P. D., Larsen, S. D., Neubig, R. R.,
and Horowitz, J. C. (2015) Inhibition of myocardin-related
transcription factor/serum response factor signaling decreases lung
fibrosis and promotes mesenchymal cell apoptosis, Am. J.
Pathol., 185, 969-986.
73.Low, B. C., Pan, C. Q., Shivashankar, G. V.,
Bershadsky, A., Sudol, M., and Sheetz, M. (2014) YAP/TAZ as
mechanosensors and mechanotransducers in regulating organ size and
tumor growth, FEBS Lett., 588, 2663-2670.
74.Vassilev, A., Kaneko, K. J., Shu, H., Zhao, Y.,
and De Pamphilis, M. L. (2001) TEAD/TEF transcription factors utilize
the activation domain of YAP65, a Src/Yes-associated protein localized
in the cytoplasm, Genes Dev., 15, 1229-1241.
75.Dupont, S., Morsut, L., Aragona, M., Enzo, E.,
Giulitti, S., Cordenonsi, M., Zanconato, F., Le Digabel, J., Forcato,
M., Bicciato, S., Elvassore, N., and Piccolo, S. (2011) Role of YAP/TAZ
in mechanotransduction, Nature, 474, 179-183.
76.Sansores-Garcia, L., Bossuyt, W., Wada, K.,
Yonemura, S., Tao, C., Sasaki, H., and Halder, G. (2011) Modulating
F-actin organization induces organ growth by affecting the Hippo
pathway, EMBO J., 30, 2325-2335.
77.Wada, K., Itoga, K., Okano, T., Yonemura, S., and
Sasaki, H. (2011) Hippo pathway regulation by cell morphology and
stress fibers, Development, 138, 3907-3914.
78.Liu, F., Lagares, D., Choi, K. M., Stopfer, L.,
Marinkovic, A., Vrbanac, V., Probst, C. K., Hiemer, S. E., Sisson, T.
H., Horowitz, J. C., Rosas, I. O., Fredenburgh, L. E.,
Feghali-Bostwick, C., Varelas, X., Tager, A. M., and Tschumperlin, D.
J. (2015) Mechanosignaling through YAP and TAZ drives fibroblast
activation and fibrosis, Am. J. Physiol., 308,
L344-L357.
79.Yu, O. M., Miyamoto, S., and Brown, J. H. (2015)
Myocardin-related transcription factor A and Yes-associated protein
exert dual control in G protein-coupled receptor- and RhoA-mediated
transcriptional regulation and cell proliferation, Mol. Cell.
Biol., 36, 39-49.
80.Liu, C. Y., Chan, S. W., Guo, F., Toloczko, A.,
Cui, L., and Hong, W. (2016) MRTF/SRF dependent transcriptional
regulation of TAZ in breast cancer cells, Oncotarget, 7,
13706-13716.
81.Speight, P., Kofler, M., Szaszi, K., and Kapus,
A. (2016) Context-dependent switch in chemo/mechanotransduction via
multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and
TGFbeta-regulated Smad3, Nat. Commun., 7, 11642.
82.Desai, A., and Mitchison, T. J. (1997)
Microtubule polymerization dynamics, Annu. Rev. Cell Dev. Biol.,
13, 83-117.
83.Weisenberg, R. C., Deery, W. J., and Dickinson,
P. J. (1976) Tubulin–nucleotide interactions during the
polymerization and depolymerization of microtubules,
Biochemistry, 15, 4248-4254.
84.Putnam, A. J., Schultz, K., and Mooney, D. J.
(2001) Control of microtubule assembly by extracellular matrix and
externally applied strain, Am. J. Physiol., 280,
C556-564.
85.Mooney, D. J., Hansen, L. K., Langer, R.,
Vacanti, J. P., and Ingber, D. E. (1994) Extracellular matrix controls
tubulin monomer levels in hepatocytes by regulating protein turnover,
Mol. Biol. Cell, 5, 1281-1288.
