REVIEW: Hemoglobin and Myoglobin as Reducing Agents in Biological Systems. Redox Reactions of Globins with Copper and Iron Salts and Complexes
G. B. Postnikova* and E. A. Shekhovtsova
Institute of Cell Biophysics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: gb_post@icb.psn.ru* To whom correspondence should be addressed.
Received April 18, 2016; Revision received September 27, 2016
In addition to reversible O2 binding, respiratory proteins of the globin family, hemoglobin (Hb) and myoglobin (Mb), participate in redox reactions with various metal complexes, including biologically significant ones, such as those of copper and iron. HbO2 and MbO2 are present in cells in large amounts and, as redox agents, can contribute to maintaining cell redox state and resisting oxidative stress. Divalent copper complexes with high redox potentials (E0, 200-600 mV) and high stability constants, such as [Cu(phen)2]2+, [Cu(dmphen)2]2+, and CuDTA oxidize ferrous heme proteins by the simple outer-sphere electron transfer mechanism through overlapping π-orbitals of the heme and the copper complex. Weaker oxidants, such as Cu2+, CuEDTA, CuNTA, CuCit, CuATP, and CuHis (E0 ≤ 100-150 mV) react with HbO2 and MbO2 through preliminary binding to the protein with substitution of the metal ligands with protein groups and subsequent intramolecular electron transfer in the complex (the site-specific outer-sphere electron transfer mechanism). Oxidation of HbO2 and MbO2 by potassium ferricyanide and Fe(3) complexes with NTA, EDTA, CDTA, ATP, 2,3-DPG, citrate, and pyrophosphate PPi proceeds mainly through the simple outer-sphere electron transfer mechanism via the exposed heme edge. According to Marcus theory, the rate of this reaction correlates with the difference in redox potentials of the reagents and their self-exchange rates. For charged reagents, the reaction may be preceded by their nonspecific binding to the protein due to electrostatic interactions. The reactions of LbO2 with carboxylate Fe complexes, unlike its reactions with ferricyanide, occur via the site-specific outer-sphere electron transfer mechanism, even though the same reagents oxidize structurally similar MbO2 and cytochrome b5 via the simple outer-sphere electron transfer mechanism. Of particular biological interest is HbO2 and MbO2 transformation into met-forms in the presence of small amounts of metal ions or complexes (catalysis), which, until recently, had been demonstrated only for copper compounds with intermediate redox potentials. The main contribution to the reaction rate comes from copper binding to the “inner” histidines, His97 (0.66 nm from the heme) that forms a hydrogen bond with the heme propionate COO– group, and the distal His64. The affinity of both histidines for copper is much lower than that of the surface histidines residues, and they are inaccessible for modification with chemical reagents. However, it was found recently that the high-potential Fe(3) complex, potassium ferricyanide (400 mV), at a 5 to 20% of molar protein concentration can be an efficient catalyst of MbO2 oxidation into metMb. The catalytic process includes binding of ferrocyanide anion in the region of the His119 residue due to the presence there of a large positive local electrostatic potential and existence of a “pocket” formed by Lys16, Ala19, Asp20, and Arg118 that is sufficient to accommodate [Fe(CN)6]4–. Fast, proton-assisted reoxidation of the bound ferrocyanide by oxygen (which is required for completion of the catalytic cycle), unlike slow [Fe(CN)6]4– oxidation in solution, is provided by the optimal location of neighboring protonated His113 and His116, as it occurs in the enzyme active site.
KEY WORDS: hemoglobin, myoglobin, leghemoglobin, copper and iron salts and complexes, redox reactions, electrostatic potentialDOI: 10.1134/S0006297916130101
Abbreviations: Hb, hemoglobin; Lb, leghemoglobin; Mb, myoglobin; MbO2, oxymyoglobin; metMb, metmyoglobin; bipy, 4,4′-bipyridine; CA-Mb, metmyoglobin carboxyamidated at histidine residues; CDTA, trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid; Cit, citrate; CM-Mb, metmyoglobin carboxymethylated at histidine residues; dmphen, 2,9-dimethyl-1,10-phenanthroline; 2,3-DPG, 2,3-diphosphoglycerate; DTA, 2,5-dithiohexane-1,6-dicarboxylate; EDTA, ethylenediaminetetraacetic acid; EP, electrostatic potential; NTA, nitrilotriacetic acid; phen, 1,10-phenanthroline; PPi, pyrophosphate; ROS, reactive oxygen species.
Oxygen transporters, hemoglobin (Hb) and muscle myoglobin (Mb), are
well-studied proteins with known three-dimensional structures. In
contrast to tetrameric Hb, Mb is responsible for intracellular oxygen
storage in muscles and its transport from the plasma membrane to
mitochondria. The Hb molecule is composed of four polypeptide
subunits – two α- and two β-chains. Each of the
subunits has a highly helical tertiary structure similar to the
structure of myoglobin (Fig. 1a). Myoglobin
has higher affinity for oxygen than hemoglobin, so it can bind oxygen
near the capillary walls (i.e. at a lower oxygen partial pressure,
PO2) and then transport it to the mitochondria. The heme
groups in both proteins are located in hydrophobic pockets in a way
that both propionate residues are directed toward the solvent. Out of
two axial Fe heme ligands, only the fifth ligand, proximal His93(F8),
belongs to the protein, while the sixth coordination site is either
free (deoxy-form), or is occupied by an O2 molecule or some
other external ligand. The redox potential of the Hb α-chain
is +110 mV; the redox potentials of the Hb β-chain and
myoglobin is +55 mV.
Fig. 1. a) Spatial structure of sperm whale Mb. b) Spatial structure of sperm whale Mb in the XY projection and location of histidine residues. “Inner” His24(B5), His36(C1), His64(E7), His82(EF5), His93(F8), and His97(FG3) residues are shown in black. The other histidine residues, titratable His12(A10), His48(CD6), His81(EF4), His113(G14), His116(G17), and His119(GH1), are accessible to the solvent.
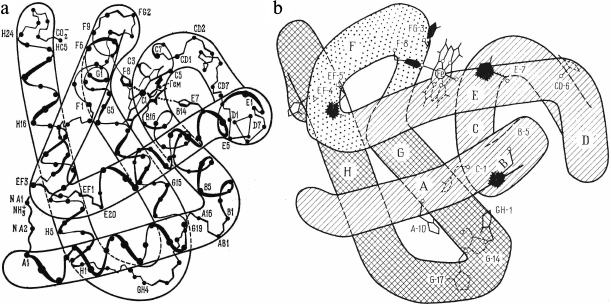
Another member of the globin family is leghemoglobin (Lb), a monomeric protein from root nodules of leguminous plants. Lb displays the highest affinity for oxygen among all globins. Its main function is maintaining low concentration of free O2 during the process of nitrogen fixation, because the nitrogenase complex of symbiotic bacteria is rapidly inactivated by O2. The 3D-structure of Lb is homologous to those of Mb and Hb α- and β-chains, however, the redox potential of Lb is much higher (+270 mV), and it contains only two histidines, proximal and distal, in the heme pocket [1, 2].
The reduced state of the heme complex in oxyglobins is essential for their function, since oxidized met-forms of these proteins (with a H2O molecule as the sixth ligand of the Fe heme) do not bind oxygen. At the same time, oxyglobins can undergo spontaneous oxidation (autooxidation) under aerobic conditions, especially at acidic pH [3-5]. Globins also easily enter redox reactions with salts and complexes of metals. Of these compounds, iron and copper salts and complexes are of interest in biology, because they participate in numerous metabolic processes in organisms [6-9].
Pollution of the environment with heavy metals leads to an increase in the metal concentrations in all living organisms. Disturbances in ionic homeostasis can result in the development of serious diseases and even death and have become a real threat for the existence of living beings [10-13]. Under aerobic conditions in vitro, transition metal compounds can undergo cyclic redox transitions in the presence of low molecular weight reductants and serve as sites for the productions of reactive oxygen species, such as O2–, HO2, and hydrogen peroxide as a final product of their disproportionation. Since HbO2 and MbO2 are abundant in cells and are good reductants of transition metal compounds, they can be directly involved in these processes, which has been demonstrated in several model systems [7, 14, 15]. It is important to note that in contrast to redox reactions in the presence of low molecular weight reductants, reactions of metal complexes with HbO2 and MbO2 do not produce ROS, due probably to the fact that the formed oxidized met-forms of these proteins display high peroxidase activity [3, 4, 16, 17].
The role of Hb and Mb as reducing agents in biological systems has attracted close attention, since it might be important for understanding the involvement of cell proteins in the maintenance of the cell redox potential and its protection from oxidative stress [15, 17, 19]. Oxidized metHb and metMb can be reduced to physiologically active HbO2 and MbO2 by low-specificity low molecular weight cell diaphorases and specialized NAD-dependent enzymes – erythrocyte NADH-dependent metHb reductase (NADH-cytochrome b5 reductase) and muscle metMb reductase. Components of the metMb reductase system include Cyt b5 of the sarcoplasmic reticulum membrane and the closely related Cyt b of the outer mitochondrial membrane [20]. Root nodules of leguminous plants contain the Lg-reducing enzyme – metLb reductase [21, 22].
The mechanism by which a metal compound reacts with a heme protein depends on the redox potential (E0) and the type of metal complex. Complexes with high redox potentials (E0, 200-600 mV) oxidize heme proteins by the simple outer-sphere electron transfer mechanism via overlapping π-orbitals of the heme and the metal complex [23, 24]:
P(Fe2+) + MzLn → P(Fe3+) + Mz–1Ln (1)
According to the theory of Marcus [25], the reaction rate in this case would be proportional mostly to the difference in the potentials and the rate of electron self-exchange of the protein and the reagent and, as a rule, does not depend on solution pH or ionic strength. The simple outer-sphere mechanism is favored by high stability constant of the metal complex and the presence in it of aromatic ligands with extended π-orbitals that can overlap with the porphyrin π-system and form a pathway for the electron transfer. When the reagent is charged, the contribution of electrostatic interactions to the reaction rate is calculated from the Wherland–Gray equation within the framework of Debye–Hückel theory.
Oxidation of heme proteins by the metal complexes with lower redox potentials (E0, ~100-150 mV) occurs via a different mechanism. Such complexes, especially those without extended π-orbitals, react mostly through preliminary binding to the protein with substitution of the metal ligands for protein groups (site-specific electron transfer) [26, 27]. Substitution of one or several ligands (water molecules in the case of a metal ion) for protein groups after specific binding to the protein changes the reagent redox potential and strengthens its electron-accepting properties. In this case, the rate of electron transfer depends on pH and ionic strength of the solution, both of which affect the reagent–protein complex formation, and should decrease with increase in the metal complex stability, because strong ligands prevent their exchange upon binding to the protein.
Of the most interest in biology is transformation of oxyglobins into nonfunctional met-forms in the presence of small amounts of metallic compounds (catalysis). Until recently, the only known case of such transformation was catalytic oxidation of HbO2, MbO2, and LbO2 by copper (II) compounds with redox potentials of 100-150 mV [28-30] by the following scheme:
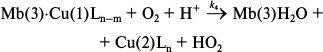 (5)
(5)
Oxidation occurs by the site-specific mechanism of electron transfer through the formation of a specific complex between the copper reagent and histidine residues of the protein. When bound, reduced copper is reoxidized by oxygen much faster than in solution, which is essential for the completion of the catalytic cycle (no catalysis is observed under anaerobic conditions).