86.Heck, J. N., Ponik, S. M., Garcia-Mendoza, M. G.,
Pehlke, C. A., Inman, D. R., Eliceiri, K. W., and Keelym, P. J. (2012)
Microtubules regulate GEF-H1 in response to extracellular matrix
stiffness, Mol. Biol. Cell, 23, 2583-2592.
87.Danowski, B. A. (1989) Fibroblast contractility
and actin organization are stimulated by microtubule inhibitors, J.
Cell Sci., 93 (Pt. 2), 255-266.
88.Ingber, D. E., and Tensegrity, I. (2003) Cell
structure and hierarchical systems biology, J. Cell Sci.,
116, 1157-1173.
89.Krendel, M., Zenke, F. T., and Bokoch, G. M.
(2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between
microtubules and the actin cytoskeleton, Nat. Cell Biol.,
4, 294-301.
90.Bartolini, F., Moseley, J. B., Schmoranzer, J.,
Cassimeris, L., Goode, B. L., and Gundersen, G. G. (2008) The formin
mDia2 stabilizes microtubules independently of its actin nucleation
activity, J. Cell Biol., 181, 523-536.
91.Gaillard, J., Ramabhadran, V., Neumanne, E.,
Gurel, P., Blanchoin, L., Vantard, M., and Higgs, H. N. (2011)
Differential interactions of the formins INF2, mDia1, and mDia2 with
microtubules, Mol. Biol. Cell, 22, 4575-4587.
92.Goode, B. L., and Eck, M. J. (2007) Mechanism and
function of formins in the control of actin assembly, Annu. Rev.
Biochem., 76, 593-627.
93.Sandbo, N., Ngam, C., Torr, E., Kregel, S., Kach,
J., and Dulin, N. (2013) Control of myofibroblast differentiation by
microtubule dynamics through a regulated localization of mDia2, J.
Biol. Chem., 288, 15466-15473.
94.Ott, C., Iwanciw, D., Graness, A., Giehl, K., and
Goppelt-Struebe, M. (2003) Modulation of the expression of connective
tissue growth factor by alterations of the cytoskeleton, J. Biol.
Chem., 278, 44305-44311.
95.Samarakoon, R., Goppelt-Struebe, M., and Higgins,
P. J. (2010) Linking cell structure to gene regulation: signaling
events and expression controls on the model genes PAI-1 and CTGF,
Cell. Signal., 22, 1413-1419.
96.Dai, P., Nakagami, T., Tanaka, H., Hitomi, T.,
and Takamatsu, T. (2007) Cx43 mediates TGF-beta signaling through
competitive Smads binding to microtubules, Mol. Biol. Cell,
18, 2264-2273.
97.Dong, C., Li, Z., Alvarez, R., Jr., Feng, X. H.,
and Clermont-Goldschmidt, P. J. (2000) Microtubule binding to Smads may
regulate TGF beta activity, Mol. Cell, 5, 27-34.
98.Fuchs, E., and Weber, K. (1994) Intermediate
filaments: structure, dynamics, function, and disease, Annu. Rev.
Biochem., 63, 345-382.
99.Herrmann, H., Bar, H., Kreplak, L., Strelkov, S.
V., and Aebi, U. (2007) Intermediate filaments: from cell architecture
to nanomechanics, Nat. Rev., 8, 562-573.
100.Gruenbaum, Y., Margalit, A., Goldman, R. D.,
Shumaker, D. K., and Wilson, K. L. (2005) The nuclear lamina comes of
age, Nat. Rev., 6, 21-31.
101.Goldman, R. D., Khuon, S., Chou, Y. H., Opal,
P., and Steinert, P. M. (1996) The function of intermediate filaments
in cell shape and cytoskeletal integrity, J. Cell Biol.,
134, 971-983.
102.Parry, D. A., and Steinert, P. M. (1999)
Intermediate filaments: molecular architecture, assembly, dynamics and
polymorphism, Quart. Rev. Biophys., 32, 99-187.