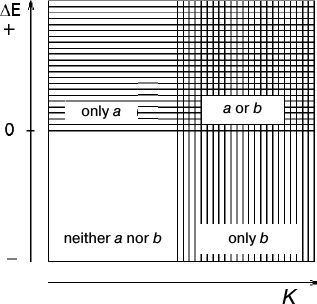
The catalytic activity of copper complexes can be predicted from their standard redox potentials and stability constants [6]. According to Fig. 2, copper complexes with low E0 and saturated ligands containing N or O donor atoms (such as EDTA, glycine, and ATP) are poor catalysts, because at high Cu+ reoxidation rate, the electron transfer will be too slow. Strong oxidants that readily react with proteins by the outer-sphere mechanism ([Cu(dmphen)2]2+, CuDTA, Cu(phen2)2+, and copper complexes with saturated ligands containing donor S atoms) cannot be efficient catalysts, because their reduced forms are poorly oxidized by oxygen according to the laws of thermodynamics. Strong catalytic activity is usually characteristic of compounds with intermediate redox potentials, e.g. Cu2+ complexes with unsaturated ligands containing O and N atoms (tyrosine, imidazole, and other nitrogen-containing heterocycles).
Although more than 20 ions and complexes of various metals (Ag, Mg, Mn, Co, Zn, Fe, Cr, etc.) have been examined for catalytic activity, none of them noticeably affected the rates of HbO2 and MbO2 oxidation [28]. However, we found that addition of small amounts of potassium ferricyanide (1 to 20% of protein concentration) to MbO2 solution resulted in complete oxidation of MbO2 [32, 33]. Hence, we demonstrated for the first time that the reaction of oxyglobin oxidation could be efficiently catalyzed by a high-potential iron complex; we have studied the mechanism of this reaction in detail.
Fig. 2. Scheme predicting the mechanisms of electron transfer between heme protein and metal complex: a – the simple outer-sphere electron transfer through the heme edge that includes nonspecific electrostatic binding to the protein (mechanisms 1 and 2). Distinguishing between mechanisms 1 and 2 requires data on the reaction rate dependence on ionic strength or direct NMR data on the reagent binding to the protein; b – the site-specific outer-sphere mechanism (mechanism 3). In the intermediate situation, the protein can react with the metal complex via both mechanisms simultaneously.
REDOX REACTIONS OF HEMOGLOBIN, MYOGLOBIN, AND LEGHEMOGLOBIN WITH
COPPER SALTS AND COMPLEXES
Copper complexes with high redox potentials (200-600 mV). Strong oxidizing compounds, [Cu(dmphen)2]2+ and CuDTA (Table 1), oxidize HbO2 at high rates. Both α- and β-chains are oxidized completely, since their redox potentials are +110 and +55 mV, respectively [6, 9]. The reaction rate is proportional to the reagent concentration up to a 10-fold reagent excess (relatively to the protein) (pH 6.15, 0.1 M MES buffer, 25°C) and does not reach saturation. The oxidation of HbO2 by copper phenanthroline complex [Cu(phen)2]2+ occurs in a similar way. The redox potential of [Cu(phen)2]2+ is lower than that of Cu(dmphen)2; however, both complexes are highly stable and have extended π-orbitals typical of chelating agents.
[Cu(dmphen)2]2+, CuDTA, and [Cu(phen)2]2+ interact with MbO2 by the same mechanism. Both proteins are oxidized in a second order reaction. The rate constants for Mb(2) oxidation with [Cu(dmphen)2]2+, CuDTA, and [Cu(phen)2]2+ are 2.8·106, 1.8·105, and 4.3·104 M–1·s–1, respectively, which correlates well with the reduction potentials of these complexes (Table 1). Decreasing O2 concentration in a solution increases the reaction rate, thereby indicating that the reaction occurs via the non-ligand form of the ferrous protein:
As it was demonstrated for [Cu(phen)2]2+, the rate constants for the oxidation of Hb and Mb oxy-derivatives are 100-300 times lower than for their deoxy-counterparts.
Table 1. Reduction potentials and stability
constants for divalent copper complexes
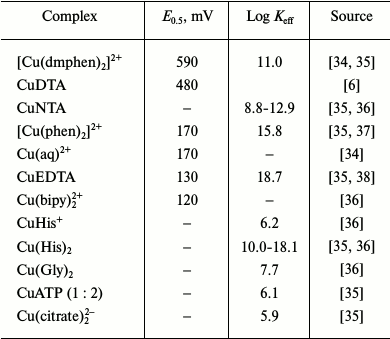
The differences in the rates of deoxyHb and deoxyMb oxidation by [Cu(phen)2]2+, [Cu(dmphen)2]2+, and CuDTA could be well described within the framework of Marcus theory, considering the contribution of electrostatic interactions from the Wherland–Gray equation. According to some authors [6], electron transfer in the reactions of these complexes with hemoglobin and myoglobin occurs via the simple outer-sphere mechanism and does not require reagent binding to the protein, as well as specific properties of the protein required for the charge transfer [6].
Oxidation of globins by copper compounds with intermediate redox potentials (100-150 mV). Hemoglobin. Oxidation of horse HbO2 by Cu2+ was discovered by Rifkind et al. [28, 39, 40]. Stoichiometric concentrations of copper (one Cu2+ per heme) oxidize 50% of HbO2 (β-chains only) very rapidly (semi-conversion time, ~3 min). At Cu2+/heme ratio less than 0.5, the reaction kinetics are biphasic and include a fast initial phase, during which all added Cu2+ is reduced, and slower (by an order of magnitude) stage that lasts until all β-chains are oxidized. The slow stage is limited by the rate of Cu+ reoxidation (catalysis).
The rate of the fast phase decreases and the rate of the slow phase increases with increasing O2 concentration. In the absence of O2, only the fast phase takes place. The oxidation rate for various Hb derivatives decreases in the order Hb(2) > HbO2 >> HbCO, which implies that the reaction occurs via formation of a non-liganded form of the ferrous protein.
The processes involved in this reaction have been investigated by comparing the kinetics of oxidation of human and horse hemoglobins [31]. It was found that at the same copper concentration (0.5 Cu2+ per heme), 50% of horse HbO2 was rapidly (within 3 min) oxidized, while human HbO2 was oxidized by only 8%. In contrast to horse Hb containing only one Cu2+-binding site (as determined by equilibrium dialysis), human Hb has two ligand-binding sites. Since the additional site has a higher affinity for copper, it probably does not participate in electron transfer, but it successfully competes for Cu2+, thereby decreasing the portion of human Hb undergoing oxidation during the fast reaction stage.
It was shown that the site responsible for the fast reaction stage in human and horse hemoglobins is probably located close to the Cys93 residue at the proximal side of the heme, because chemical modification of the Cys93 SH-group by iodoacetamide or N-ethylmaleimide completely inhibits HbO2 oxidation, but it suppresses copper binding by only 20%. Apparently, Cys93 does not play an important role in Cu2+ binding to the native protein, but rather participates in electron transfer from the heme Fe to the closely bound copper.
It has been demonstrated by various methods that Cu2+ binding by the native protein is mediated mostly by surface histidine residues [28-31]. An additional binding site in human and rabbit hemoglobins with higher affinity for copper may involve the β-chain His2 or His116 residues (in horse Hb, these residues are substituted by Glu and Arg, respectively). Studies of mutant human HbA2 with the His116→Arg substitution in the β-chain suggested that the copper-binding residue is His β-2, because HbA2 oxidation rates at different copper concentrations and its copper-binding properties were identical to those of the intact human (and rabbit) Hb.
According to Rifkind et al., the rate of oxidation of horse HbO2 by copper ions slows in the presence of Zn2+, which competes with Cu2+ for histidine binding. Also, the presence of Zn2+ decreases 4-5-fold the amount of human HbO2 oxidized during the fast phase [31]. However, later studies demonstrated that Zn2+ does not affect the rate of HbO2 oxidation in the presence of Cu2+ [41]. According to data of equilibrium dialysis and EPR analysis, copper and zinc ions have similar affinity for Hb, but they bind to different sites on the surface of the protein [31, 40].
The rates of metHb formation in the presence of various ligands change in the order aquo = Cit = ATP > Phen > His = NTA >> EDTA. Copper complexes with weak biological chelators (citrate and ATP) (Table 1) are characterized by the same rates as Cu(aq)2+; more stable complexes react more slowly, which corresponds to the site-specific mechanism. The fact that Hb oxidation by CuNTA is completely inhibited by modification of the β-chain Cys93 SH-group with N-ethylmaleimide (NEM) also suggests the site-specific mechanism of the oxidation reaction.
Myoglobin. The kinetics of oxidation of sperm whale oxymyoglobin by copper ions has been studied at pH 4.8-7.5 within the temperature range 10-40°C at different protein/reagent ratios [29]. Sperm whale Mb has 12 histidine residues (Fig. 1b). Five of these residues are located within the protein globule and are inaccessible to the solvent; the other seven histidines are located on the protein surface at different distances from the heme. These residues can be protonated and bind copper with different affinities (Table 2).
Table 2. Ionization pK values and
distances to heme Fe atom for protonated histidine residues in
myoglobins
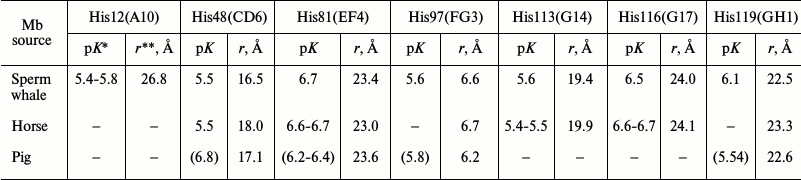
* pK values are from [48] and [49]. pK values in parentheses are from [50].
** Distances from heme Fe atom to histidine ND1 nitrogen atom were
calculated with the MOLMOL 2.5.1 program using Mb coordinates from the
NCBI database.
The dependence of the initial rate of oxidation of sperm whale MbO2 (V0) on copper concentration is rather complex. When 1/V0 is plotted against 1/[Cu2+], the resulting graph is a broken line composed of two linear fragments within low (from 0.1 to 1) and high (from 1 to 12) [Cu2+]/[MbO2] ratios, corresponding to low and high V0 values, respectively [29]. The parameters of activation also suggest that two parallel processes take place during MbO2 oxidation with copper that are related to structural rearrangements in the protein molecule.
In all cases, the pH dependence of the initial rate of metMb formation has sigmoidal shape with pKeff 5.6-6.0, which, according to the authors, reflects ionization of the His119 residue not involved in copper binding (Table 2). Analysis of the experimental and published data led to the conclusion that sperm whale Mb has only one site near His12 residue (26.8 Å from the heme Fe and 8.9 Å from His119) that binds Cu2+ with high affinity (Kd, 3.4·105 M–1). The Mb molecule also contains 5 to 7 low-affinity (Kd, ~2.1·103 M–1) copper-binding sites that do not participate in the redox process [29]. This conclusion is mostly based on X-ray data that demonstrated that sperm whale Mb has only one high-affinity copper-binding site close to His12(A10) and one high-affinity zinc-binding site close to His119(GH1) [42]. However, it contradicts the data of equilibrium dialysis and NMR spectroscopy on the presence of several high-affinity copper-binding sites in the molecule [8, 43]. Moreover, a hypothesis on predominant contribution of Cu2+ that binds close to His12 does not correlate with the fact demonstrated by the authors themselves that CM-MbO2 carboxymethylated at all surface histidine residues is oxidized with the same efficiency as the intact protein. Apparently, MbO2 oxidation by copper salts might occur via different mechanisms depending on which histidines bind the reagent and on the stability of the formed complex.
The kinetics of oxidation of sperm whale MbO2 in the presence of up to 300-fold excess of Cu(aq)2+, Cu(Gly)2, Cu(His)2, CuNTA, or CuEDTA is of a first order reaction (by protein) [8]. The rate of metMb formation reaches saturation at approximately 50-fold excess of the reagent and decreases with increasing copper complex stability. For Cu(Gly)2, Cu(His)2, CuNTA, and CuEDTA, logkobs (s) depends linearly on logK (M–1), which correlates well with the site-specific mechanism of electron transfer. Substituents with high affinity for metal prevent the exchange with protein ligands upon binding with the protein and, therefore, decrease the reaction rate.
Oxidation of sperm whale MbO2 by CuNTA slows in the presence of ZnNTA and NiNTA, which proves the existence of specific sites for the interaction of the reagent with myoglobin. To prevent ligand exchange and formation of mixed complexes to avoid the overcomplication of data analysis, the same complex ligand forms were used.
The rate of MbO2 oxidation by Cu(Gly)2 increases with increasing ionic strength and decreasing medium pH. Since Cu(Gly)2 bears no charge, this indicates that the reactive copper forms in solution are either Cu(Gly)+ or Cu2+, but not electrically neutral Cu(Gly)2. This can explain the observed saturation of the concentration dependence curve, because the reaction could be completely inhibited by excess glycine and redox-inactive Ni(Gly)2 and Zn(Gly)2.
Leghemoglobin. Studies of LgO2 oxidation in the presence of copper ions at equimolar [LbO2]/[Cu2+] ratio in the pH interval 4.8-7.5 [44] showed that the dependence of the initial rate of LgO2 oxidation (V0) on pH is not sigmoid (as in the case of MbO2) but resembles the curve for the LbO2 autooxidation, with a sharp increase at pH < 6. It is possible that Cu2+ binds to the distal His61 residue, which in contrast to distal His64 in myoglobin does not form a H-bond with the sixth ligand O2 of the heme Fe, but is oriented toward the solvent [2].
In the presence of an equimolar amount of Cu2+, the rate of metLb formation is only ~2.5 times higher than the rate of LbO2 autooxidation (for comparison, under the same conditions, Cu2+ increases the rate of MbO2 oxidation 45-fold). A 16-fold increase in the copper concentration does not affect the reaction rate (at pH 5.0). Although the authors suggest that the site-specific mechanism of electron transfer is active, we cannot exclude that copper binding to the distal His residues simply activates LbO2 autooxidation due to changes in the conformation of the heme pocket, especially at acidic pH values, because the rate of LbO2 autooxidation is much higher than that of animal globins.
As determined in studies of LbO2 oxidation by differently charged copper compounds (Cu2+, CuHis+, Cu(His)2, Cu(Gly)2, and CuNTA) in the presence of a 70-fold excess of the reagent [44], the rate constant for the pseudo first order reaction of LbO2 oxidation by Cu2+ is 11.37·10–3 s–1; the rate constants for CuHis (1 : 1), CuGly (1 : 2), CuNTA (1 : 1), and CuHis (1 : 2) are 8.67·10–3, 3.98·10–3, 0.36·10–3, and 0.02·10–3 s–1, respectively (LbO2 concentration, 20 µM; Cu2+ concentration, 1.4 mM; pH 5.6; ionic strength, 0.1). Reaction rate depends on the number of ligands bound and E0 value of the complexes. Thus, CuHis+ oxidizes LbO2 ~400 times faster than Cu(His)2. The effective stability constants of the complexes increase in the order CuHis+ < Cu(Gly)2 < CuNTA < Cu(His)2. The observed inverse correlation between the reaction rates and the effective stability constants of the copper complexes suggests preliminary binding of the reagents to the protein and, according to the authors, proves the site-specific mechanism of electron transfer.
The rate of LbO2 oxidation by Cu(Gly)2 increases 80-fold with pH decrease from 7.6 to 5.6. The character of the pH-dependence could possibly be explained by the fact that the ratio between different glycine–copper complexes with different redox potentials strongly depends on pH. Thus, the ligand-free Cu2+ form dominates at pH < 4; while completely saturated with ligands neutral Cu(Gly)2 dominates at pH > 7. At pH 4-7, there is also Cu(Gly)+, which reaches, respectively, its maximum content at pH 5. The concentrations of Cu2+ and Cu(Gly)+ and of Cu(Gly)+ and Cu(Gly)2 are the same at pH 4 and 6 (isosbestic points). At pH 7.6-5.6, ionic strength does not noticeably affect the reaction rate, thereby indicating that either the redox-active form of the complex is not charged or that charged forms do not bind to the protein.
The kinetic curves are typical for Michaelis–Menten kinetics of an enzyme–substrate complex with saturation. Based on Lineweaver–Burk plots, Km = 0.1 mM and Vmax = 4.3 µM·min–1. Redox-inactive Zn(Gly)2 and Ni(Gly)2 compete with copper for LbO2 binding (as well as glycine added at the same concentration) and completely inhibit LbO2 oxidation. Therefore, both inhibition and saturation of the concentration curve could be explained by the formation of more stable low-potential Cu(Gly)3 and Cu(Gly)4 complexes. Since LbCO reacts with Cu(Gly)2 350-fold more slowly than LbO2, the reaction apparently occurs via deoxyLb formation.
Binding of copper and zinc compounds with myoglobin and leghemoglobin. According to data of equilibrium dialysis [43], sperm whale metMb can bind up to six bivalent copper ions. Three binding sites have high affinity for copper and can be saturated at [Cu2+]/[Mb] ratio of 1 to 4 (Kd, 105-106 M–1). The high affinity for copper is related to the formation of a chelate complex in which the metal ligands can be also other than His residues, Lys, Asp, Glu, Gln, located close enough and possess appropriate spatial orientation [42]. At [Cu2+]/[Mb] ratio of 10, all six binding sites are saturated. The affinity of copper ions for Mb depends on pH, because protons can compete with Cu2+ for binding to the protein.
Redox-inactive Zn2+ forms complexes with metMb at the same sites as Cu2+ but with lower affinity. Thus, in the presence of a 20-fold excess of Zn2+, when all six sites are occupied, zinc is easily substituted at three sites upon addition of 1 to 4 copper equivalents [43]. Only one Zn2+ remains bound to metMb, i.e. competes with copper for the binding site, even in the presence of a 10-fold excess of Cu2+. Different affinities of Cu2+ and Zn2+ for the same binding sites in Mb could be explained by different structures of the formed complexes [45]. At [Zn2+]/[Mb] ratio up to 5 : 1 (pH 6), only one site near His119 is saturated (Kd, 4.4·105 M–1) [46].
Sperm whale metMb crystals were analyzed by differential Fourier synthesis. The crystals were incubated in the presence of 80-fold excess of CuCl2 or 3-4-fold molar excess of Zn(CH3COO)2 at pH 6 [42]. The authors found only one copper-binding site close to His12(A10) and one zinc-binding site close to His119(GH1). Supposedly, in both cases the metal ions form coordination bonds with functional groups of the same closely located Lys16(A14) and Asp122(GH4) residues, which explains the high affinity of this site for Cu2+ and Zn2+ and the competition between the ions for its binding. This correlates well with the equilibrium dialysis data for zinc, but contracts data suggesting that sperm whale Mb has three sites with high affinity for copper. Binding of Cu2+ (10- to 40-fold excess) to metMb causes loss of protein tertiary structure and changes the spectral properties of the protein within 2-6 h due to heme dissociation [11].
Complex formation between CuNTA and sperm whale metMb (pH 5.4) or MbCO (pH 4.7) was studied by high-resolution NMR, and the sites of CuNTA binding to the protein were identified [8]. The C2H and C4H resonance signals widened more for His113, His116, and His48 than for His12 and His119 residues. The authors concluded that the first three residues bind copper more tightly. All the identified residues, except His48, are located far from the heme (Table 2).
Localization of Cu(Gly)2 in complexes with sperm whale and horse metmyoglobins was studied by high-resolution NMR spectroscopy [30, 47]. Based on the assignments by Cocco et al. [48], it was concluded that resonance signals of four histidine residues, His113, His116, His48, and His81, are widened in the spectra of both proteins. The resonance signals of His119 and “inner” His24, that forms a hydrogen bond with His119, widened only slightly. Since the distance between His113 and H116, that are present in both horse and sperm whale myoglobins, is only 0.62 nm, it could not be excluded that the widening of their signals reflects copper binding to one of the residues. When Cu(Gly)2 was added to chemically modified sperm whale CM-metMb, in which all surface histidines were carboxymethylated with bromoacetate, no signal widening in the NMR spectra was observed, thereby suggesting that the reagent did not bind to the modified histidines [30, 47, 50].
In the presence of low CuSO4 concentrations or higher CuNTA concentrations (10 and 50% of protein concentration, respectively), the signal of distal His61 in the high-resolution NMR spectra of soybean LbCO widens considerably, which confirms that copper binds to this residue [44].
During the last few years, the ability of histidine residues in Mb to bind ions and complexes of copper and other metals has been used for Mb site-specific proteolytic cleavage under mild conditions [51] and for investigating the properties of Mb adsorption on phospholipid monolayers [52].
Contribution of copper complexes with different location in the protein molecule to redox activity of oxymyoglobin. To estimate the effect of the location of copper-binding sites on electron transfer and to reveal the correlation between complex stability and contribution of the binding site to total reaction rate, Cu(Gly)2-catalyzed oxidation was studied for the native sperm whale MbO2 and its mutants in which His113, His116, and His48 residues with the highest affinity for copper according to NMR data were replaced with Ala, Asp, and Ala, respectively [53]. Within the [Cu(Gly)2]/[MbO2] ratio interval of 1 to 20, the biggest effect (31% decrease in reaction rate) was observed for the His48→Ala mutant, whereas His116 substitution by Asp slowed the reaction by only 7%. Additional substitution in this mutant, His113 by Ala, did not affect the reaction rate. Therefore, His113 is not involved in the redox activity of the protein, even if its affinity for Cu(Gly)2 is the highest among the studied residues [8]. Similarly, human Hb β-2 histidine is capable of the formation of high-affinity complexes with copper, but it does not participate in redox reactions [31, 40].
Oxidation of sperm whale, horse, and pig MbO2 has also been studied. These proteins have similar spatial structures and redox potentials but differ in the number of surface histidine residues [54-56]. Amino acid residues that interact with the heme are conserved in all three proteins. However, horse Mb lacks His12 that is present in sperm whale Mb, while in pig Mb, His12, His113, and His116 are replaced by Gln residues. Analysis of primary and tertiary structures of these myoglobins revealed that residues near histidine residues are very similar, i.e. complexes with the same structure and affinity for copper should form on copper-binding in all three proteins.
It was found that the oxidation patterns of sperm whale and horse MbO2 proteins differ from that of pig MbO2. In the presence of one or less than one Cu2+ equivalents, the sperm whale and horse proteins are completely converted into metMb in a slow catalytic reaction. Under the same conditions, only a small portion (10-15%) of pig MbO2 undergoes oxidation within 1 min (rapid phase), while the rest of the protein remains unoxidized.
Since the kinetic properties of sperm whale and horse MbO2 are similar, and His12 in the horse Mb is replaced by Gln, we conclude that the contribution of His12 to the redox reaction is insignificant. It is obvious that catalysis is explained by Cu2+ binding to His116 and His113 residues (that are absent in pig Mb). Such binding would create conditions for rapid reoxidation of the bound reduced copper (Cu+), which would promote full oxidation of MbO2. This process should also dominate at low Cu2+ concentrations, when these sites are saturated.