103.Colucci-Guyon, E., Portier, M. M., Dunia, I.,
Paulin, D., Pournin, S., and Babinet, C. (1994) Mice lacking vimentin
develop and reproduce without an obvious phenotype, Cell,
79, 679-694.
104.Eckes, B., Colucci-Guyon, E., Smola, H.,
Nodder, S., Babinet, C., Krieg, T., and Martin, P. (2000) Impaired
wound healing in embryonic and adult mice lacking vimentin, J. Cell
Sci., 113 (Pt. 13), 2455-2462.
105.Mor-Vaknin, N., Legendre, M., Yu, Y., Serezani,
C. H., Garg, S. K., Jatzek, A., Swanson, M. D., Gonzalez-Hernandez, M.
J., Teitz-Tennenbaum, S., Punturieri, A., Engleberg, N. C., Banerjee,
R., Peters-Golden, M., Kao, J. Y., and Markovitz, D. M. (2013) Murine
colitis is mediated by vimentin, Sci. Rep., 3, 1045.
106.Dos Santos, G., Rogel, M. R., Baker, M. A.,
Troken, J. R., Urich, D., Morales-Nebreda, L., Sennello, J. A.,
Kutuzov, M. A., Sitikov, A., Davis, J. M., Lam, A. P., Cheresh, P.,
Kamp, D., Shumaker, D. K., Budinger, G. R., and Ridge, K. M. (2015)
Vimentin regulates activation of the NLRP3 inflammasome, Nat.
Commun., 6, 6574.
107.Eckes, B., Dogic, D., Colucci-Guyon, E., Wang,
N., Maniotis, A., Ingber, D., Merckling, A., Langa, F., Aumailley, M.,
Delouvee, A., Koteliansky, V., Babinet, C., and Krieg, T. (1998)
Impaired mechanical stability, migration and contractile capacity in
vimentin-deficient fibroblasts, J. Cell Sci., 111 (Pt.
13), 1897-1907.
108.Challa, A. A., and Stefanovic, B. (2011) A
novel role of vimentin filaments: binding and stabilization of collagen
mRNAs, Mol. Cell. Biol., 31, 3773-3789.
109.Krupsky, M., Kuang, P. P., and Goldstein, R. H.
(1997) Regulation of type I collagen mRNA by amino acid deprivation in
human lung fibroblasts, J. Biol. Chem., 272,
13864-13868.
110.Ricupero, D. A., Poliks, C. F., Rishikof, D.
C., Cuttle, K. A., Kuang, P. P., and Goldstein, R. H. (2001)
Phosphatidylinositol 3-kinase-dependent stabilization of alpha1(I)
collagen mRNA in human lung fibroblasts, Am. J. Physiol.,
281, C99-C105.
111.Rishikof, D. C., Kuang, P. P., Poliks, C., and
Goldstein, R. H. (1998) Regulation of type I collagen mRNA in lung
fibroblasts by cystine availability, Biochem. J., 331
(Pt. 2), 417-422.
112.Cai, L., Fritz, D., Stefanovic, L., and
Stefanovic, B. (2010) Binding of LARP6 to the conserved 5¢
stem-loop regulates translation of mRNAs encoding type I collagen,
J. Mol. Biol., 395, 309-326.
113.Challa, A. A., Vukmirovic, M., Blackmon, J.,
and Stefanovic, B. (2012) Withaferin-A reduces type I collagen
expression in vitro and inhibits development of myocardial
fibrosis in vivo, PloS One, 7, e42989.
114.Bargagna-Mohan, P., Hamza, A., Kim, Y. E.,
Khuan Abby Ho, Y., Mor-Vaknin, N., Wendschlag, N., Liu, J., Evans, R.
M., Markovitz, D. M., Zhan, C. G., Kim, K. B., and Mohan, R. (2007) The
tumor inhibitor and antiangiogenic agent Withaferin A targets the
intermediate filament protein vimentin, Chem. Biol., 14,
623-634.
115.Zhang, Y., and Stefanovic, B. (2016) Akt
mediated phosphorylation of LARP6; critical step in biosynthesis of
type I collagen, Sci. Rep., 6, 22597.