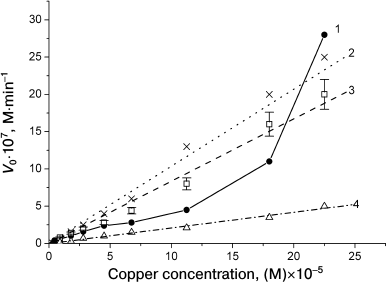
The concentration dependences of the reaction rates at [Cu2+]/[MbO2] ratios from 0.2 to 10 are similar for horse and sperm whale MbO2 and have a complex shape [29], because there is no additivity in the reaction rate increase with increasing copper ion concentration (Fig. 3, curve 1). The reaction rate slowly increases with increasing [Cu2+]/[MbO2] ratio up to 3 : 1, reaches a plateau at ratio 5 : 1, and then increases drastically. The pronounced sigmoid shape of the pH dependence of MbO2 oxidation rate in the presence of one Cu2+ equivalent indicates that the rate is affected by ionization of a group with pKeff 6.3-6.4 for sperm whale MbO2 and pKeff 6.7-6.8 for horse MbO2, i.e. by ionization of His116 (Table 2). In the presence of five-fold molar excess of Cu2+, the pH dependence shifts toward acidic pH, which suggests the involvement of more than one histidine residues. Note that at 10-fold Cu2+ excess, when all surface histidines are saturated, the concentration dependence still does not reach saturation (Fig. 3, curve 1).
Fig. 3. Dependence of oxidation rates of native sperm whale MbO2 (1), CM-MbO2 (2), and CA-MbO2 (3) on copper ion concentration at [Cu2+]/[protein] ratios from 0.2 to 10 and dependence of the oxidation rate of native MbO2 on Cu(Gly)+ concentration at the same Cu/protein ratios (4). Protein concentration, 2.25·10–5 M; 0.01 M Tris-maleate buffer, pH 7.5, 20°C. Error bars are shown for curve 3 only to avoid overcomplication of the figure [47].
Addition of zinc ions to the reaction mixture at [Zn2+]/[MbO2] ratios up to 5 : 1 did not produce any pronounced effect on the reaction rate. Since only one MbO2 site near His119 is saturated at such [Zn2+] concentrations (binding constant, 4.4·105 M–1 at pH 6 [46]), neither His119 nor His12 are involved in the catalysis.
Rapid oxidation of 10-15% of pig MbO2 (rapid phase) is not accompanied by the catalysis, since, obviously, no reoxidation of reduced copper takes place. Apparently, Cu+ easily dissociates into the solution, where it undergoes slow proton-assisted oxidation at acidic pH values [57]. Indeed, further slight oxidation of pig MbO2 is observed after the rapid phase only at pH 5. Out of four histidine residues, His48(CD6), His81(EF4), His97(FG3), and His119(GH1), that are conserved in all the studied myoglobins, probably only His48 and His97 bind copper and are responsible for the rapid phase of the reaction. These residues are most closely located to the heme (Table 2). His97, that is only 6.2 Å from the heme, should contribute most to the reaction. The rapid phase of human and horse HbO2 oxidation by Cu2+ correlates well with copper binding to the β-chain His97(FG4) residue, which is analogous to His97(FG3) located close to Cys93 at the proximal side of the heme in myoglobin.
Oxidation in the presence of Cu2+, Cu(Gly)+, and Cu(Gly)2 has been studied for native sperm whale MbO2 and its derivatives that were fully alkylated at all accessible histidine residues with sodium bromoacetate (CM-MbO2) and iodoacetamide (CA-MbO2) [30, 47]. The effects of reagent concentration, pH, ionic strength, and Zn2+ (that competes with copper for the binding to histidine residues) were studied. In contrast to the oxidation of native MbO2, the reaction rates of CM-MbO2 and CA-MbO2 oxidation at [Cu2+]/[protein] ratios from 0.2 to 10 were similar (the difference was within the experimental error) and increased proportionally to increase in reagent concentration (Fig. 3, curves 2 and 3). The concentration dependences of CM-MbO2 and CA-MbO2 reactions with Cu(Gly)+ and Cu(Gly)2 copper–glycine complexes at [reagent]/[protein] ratios from 0.2 to 10 remain linear, although the reaction rates decrease (Fig. 3, curve 4). The oxidation rates of all three proteins in the presence of Cu(Gly)+ decrease two-fold on average compared to the reaction rates with CuCl2. In the presence of Cu(Gly)2, the reaction rates decreased 3-4-fold.
Note that within this range of copper ion concentrations, the oxidation rates of modified MbO2 are equal or even exceed 2-3-fold the oxidation rate of native MbO2. Together with the absence of saturation, this indicates that the major contribution to the reaction was made not by the surface, but by “inner” histidines, primarily, His97, and probably, distal His64, that were not alkylated by the modifying reagents. In favor of this – the pH dependences of CM-MbO2 and CA-MbO2 oxidation, as well as pH dependence of native MbO2 oxidation in the presence of 10-fold excess of Cu2+. These curves have similar shapes and represent an average between monotonous and sigmoidal curves with pKeff < 6. At high (10-50-fold) excess of Cu(Gly)2, the concentration curves for MbO2 and CM-MbO2 reach saturation, which suggests reagent binding to the protein. The reaction rate for CM-MbO2 in this case is 2-3-fold lower than for the native protein.
Therefore, MbO2 oxidation by copper reagents can occur via different mechanisms and depends on which histidine residues bind the reagent, as well as on the stability of the formed complex. At low (up to equimolar) concentrations, a slow catalytic process dominates that is mediated by preferential copper binding in the region of His113 and His116, i.e. residues that possess the highest affinity for copper and compete with His97 for copper binding. The affinity of His97 for Cu2+ should be considerably lower than that of His113 and Hs116, because His97 forms an H-bond with the heme COO– group and is only partially exposed to the solvent [49, 50]. Nevertheless, the data on the CM-MbO2 and CA-MbO2 oxidation show that complex formation between the reagent and His97 markedly contributes to the reaction rate under these conditions, but its contribution becomes major in the presence of larger than five-fold excess of copper. The β-chains of Hb also contain His116(G18) residues that are analogous to His116(G17) in Mb. Both His97 and His116 are absent in the α-chains, which are resistant to the oxidation by copper.
Oxymyoglobin conversion into its oxidized met-form occurs via the site-specific mechanism of electron transfer through preliminary binding of the reagent to the protein histidines. However, complex formation between copper and His48, His113, and His116 residues that have the highest affinity for copper and are located on the protein surface 1.8-2.7 nm from the heme add very little (below 35%) to the total rate of the process, especially in the presence of excess (8-10-fold) amounts of the reagent. The contribution of these histidines (and probably His81 and His119, but not His12) becomes noticeable only at low (below five-fold molar excess) concentrations of the reagent, which is confirmed by the sigmoid shape of the pH dependence with transition at pK 6.5-6.7. The major contribution to the rate of the studied redox reaction should come from the formation of copper complexes with the “inner” histidines – His97, which is located 0.66 nm from the heme and forms a H-bond with the heme propionate COO– group, and distal His64. Both residues are inaccessible to modification by chemical reagents, and their affinity for copper is much lower than that of the surface histidines.
REDOX REACTIONS OF GLOBINS WITH IRON COMPLEXES
Oxidation of globins by potassium ferricyanide. Hemoglobin and myoglobin. Potassium ferricyanide, K3[Fe(CN)6], is widely used for rapid oxidation of ferrous derivatives of various heme proteins into the corresponding ferric forms. The mechanism of oxidation of deoxy- and oxy-derivatives of human Hb and Mb was firstly studied by Antonini et al. by the stopped-flow method [58]. They showed that one [Fe(CN)6]3– equivalent oxidizes one protein equivalent (per subunit). In the presence of excess reagent, the rate of HbO2 oxidation is typical of a first order reaction. The rate is higher at acidic pH values and increases linearly with decrease in O2 concentration in the solution. The kinetics of deoxyhemoglobin, Hb(2), oxidation differs from that of a simple bimolecular reaction. The rate constant strongly depends on pH (7·104 M–1·s–1 at pH 6 vs. 0.8·104 M–1·s–1 at pH 9.2). Analysis of kinetic data for the tetrameric Hb is complicated because of the difference in the redox potentials of α- and β-chains (E0, +110 and +55 mV, respectively) and conformational transitions in the tetramer that are induced by ligands and medium pH [58, 59].
Unlike Hb(2), oxidation of deoxymyoglobin, Mb(2), by ferricyanide is a simple bimolecular reaction [58]. The second order rate constant only slightly depends on pH, being 2·106 M–1·s–1 at pH 6.0 and 1.4·106 M–1·s–1 at pH 9.2. Under the same conditions, the rate constant for Mb(2) is ~100 times higher than that for Hb(2), which correlates well with the differences in the redox potentials of these proteins. Oxidation of MbO2 by excess [Fe(CN)6]3– is a first order reaction (in myoglobin), and its rate depends on the O2 concentration. When extrapolated to the zero O2 concentration, the rate constant corresponds to the constant of O2 binding to the protein. Based on data on the interaction of Mb(2) and Hb(2) with various ligands and O2 replacement with CO, it was suggested that electron transfer in the reaction of these proteins with ferricyanide occurs by the inner-sphere mechanism.
However, it was demonstrated later [8] that the data of Antonini et al. [58] showed in fact that HbO2 and MbO2 oxidation by ferricyanide occurs via non-liganded forms of the proteins:
Reaction (9) is very fast (k2 = 2.76·106 M–1·s–1 at pH 5.7), while dissociation of bound O2 (reaction (8)) is the rate-limiting stage. Despite low concentration of Mb(2) in a solution (because of its high affinity for oxygen (k1 = 107 M–1·s–1, k–1 = 10 s–1)), it should react ~1000 times faster than MbO2 due to the large difference in redox potentials for the Mb(2)/metMb and MbO2/metMb pairs (+55 and above +400 mV, respectively) as high deoxyMb affinity for O2 stabilizes the oxy-form of the protein.
The rate of reaction (9) depends only slightly on pH at pH 5.7-9.0, with an increase at pH < 7.0 (pKeff < 6.2) [23, 60]. The effect of pH on k2 might be related the protonation of one or both heme propionates (with pK ≤ 5.3) that are oriented toward the solvent. Neutralization of heme propionates at pH < 7.0 should promote Mb interaction with [Fe(CN)6]3–. Besides, the same effect of pH might be related to the protonation of the His97 residue (pK 5.6) close to the heme. It was suggested that in this case, electron transfer occurs via the simple outer-sphere mechanism through the heme edge exposed to the solvent, similarly to ferrocytochrome c oxidation by ferricyanide [23].
It should be noted that the reaction of ferricyanide with MbO2 and HbO2, as well as with Cyt c, can be affected by electrostatic interactions, although the dependence of the oxidation rates of these proteins on ionic strength has not yet been investigated. Studies on the oxidation of dimeric HbO2 from the mollusk Scapharca innaequivalvis by ferricyanide [61] showed that the ferrocyanide anion [Fe(CN)6]4– formed in the reaction remains bound to the protein. The binding site is close to Cys92(F2) residue in each subunit and, most probably, is formed by a cluster of positively charged Lys96, Arg53, Lys65, and Arg67 residues located near the proximal His101(F11).
Leghemoglobin. Oxidation of soybean LbO2 by potassium ferricyanide (second order rate constant at pH 6.6, ~42 M–1·s–1) was studied [44]. The dependence of the initial reaction rate on [Fe(CN)6]3– concentration is linear. No saturation is achieved up to a 10-fold excess of the reagent. The rate of LbO2 oxidation by ferricyanide increases with decreasing pH within the pH range of 5 to 8 and does not depend on the ionic strength. The reaction is not inhibited by Ni or Zn salts and complexes. Taken together, these data suggest that the reaction proceeds via the simple outer-sphere electron transfer through the heme edge without preliminary binding of [Fe(CN)6]3– anion to the protein.
Redox reaction of globins with carboxylate and phosphate iron complexes. Hemoglobin. The role of HbO2 as an aerobic biological reductant of trivalent iron was proposed based on the observation that HbO2 can mediate iron transfer from transferrin to the Fe2+ chelator 2,2-bipyridine [62]. The hypothesis was then confirmed in rabbit erythrocytes and reticulocytes. In these cells, the intracellular levels of reduced iron were maintained only in the presence of HbO2, and transmembrane Fe2+ transport correlated with Fe3+ reduction by oxyhemoglobin [62]. HbO2 and MbO2 were found to reduce ATP–Fe3+ complexes in several model systems, which suggested that O2-transporting proteins can also act as strong reducing agents and also suggested a new function (Fe chelation) for ATP, that had previously been considered only as an energy intermediate [7, 63, 64].
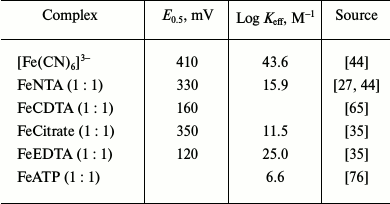
The ability of human HbO2 to reduce complexes of trivalent iron with NTA, EDTA, ATP, 2,3-DPG, citrate, and pyrophosphate (PPi) has been studied at different [Fe]/[chelator] ratios both in the presence and absence of the second chelator bipyridine (bipy) [7, 63, 64]. The reaction rate was registered from the spectra of the protein met-form and the bipy-reduced Fe complex (pH 7.0, 0.14 M HEPES, 37°C). The reaction is not inhibited by superoxide dismutase, which indicates that no formation of O2– radical takes place, since O2– can act as a reductant [7]. Under the same conditions, the rate of HbO2 reaction with FeNTA is considerably lower than with potassium ferricyanide (half-oxidation times, 240 and 15 s, respectively). The reaction rate decreases in the order FeNTA > FeATP > FeEDTA > FeCitrate. For FeNTA and FeEDTA, the reaction rates in the presence and the absence of bipy are the same; in all other cases, bipy increases the reaction rate ~3-fold. In the absence of O2, the rate of Hb(2) oxidation by FeNTA (2.2·103 M–1·s–1) is three orders of magnitude higher than of HbO2, i.e. the limiting stage of the reaction is seemingly the dissociation of bound O2. The activation energies of HbO2 oxidation with FeEDTA, FeNTA, FeCitrate, and FeATP at 37°C (as determined from the reaction temperature-dependence curves) were 28.1 ± 0.4, 24.0 ± 2.8, 22.0 ± 1.0, and 33.4 ± 0.2 kcal/mol, respectively, i.e. varied within 22.1 to 33.4 kcal/mol and to higher values for the iron complexes with EDTA and ATP.
The concentration curves for the reaction of HbO2 with FeNTA reach saturation. According to Michaelis–Menten kinetics, Km = 2.4 mM and Vmax ~ 10–6 M–1·s–1 [63]. The bimolecular reaction rate constant stays the same up to 2.5-fold excess of the reagent and then decreases slightly. The reaction rate drops abruptly with pH increase from 6 to 8, a phenomenon that the authors explain by increase in Hb affinity for oxygen. At the same time, it is believed [8] that an increase in the rate of MbO2 oxidation by neutral FeNTA with pH decrease from 8 to 5 is due to the changes in the ratio between the main forms of the reagent. At pH 5-8, the major reagent form is the charged dimer (FeNTAOH)2–; the other form that is present in insignificant amounts is negatively charged FeNTA(OH)–. At pH > 8, FeNTA(OH)22– prevails in the solution. All three forms are redox-inactive, in contrast to the redox active uncharged FeNTA, whose concentration considerably increases only at acidic pH (pH < 5).
An increase in ionic strength from 0 to 0.5 (pH 7.0) results in a noticeable decrease in the rate of the reaction of FeNTA with HbO2, thereby suggesting the presence of a positively charged site in the protein essential for the binding of negatively charged reagents [7, 63]. However, other studies showed [64] that changes in the ionic strength (NaCl concentration, from 0 to 0.25 mM) only slightly affected the rate constants of the reactions of HbO2 with FeNTA and FeEDTA. The rate constants increased less than two-fold with NaCl concentration increase to 140 mM and then decreased slightly. These effects exclude the contribution of electrostatic interactions and are related most probably to the effect of salt on the protein conformation, affinity for the ligand, and/or redox potential. Note that the redox potential of the Fe(3)NTA/Fe(2)NTA pair increases at acidic pH, thereby increasing the driving force of the reaction [8].
Of all biological complexes of iron, only FeATP displays the same efficiency as FeNTA in the redox reaction with HbO2, all other complexes are less efficient [64]. The nature of Fe complexes with ATP and citrate in a solution depends on the ratio between the components. At 1 : 1 ratio, these complexes have multinuclear structures that depolymerize only at [Fe]/[ATP] ratios of 1 : 20 or 1 : 40. At [Fe]/[ATP] ratio of 1 : 20, the rate of HbO2 oxidation (8.2·10–3 min–1) is 1.5 times higher than at equimolar ratio between the complex components (5.1·10–3 min–1) (pH 7.2, 140 mM NaCl). Under the same conditions, the rate of reaction of FeCitrate with HbO2 decreases two times – from 5.4·10–3 to 2.8·10–3 min–1.
Some authors believe that electron transfer in the reactions of HbO2 with FeNTA, FeEDTA, FeATP, and FeCitrate occurs via the site-specific mechanism [7, 63], while others suggest the simple outer-sphere electron transfer through the heme edge as in structurally similar MbO2 and Cyt b5 and do not exclude the possibility of weak nonspecific binding of the charged reagent to the protein [64]. The binding of iron complexes to HbO2 and the site-specific mechanism of electron transfer have been confirmed by the observed correlation between the reaction rate, reagent redox potential, and the stability constant of the complex (Table 3), as well as by the fact that addition of a 10-fold excess of redox-inactive NiNTA inhibits HbO2 oxidation by FeNTA by 75% [63]. However, this disagrees with the observation that ZnNTA and MnNTA have virtually no effect on the reaction rate, although the strength of Hb binding of the metals decreases in the order Zn > Cd > Ni > Co > Mn, with the binding constant for Ni being considerably lower than the binding constant for Zn [39].
Table 3. Reduction potentials and stability
constants of Fe3+ complexes
Myoglobin. The kinetics of metMb formation from sperm whale ferromyoglobin in the presence of FeNTA excess at pH 7.0 was studied [8]. Even though the reaction ability of Mb forms decreases in the order Mb(2) > MbO2 > MbCO, kinetic analysis showed that in all the reactions, the reacting protein form was ligand-free Mb(2). The rate of MbO2 oxidation with FeNTA considerably increases at pH < 7.0, which, as in the case of Cyt b5, could be explained by an increase in the concentration of the redox-active uncharged form of the reagent and by the increase in the reaction driving force, because the redox potential of the Fe(3)NTA/Fe(2)NTA pair increases at acidic pH. The activation energy of the reaction is 32 kcal/mol as determined from the temperature dependence. Since Mb(2) and Hb(2) have considerably lower energy of activation in this type of reactions (from 5 to 15 kcal/mol) [65, 66], the high value of this parameter corroborates the conclusion that ligand dissociation from MbO2 is a rate-limiting step of the process, and the value of 32 kcal/mol is a sum of the energies of activation of two stages – O2 dissociation and the heme oxidation.
The rate of oxidation of MbO2 by FeNTA does not depend on the ionic strength [8]. Redox-inactive inhibitors (NiNTA and ZnNTA) do not affect the oxidation rate, which indicates that no reagent binding to the protein takes place. NMR titration of MbCO by FeNTA revealed no widening of the protein resonance signals up to very high FeNTA concentrations, i.e. no specific reagent-binding sites on the protein surface was identified. All these facts suggest the simple outer-sphere electron transfer through the heme edge as the most probable mechanism of the reaction of MbO2 with FeNTA.
The kinetics of reduction of horse metMb with FeEDTA2– was studied at different pH, temperature, and ionic strength values in the presence of reagent excess to provide the pseudo first reaction order in the protein [67]. The bimolecular reaction rate constants vary from 28 M–1·s–1 at pH 6.0 to 9
M–1·s–1 at pH 8.5 (0.5 M phosphate buffer, 25°C). The shape of the pH dependence within the pH interval from 5.5 to 8.5 with pKeff ~ 5.8 is similar to the pH dependence of oxidation of horse MbO2 by ferricyanide. An increase in the ionic strength (pH 7.0) inhibits the reaction of metMb with FeEDTA2–, the effect is most pronounced at I < 0.1. Since the horse Mb molecule is not charged at pH 7.0, this can indicate contribution of local electrostatic interactions to the mechanism of electron transfer [60, 67].
Using different reagent and O2 concentrations, the authors [65] studied the kinetics of reversible oxidation of horse metMb with FeCDTA2– and of oxidation of horse MbO2 with FeCDTA–1, which are similar in their structure and redox potential to the FeEDTA1–/2– pair [65]. The reaction scheme is the following:
metMb + FeCDTA2– + O2 → MbO2 + FeCDTA– (10)
MbO2 ↔ Mb(2) + O2 (11)
Mb(2) + FeCDTA– → met-Mb + FeCDTA2– (12)
It was shown that reactions (10) and (12) follow second order kinetics up to very high reagent concentrations (over 1000-fold excess relative to the protein). There is no saturation, and the reaction rate is proportional to the difference in the potentials of the reagents. The bimolecular rate constant of the oxidation reaction (kox) is 1.48·102 M–1·s–1 (0.1 M phosphate buffer, pH 6.8, 25°C). It does not depend on pH, and it corresponds to the equilibrium constant of 0.21. The rate of metMb reduction (kred) with FeCDTA2– (reaction (10)) is 28 M–1·s–1 (0.1 M phosphate buffer, pH 6.8, 25°C). It decreases considerably at pH > 8.0, when metMb undergoes the met–hydroxy transition (pKa 8.86) due to ionization of the ligand-bound water molecule. HydroxyMb, Mb(3)OH– (E0, 30 mV), is reduced much more slowly than Mb(3)H2O (E0, +55 mV), which correlates with the kred values of 31 and 4 M–1·s–1 at pH 7.0 and 9.5, respectively.
KCN produces a strong inhibitory effect on both the equilibrium and the rate of reduction of metMb with FeCDTA2–. The ligands CN– and OH– bind more tightly to metMb than H2O. At the same time, they bind more weakly or do not bind at all to Mb(2). For this reason, they should stabilize metMb relative to Mb(2) and decrease the rate of reduction. Although the proposed reaction scheme assumes involvement of both ligand-free and liganded Mb in the electron transfer, the data obtained at different CN– and FeCDTA2– concentrations suggest that in this case, as well as in redox reactions of ferromyoglobins, preliminary dissociation of the ligand occurs. The reduction of metMb with FeCDTA2– and with smaller by size FeEDTA2– occurs at the same rate and with the same activation parameters, which suggests that in both cases electrons are transferred by the simple outer-sphere mechanism through the partially exposed heme edge [65, 67].
Leghemoglobin. Studies of oxidation of soybean LbO2 by FeNTA [44] showed that this reaction is much slower than its reaction with ferricyanide (second order reaction constants at pH 6.6 of 1 and 42 M–1·s–1, respectively, in 50 mM HEPES/50 mM MES buffer at 20°C). The initial reaction rate reaches saturation with increase in FeNTA concentration, thereby indicating that during electron transfer the reagent binds to a specific site on the protein surface. The reaction parameters were determined by the classical Michaelis–Menten kinetics from the concentration dependence of the reaction rate: Km = 1.7 mM, Vmax = 21.7 µM·min–1. Addition of five-fold excess of ZnCl2 (relative to FeNTA) inhibits the reaction by 65%, which suggests that zinc and iron ions compete for the binding with the protein. However, Ni salts and complexes do not affect the reaction rate.
The rate of LbO2 oxidation with FeNTA noticeably decreases with increasing pH, which is most probably due to increase in the reaction driving force (as in the case of MbO2 oxidation with the same reagent) [8]. However, in contrast to MbO2 oxidation with FeNTA and ferricyanide, the rate of reaction of LbO2 with FeNTA decreases by ~50% with increase in ionic strength from 0.01 to 0.1 at pH 7.6, but remains the same at lower pH values. This could not be explained by electrostatic interactions between the protein and the reagent, especially in view of the earlier mentioned fact that uncharged FeNTA is redox-active, while negatively charged FeNTA(OH)– and FeNTA(OH)22– forms are redox-inactive.
NMR studies showed that the presence of equimolar concentration of FeNTA (pH 7.0) results in widening of proton resonance signals of distal His61 and of two signals assigned to Lys residues. This indicates that charged iron complexes bind to one or two sites of the Lb molecule, most probably to a cluster of Lys57, Lys64, and Lys95 residues close to the heme. It is possible that distal His61, which is oriented toward the solvent, is also involved in the binding. The authors concluded that in contrast to LbO2 redox reaction with [Fe(CN)6]3–, FeNTA oxidizes LbO2 via the site-specific mechanism.
Therefore, oxidation of oxygen-transporting proteins, such as HbO2 and MbO2, by various iron complexes occurs mostly by the simple outer-sphere mechanism of electron transfer through partially exposed heme edge irrespectively of the redox potential of the complex. If the reagent is charged, it can bind nonspecifically to the protein due to electrostatic interactions (as in the case of HbO2) [8, 64]. The differences in rate constants for HbO2 and MbO2 could be explained by differences in their redox potentials (+170 and +55 mV, respectively) and by shorter distance between the chelator metal and heme Fe in Mb. However, redox reactions of LbO2 with FeNTA most probably occur by the site-specific mechanism of electron transfer [44]. But the data do not exclude the simple outer-sphere mechanism of electron transfer even in this case, since the same reagents oxidize structurally similar MbO2 and Cyt b5 by the simple outer-sphere mechanism (the reaction rates correlate with the difference in the potentials of the reagents and with the rates of their self-exchange according to the theory of Marcus). Besides, the binding of charged iron complexes to one or two sites in Lb most probably happens with the involvement of a cluster of three lysine residues (Lys57, Lys64, and Lys95) located close to the heme.
Note that direct interaction of the studied iron complexes with the heme Fe atom in globins (inner-sphere electron transfer) could be completely excluded, since the size and structure of these complexes prevent their entering into the heme pocket of the protein. The inner-sphere mechanism of electron transfer with bridge formation, i.e. when the reagent binds to the heme Fe via EDTA4– bridge or heme ligand, is contradicted by the data on the reduction of Mb(3)CN– and Mb(3)OH– by this reagent. Mb(3)CN– and Mb(3)OH– should react faster than metMb, because the CN– and OH– anions are better bridge groups than water molecules [65].
Oxidation of native and modified oxymyoglobins catalyzed by ferrocyanide ions. We were the first to demonstrate that addition of small amounts of potassium ferricyanide to MbO2 solution results in complete oxidation of MbO2, i.e. that a high-potential iron complex can act as an efficient catalyst in the oxyglobin oxidation reaction [32, 33, 68-70]. The reaction involves two simultaneous processes: fast oxidation by ferricyanide of some amount of MbO2 via interaction with the heme by the simple outer-sphere mechanism, and slower conversion of the rest of MbO2 into metMb by the catalytic mechanism. The catalytic process is a first order reaction with kexp = 0.18 ± 0.01 min–1. Only two Mb forms are present in the solution at all the times, MbO2 and metMb, which is proved by the existence of two isosbestic points – at 523 and 592 nm.
We found that the catalyst in the MbO2 oxidation reaction is ferrocyanide anion [Fe(CN)6]4– that is formed in the first process (reaction (13)). The catalytic process includes [Fe(CN)6]4– binding to the protein (reaction (13)), proton-assisted oxidation of the bound [Fe(CN)6]4– by dissolved O2 (reaction (14)), and formation of the final reaction products (reaction (15)):
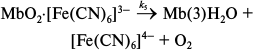 (15)
(15)Reaction (15) goes through several stages, so k5 is an effective constant that includes several individual constants, both kinetic and equilibrium ones. All these reactions are fast and do no limit the rate of the process [33].
Solving the system of equations (13)-(15) within the framework of formal kinetics of homogenous catalysis under conditions of stationary reactions allowed to calculate the equilibrium and kinetic constants, Kd = k–3/k3 and k4, from the experimental data at two pH values – 6.4 and 7.3 (Table 4). When [[Fe(CN)6]4–] << [MbO2], both constants could be calculated independently from Eq. (16), that describes well the observed linear dependence of the initial reaction rate on the ferrocyanide and proton concentrations:
V0 = k4 [MbO2]0 [[Fe(CN)6]4–]0 [O2][H+]/Kd (16)
where Kd = k–3/k3, and [O2] is constant and equals to 0.32 mM.
Table 4. Equilibrium and kinetic parameters
for oxymyoglobin oxidation catalyzed by ferrocyanide ions [68, 69]
* Calculated using Kd at pH 6.4 in the presence
of ferrocyanide excess.
** Calculated using k4[O2] =
1.03·106 M–1·min–1
in the presence of ferrocyanide excess.
When [[Fe(CN)6]4–] >> [MbO2], the dependence of V0 on [[Fe(CN)6]4–] is like that typical for enzymatic kinetics:
V0 = k4 [MbO2]0 [[Fe(CN)6]4–]0 [O2][H+]/Kd + [[Fe(CN)6]4–]0 (17)
Oxidation of sperm whale, horse and pig native MbO2 in the presence of [Fe(CN)6]4– was compared. These proteins have similar spatial structures and redox potentials, but differ in the number of surface histidine residues and in the total molecule charge. The oxidation of MbO2(His119→Asp) mutant and chemically modified sperm whale CM-MbO2, in which all accessible histidines were alkylated with sodium bromoacetate, was also studied [68-70]. The dependence of the reaction rate on the catalyst concentration, pH, ionic strength of the medium was investigated as well as and complex formation between MbO2 and redox-inactive Zn2+. Distributions of the electrostatic potential around the studied myoglobins at pH 5-8 and steric properties of Mb surface were analyzed to identify pockets large enough to accommodate [Fe(CN)6]4– anion.
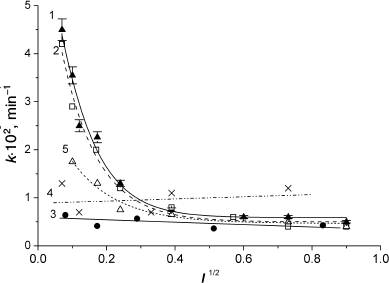
When the amount of the catalyst was much lower than that of the protein (1 to 20% of protein concentration), the dependence of the oxidation rate on the catalyst concentration was linear for all the proteins. Sperm whale and horse MbO2 proteins were oxidized in almost identical manner (Fig. 4, curves 1 and 2). Pig MbO2 and CM-MbO2 (Fig. 4, curves 3 and 4) were oxidized at slower rates, while the rate of oxidation of mutant MbO2(His119→Asp) was somewhat intermediate (Fig. 4, curve 5). At [Fe(CN)6]4– concentrations ranging from equimolar to 50-fold excess relative to the protein, the oxidation rates of sperm whale and horse native MbO2 were almost the same and reached saturation at [[Fe(CN)6]4–]/[MbO2] ratios exceeding 30 : 1 (Fig. 5, curves 1 and 2). The oxidation rates for pig MbO2 and sperm whale CM-MbO2 were almost identical; however, they were five-fold lower and did not reach saturation within the studied range of [Fe(CN)6]4– concentrations (Fig. 5, curves 3 and 4). The rate of oxidation of MbO2(His119→Asp) was 2-3-fold lower than that of the native sperm whale MbO2 (Fig. 5, curve 5).
Within the pH interval from 5 to 8, the oxidation rates of horse and sperm whale MbO2 in the presence of 5% ferrocyanide increased significantly at pH < 7.0 (Fig. 6, curves 1 and 2). This effect was less pronounced for MbO2(His119→Asp) (Fig. 6, curve 5). The oxidation rates of pig MbO2 and CM-MbO2 were extremely low over the entire studied pH range and did not depend on pH (Fig. 6, curves 3 and 4). Increase in ionic strength from 0 to 0.55 strongly inhibited oxidation of sperm whale and horse MbO2, especially at I = 0-0.1 (Fig. 7, curves 1 and 2), which indicates an important role of electrostatic interactions in the reaction mechanism. Note that at I > 0.1, the reaction rate was low at all studied pH values (from 5 to 8). The dependence of MbO2(His119→Asp) oxidation rate on the ionic strength at I < 0.1 was considerably less pronounced (Fig. 7, curve 5). The rates of pig MbO2 and sperm whale CM-MbO2 in the presence of potassium ferrocyanide were virtually independent of the ionic strength (Fig. 7, curves 3 and 4), remaining low within the whole range of ionic strength values. An slight increase in the CM-MbO2 oxidation rate at high I values was probably related to the fact that screening of charges of the negatively charged protein and reagent facilitates their interaction.
Fig. 4. Dependence of oxidation rates of sperm whale native MbO2 (1), horse MbO2 (2), pig MbO2 (3), sperm whale CM-MbO2 (4), and sperm whale MbO2(His119→Asp) mutant (5) on [Fe(CN)6]4– concentration when the catalyst concentration is lower than the protein concentration; 0.01 M Tris-maleate buffer (pH 5.2) at 20°C. Protein concentration, 2.25·10–5 M [68, 69].
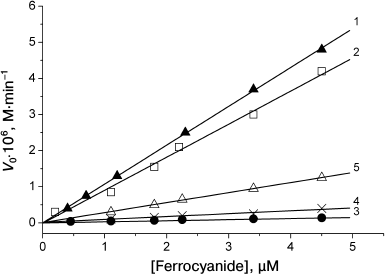
Fig. 5. Dependence of the oxidation rates of sperm whale native MbO2 (1), horse MbO2 (2), pig MbO2 (3), sperm whale CM-MbO2 (4), and sperm whale MbO2(His119→Asp) mutant (5) on [Fe(CN)6]4– concentration in the presence of excess catalyst amounts. 0.01 M Tris-maleate buffer (pH 6.4) at 20°C. Protein concentration, 2.25·10–5 M [68, 69].
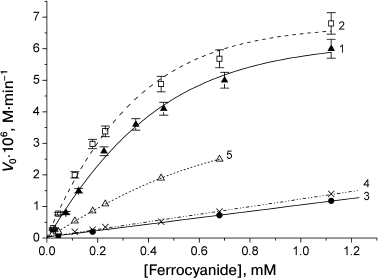
Fig. 6. pH dependence of the oxidation rates of sperm whale native MbO2 (1), horse MbO2 (2), pig MbO2 (3), sperm whale CM-MbO2 (4), and sperm whale MbO2(His119→Asp) mutant (5) in the presence potassium ferrocyanide (5% of the protein concentration, 1.1·10–6 M) in 0.01 M Tris-maleate buffer at 20°C. Protein concentration, 2.25·10–5 M [68, 69].
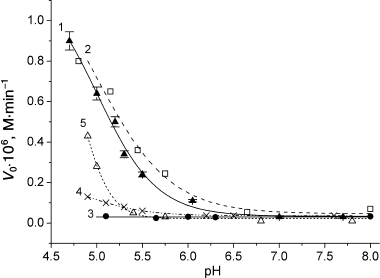
Fig. 7. Effect of ionic strength on the oxidation rates of sperm whale native MbO2 (1), horse MbO2 (2), pig MbO2 (3), sperm whale CM-MbO2 (4), and sperm whale MbO2(His119→Asp) (5) in the presence of potassium ferrocyanide (5% of protein concentration, 1.1·10–6 M) in 0.01 M Tris-maleate, KCl (pH 5.1) at 20°C. Protein concentration, 2.25·10–5 M [68, 69].
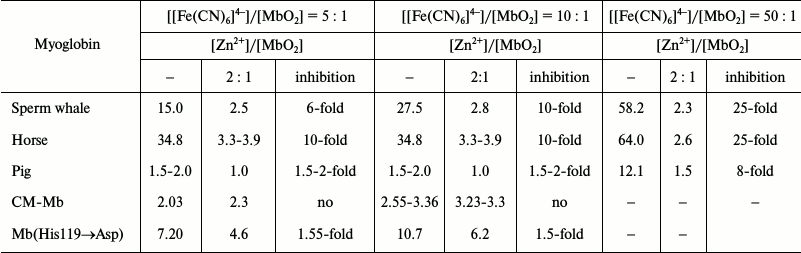
Complex formation between one of the surface histidine residues, His119(GH1), in the sperm whale, horse and pig MbO2 and zinc ion at [Zn2+]/[MbO2] ratio 2 : 1 [42, 71] completely suppresses [Fe(CN)6]4–-catalyzed oxidation of all myoglobins even at 50-fold excess of the catalyst (Table 5). Therefore, efficient catalysis requires specific binding of [Fe(CN)6]4– anion in the region of His119 residue. Zinc does not prevent fast MbO2 oxidation by ferricyanide that occurs through the heme, but no catalytic reaction with the formation of metMb takes place afterwards. The absence of inhibitory effect of Zn2+ on oxidation of MbO2(His119→Asp) and CM-MbO2 (Table 5) is due to the replacement of the Zn2+-interacting His119 residue with negatively charged Asp in the mutant myoglobin or its modification with anionic bromoacetate in CM-MbO2.
Table 5. Effect of Zn2+ on
oxidation rates (V0 ×
107, M·min–1) of sperm whale,
horse, and pig MbO2, carboxymethylated CM-MbO2,
and MbO2(His119→Asp) at various
[[Fe(CN)6]4–]/[MbO2] ratios in
0.01 M Tris-maleate buffer, pH 6.4, 20°C [68, 69]
The selectivity of [Fe(CN)6]4– binding in the region of His119 is due to high positive local electrostatic potential at this region of the Mb molecule at pH < 7.0 and to the existence of a “pocket” of sufficient volume formed by Lys16, Ala19, Asp20, His24, and Arg118 for insertion of ferrocyanide anion that can take place, when His119 is in an “open” conformation [68-70]. The affinity of [Fe(CN)6]4– for myoglobin (Kd) increases almost 10-fold at acidic pH due to the EP increase at the site of anion binding. At the same time, the existence of negative EP at the same site in the case of CM-MbO2 correlates well with a significant decrease in the [Fe(CN)6]4– affinity for myoglobin and explains low reaction rate even in the presence of excess catalyst (Table 4). At pH 5.2, the potential at the site of [Fe(CN)6]4– binding in the MbO2 (His119→Asp) mutant becomes negative and retains its initial value only in the region of His113 and His116.
The fact that inhibition of catalysis by Zn2+ binding near His119 takes place, though positive EP at this site does not change (or even increases), indicates an important role of steric interactions and of optimal dynamics of the protein groups in the [Fe(CN)6]4– binding and in the “active complex” formation. The affinity of zinc to His119 is ~100 times higher than that of ferrocyanide, since Zn2+ forms a chelate complex that involves, in addition to His119, functional groups of Lys16(A14) and Asp122(GH4) [35, 36]. Zinc binding results in the inability of His119 to acquire an “open” conformation and makes the “pocket” inaccessible for ferrocyanide [68-70].
Protonation of His113 and His116 residues located close to His119 also plays and important role in the mechanism of the ferrocyanide catalysis. On one hand, it promotes electrostatic binding of [Fe(CN)6]4– anion; on the other, it speeds up reoxidation of the anion by dissolved O2 (reaction (14)) due to optimal colocation of both reagents and a proton, as it is realized in the enzyme active site. In pig Mb, His113 and His116 are replaced by Gln, and no catalysis takes place, even though the positive potential at the [Fe(CN)6]4–-binding site remains the same and the configuration of the anion-binding pocket is preserved, so that Kd for ferrocyanide is similar to that for ferrocyanide binding to the sperm whale and horse myoglobins (Table 4).
Potassium ferrocyanide and other salts of divalent iron are known to be poorly oxidized by oxygen in solutions, even at acidic pH [37, 38, 72, 73]. However, when bound to sperm whale and horse native MbO2, ferrocyanide is rapidly oxidized under mild conditions (k4, Eq. (14)) (Table 4). The specific [Fe(CN)6]4–-binding to Mb at a site close to His119 is believed to underlie the catalytic effect of ferricyanide in Mb reaction with luminol, which increases 3.5-4.0-fold the sensitivity of chemiluminescence analysis of different ligand forms of myoglobin [74, 75].
CONCLUSION
Respiratory proteins, hemoglobin and myoglobin, can reduce biological complexes of copper and iron and can play an important role in maintaining cell redox potential and protecting cells against oxidative stress.
Complexes of Cu2+ with high redox potentials (E0, 200-600 mV) and high stability constants, such as [Cu(phen)2]2+, [Cu(dmphen)2]2+, and CuDTA oxidize ferrous derivatives of globins by the simple outer-sphere electron transfer mechanism though overlapping π-orbitals of the heme and metal complex. According to Marcus theory, the redox reaction rate correlates mostly with the difference in the potentials of the reagents and the rates of their self-exchange.
Weaker oxidants with E0 = 100-150 mV, such as Cu2+, CuEDTA, CuNTA, CuCitrate, CuATP, and CuHis react via preliminary binding to protein histidine residues with exchange of metal ligands for protein groups (the site-specific electron transfer). In this case, the redox reaction rate depends on pH and ionic strength of the solution, which affect the protein–reagent complex formation and should decrease with increase in stability of the metal complex.
The major contribution to the rate of MbO2 oxidation by copper compounds comes from copper complex formation with the “inner” histidine residues, His97 (0.66 nm from the heme) that forms a H-bond with the heme propionate COO– group, and distal His64. Complex formation between copper compounds and surface His48, His113, and His116 residues (1.8-2.7 nm from the heme) that have the highest affinity for copper contributes only slightly (less than 35%) to the total reaction rate. Copper-oxidized hemoglobin β-chains also contain His97 and His116(G18), analogous to the His116(G17) of myoglobin, but both His116 and His97 are absent in the α-chains, which are resistant to copper-catalyzed oxidation.
In contrast to redox reactions with copper reagents, oxidation of MbO2 and β-chains of HbO2 by potassium ferricyanide and trivalent iron complexes with NTA, EDTA, CDTA, ATP, 2.3-DPG, citrate, and PPi occurs mostly via the simple outer-sphere electron transfer mechanism through the exposed heme edge.
Conversion of functionally active oxyhemoglobins into nonreactive oxidized met-forms in the presence of small amounts of metallic compounds (catalysis) is of particular interest in biology. Earlier, this process had been known only for copper ions and complexes with intermediate redox potentials. A slow catalytic process dominates at low copper concentrations and is mediated by complex formation between copper and His113 and His116 residues. Both these residues are absent in pig MbO2, for which no catalysis has been observed.
It was demonstrated for the first time that high-potential complex of iron, potassium ferricyanide (E0, 400 mV), at a concentration of 5-20% of MbO2 concentration, catalyzes complete oxidation of MbO2 to metMb. The mechanism of catalysis includes (i) the complex formation between ferrocyanide anion and MbO2 in the region of His119 due to the presence of a high positive local potential, a cavity formed by Lys16, Ala19, Asp20, His24, and Arg118, that is the big enough to accommodate [Fe(CN)6]4–, and (ii) the rapid proton-assisted reoxidation of the bound ferrocyanide by dissolved oxygen due to the optimal localization of adjacent protonated His113 and His116 residues (similar to the processes occurring at the enzyme active site).
It should be noted that even though the reaction mechanism for MbO2 oxidation in the presence of Cu2+ and [Fe(CN)6]4– follows the common site-specific outer-sphere electron transfer mechanism, the exact molecular processes for these two ions differ in the type of formed catalytic complexes and their location in the protein structure. The efficiency of ferrocyanide catalysis at acidic pH (5.0) is usually 3-5 times higher than that of the copper one. At neutral pH (pH 6.0), these efficiencies are very similar (1.5-2.0-fold difference). At alkaline pH (pH > 7.0), copper catalysis is 10 times more efficient than catalysis by [Fe(CN)6]4–, because [Fe(CN)6]4– almost does not bind to the protein under alkaline conditions.
REFERENCES
1.Antonin, E., and Brunori, M. (1971) Hemoglobin
and Myoglobin in Their Reactions with Ligands, in Frontiers in
Biology, Amsterdam-London, p. 405.
2.Arutyunyan, E. G., Safonova, T. N., Obmolova, G.
V., Teplyakov, A. V., Popov, A. N., Rusakov, A. A., Rubinskii, S. V.,
Kuranova, I. P., and Vainshtein, B. K. (1990) Crystal structure of
oxyleghemoglobin at a 1.7 Å resolution, Bioorg. Khim.,
16, 293-302.
3.Shikama, K. (1998) The molecular mechanism of
autoxidation for myoglobin and hemoglobin: a venerable puzzle, Chem.
Rev., 98, 1357-1373.
4.Brantley, R. E., Smerdon, S. J., Wilkinson, A. J.,
Singleton, E. W., and Olson, J. S. (1993) The mechanism of
autooxidation of myoglobin, J. Biol. Chem., 268,
6995-7010.
5.Allen, K. E., and Cornforth, D. P. (2006) Myoglobin
oxidation in a model system as affected by nonheme iron and chelating
agents, J. Agric. Food Chem., 54, 10134-10140.
6.Augustin, M. A., and Yandell, J. K. (1979)
Oxidation of heme proteins by copper(II) complexes. Rates and mechanism
of the copper catalyzed autoxidation of cytochrome c, myoglobin
and hemoglobin, Inorg. Chim. Acta, 37, 11-18.
7.Eguchi, L. A., and Saltman, P. (1984) The aerobic
reduction of Fe(III) complexes by hemoglobin and myoglobin, J. Biol.
Chem., 259, 14337-14338.
8.Hegetschweiler, K., Saltman, P., Dalvit, C., and
Wright, P. E. (1987) Kinetics and mechanisms of the oxidation of
myoglobin by Fe(III) and Cu(II) complexes, Biochim. Biophys.
Acta, 912, 384-397.
9.Eguchi, L. A., and Saltman, P. (1987) Kinetics and
mechanisms of metal reduction by hemoglobin. 2. Reduction of copper(II)
complexes, Inorg. Chem., 26, 3669-3672.
10.Hura, C., Palamaru, I., and Hura, B. (2002)
Assessment of some heavy metals in the maternal body, risk in cancer
disease, in Metal Ions in Biology and Medicine: Proc. 7th Int. Symp.
on Metal Ions in Biology and Medicine (Khassanova, L., Collery,
Ph., Maymard, I., Khassanova, Z., and Etienne, J.-C., eds.) John Libbey
Eurotext, St. Petersburg, Vol. 7, pp. 621-624.
11.Mauk, M. R., Rosell, F. I., and Mauk, A. G.
(2009) Metal ion facilitated dissociation of heme from b-type
heme proteins, J. Am. Chem. Soc., 131, 16976-16983.
12.Clopton, D. A., and Saltman, P. (1997)
Copper-specific damage in human erythrocytes exposed to oxidative
stress, Biol. Trace Elem. Res., 56, 231-240.
13.Gunther, M. R., Sampath, V., and Caughey, W. S.
(1999) Potential roles of myoglobin autoxidation in myocardial
ischemia-reperfusion injury, Free Radic. Biol. Med., 26,
1388-1395.
14.Stadtman, E. R., and Oliver, C. N. (1991)
Metal-catalyzed oxidation of proteins, J. Biol. Chem.,
266, 2005-2008.
15.Van Dyke, B. R., and Saltman, P. (1996)
Hemoglobin: a mechanism for the generation of hydroxyl radicals,
Free Radic. Biol. Med., 20, 985-989.
16.Sievers, G., and Ronnberg, M. (1978) Study of the
pseudoperoxidative activity of soybean leghemoglobin and sperm whale
myoglobin, Biochim. Biophys. Acta, 533, 293-301.
17.Puppo, A., Rigaud, G., Job, D., Ricard, G., and
Zeba, B. (1980) Peroxidase content of soybean root nodules, Biochim.
Biophys. Acta, 614, 303-312.
18.Flogel, U., Godecke, A., Klotz, L.-O., and
Schrader, J. (2004) Role of myoglobin in the antioxidant defense of the
heart, FASEB J., 18, 1156-1158.
19.Widmer, C. C., Pereira, C. P., Gehrig, P.,
Vallelian, F., Schoedon, G., Buehler, P. W., and Schaer, D. (2010)
Hemoglobin can attenuate hydrogen peroxide-induced oxidative stress by
acting as an antioxidative peroxidase, Antioxid. Redox Signal.,
12, 185-198.
20.Arihara, K., Cassens, R. G., Greaser, M. L.,
Luchansky, J. B., and Mozdziak, P. E. (1995) Localization of
metmyoglobin-reducing enzyme (NADH-cytochrome b5
reductase) system components in bovine skeletal muscle, Meat
Sci., 39, 205-213.
21.Topunov, A. F., Melik-Sarkisyan, S. S., Lysenko,
L. A., Karpilenko, G. P., and Kretovich, V. L. (1980) Properties of
metleghemoglobin reductase from lupine root nodules, Biokhimiya,
45, 2053-2058.
22.Topunov, A. F., and Golubeva, L. I. (1989)
Reductases reducing oxygen-transporting hemoproteins: hemoglobin,
myoglobin, and leghemoglobin, Usp. Biol. Khim., 30,
239-252.
23.Zhang, B.-J., Smerdon, S. J., Wilkinson, A. J.,
and Sykes, A. G. (1992) Oxidation of residue 45 mutant forms of pig
deoxymyoglobin with [Fe(CN)6]3–, J.
Inorg. Biochem., 48, 79-84.
24.Dunn, C. J., Rohlfs, R. J., Fee, J. A., and
Saltman, P. (1999) Oxidation of deoxymyoglobin by
[Fe(CN)6]3–, J. Inorg. Biochem.,
75, 241-244.
25.Marcus, R. A., and Sutin, N. (1985) Electron
transfers in chemistry and biology, Biochim. Biophys. Acta,
811, 265-322.
26.Margalit, R., Pecht, I., and Gray, H. B. (1983)
Oxidation-reduction catalytic activity of a pentaammineruthenium (III)
derivative of sperm whale myoglobin, J. Amer. Chem. Soc.,
105, 301-302.
27.Reid, L. S., Gray, H. B., Dalvit, C., Wright, P.
E., and Saltman, P. (1987) Electron transfer from cytochrome
b5 to iron and copper complexes, Biochemistry,
26, 7102-7107.
28.Rifkind, J. M. (1974) Copper and the autoxidation
of hemoglobin, Biochemistry, 13, 2475-2481.
29.Khristova, P. K., Devedzhiev, Ya. D., Atanasov,
B. P., and Volkenshtein, M. V. (1980) Studies of electron transfer in
hemoproteins. IV. Sperm whale oxymyoglobin oxidation catalyzed by
copper ions, Mol. Biol. (Moscow), 14, 1088-1097.
30.Postnikova, G. B., Moiseeva, S. A., and
Shekhovtsova, E. A. (2010) The main role of inner histidines in the
molecular mechanism of myoglobin oxidation catalyzed by copper
compounds, Inorg. Chem., 49, 1347-1354.
31.Rifkind, J. M., Lauer, L. D., Chiang, S. C., and
Li, N. C. (1976) Copper and the oxidation of hemoglobin: a comparison
of horse and human hemoglobins, Biochemistry, 15,
5337-5343.
32.Moiseeva, S. A., Postnikova, G. B., and
Sivozhelezov, V. S. (2000) Sperm whale oxymyoglobin oxidation catalyzed
by ferrocyanide ions: kinetics and mechanism, Biophysics
(Moscow), 45, 988-997.
33.Moiseeva, S. A., Postnikova, G. B., and
Sivozhelezov, V. S. (2001) Kinetics and mechanism of oxymyoglobin
oxidation catalyzed by potassium ferrocyanide, J. Phys. Chem.
(Moscow), 75, 1504-1510.
34.Hughes, M. N. (1981) The Inorganic Chemistry
of Biological Processes, Wiley, New York, pp. 125-187.
35.Martell, A. E. (1982) Critical Stability
Constants, Plenum, New York, pp. 1-5.
36.Martell, A. E. (1981) Development of Iron
Chelators for Clinical Use (Martell, A. E., Anderson, W. F., and
Badman, D. G., eds.) Elsevier, New York, p. 67.
37.Buckingham, D. A., and Sargeson, A. M. (1964)
Chelating Agents and Metal Chelated (Dwyer, F. P., and Mellor,
D. P., eds.) Academic Press, New York, p. 237.
38.Garvan, F. L. (1964) Chelating Agents and
Metal Chelated (Dwyer, F. P., and Mellor, D. P., eds.) Academic
Press, New York, p. 283.
39.Rifkind, J. M. (1981) Copper and the oxidation of
hemoglobin, in Metal Ions in Biological Systems (Sigel, H., and
Dekker, M., eds.) New York, Vol. 12, pp. 192-232.
40.Rifkind, J. M. (1979) Oxidation of (horse)
hemoglobin by copper: an intermediate detected by electron spin
resonance, Biochemistry, 18, 3860-3865.
41.Winterbourn, C. C., and Carrell, R. W. (1977)
Oxidation of human hemoglobin by copper. Mechanism and suggested role
of the thiol group of residue β-93, Biochem. J.,
165, 141-148.
42.Banaszak, L. J., Watson, H. C., and Kendrew, J.
C. (1965) The binding of cupric and zinc ions to crystalline sperm
whale myoglobin, J. Mol. Biol., 12, 130-137.
43.Breslow, E., and Gurd, F. R. N. (1963)
Interaction of cupric and zinc ions with sperm whale metmyoglobin,
J. Biol. Chem., 238, 1332-1342.
44.Bakan, D. A., Saltman, P., Theriault, Y., and
Wright, P. E. (1991) Kinetics and mechanisms of reduction of Cu(2) and
Fe(3) complexes by soybean leghemoglobin α, Biochim. Biophys.
Acta, 1079, 182-196.
45.Jameson, R. F. (1981) Coordination chemistry of
copper with regard to biological systems, in Metal Ions in
Biological Systems (Sigel, H., ed.) Marcel Dekker, New York, pp.
1-30.
46.Postnikova, G. B., and Tselikova, S. V. (1987)
Electron transfer in hemoproteins. IX. The effect of zinc ions on the
rate of oxymyoglobin oxidation by ferricytochrome c, Mol.
Biol. (Moscow), 21, 1040-1049.
47.Shekhovtsova, E. A., and Postnikova, G. B. (2008)
Mechanism of oxymyoglobin oxidation by coper ions: myoglobins
carboxymethylated and carboxyamidated at histidine residues,
Biophysics (Moscow), 53, 562-572.
48.Cocco, M. J., Kao, Y. H., Phillips, A. T., and
Lecomte, J. T. J. (1992) Structural comparison of apomyoglobin and
metaquomyoglobin: pH titration of histidines by NMR spectroscopy,
Biochemistry, 31, 6481-6491.
49.Bashford, D., Case, D. A., Dalvit, C., Tennant,
L., and Wright, P. E. (1993) Electrostatic calculations of side-chain
pK values in myoglobin and comparison with NMR data for
histidines, Biochemistry, 32, 8045-8056.
50.Carver, J. A., and Bradbury, J. H. (1984)
Assignment of 1H NMR resonances of histidine and other
aromatic residues in met-, cyano-, oxy- and (carbon monoxy)myoglobins,
Biochemistry, 23, 4890-4905.
51.Zhang, L., Mei, Y., Zhang, Yu., Li, S., Sun, X.,
and Zhu, L. (2003) Regioselective cleavage of myoglobin with copper(2)
compounds at neutral pH, Inorg. Chem., 42, 492-498.
52.Kent, M. S., Yim, H., and Sasaki, D. Y. (2005)
Adsorption of myoglobin to Cu(2)-IDA and Ni(2)-IDA functionalized
Langmuir monolayers: study of the protein layer structure during the
adsorption process by neutron and X-ray reflectivity, Langmuir,
21, 6815-6824.
53.Van Dyke, B. R., Bakan, D. A., Glover, K. A. M.,
Hegenauer, J. C., Saltman, P., Springer, B. A., and Sligar, S. G.
(1992) Site-directed mutagenesis of histidine residues involved in
Cu(II) binding and reduction by sperm whale myoglobin, Proc. Natl.
Acad. Sci. USA, 89, 8016-8019.
54.Moiseeva, S. A., and Postnikova, G. B. (2001)
Mechanism of oxidation of oxymyoglobin by copper ions: comparison of
sperm whale, horse, and pig myoglobins, Biochemistry (Moscow),
66, 780-787.
55.Rousseaux, J., Dautrevaux, M., and Han, K. (1976)
Comparison of the amino acid sequence of pig heart myoglobin with other
ungulate myoglobins, Biochim. Biophys. Acta, 439,
55-62.
56.Goraev, E. V., Postnikova, G. B., Moiseeva, S.
A., and Shekhovtsova, E. A. (2002) Oxidation of respiratory proteins by
metals. Catalytic oxidation of oxymyoglobin by copper ions: kinetics
and mechanism, in Metal Ions in Biology and Medicine: Proc.
Seventh Int. Symp. on Metal Ions in Biology and Medicine (Khassanova,
L., Collery, Ph., Maymard, I., Khassanova, Z., and Etienne, J.-C.,
eds.) John Libbey Eurotext, St. Petersburg, Vol. 7, pp. 68-72.
57.Gray, R. D. (1969) The kinetics of oxidation of
copper(I) by molecular oxygen in perchloric acid-acetonitrile solution,
J. Amer. Chem. Soc., 91, 56-62.
58.Antonini, E., Brunori, M., and Wyman, J. (1965)
Studies on the oxidation-reduction potentials of heme proteins. IV. The
kinetics of oxidation of hemoglobin and myoglobin by ferricyanide,
Biochemistry, 4, 545-551.
59.Brunori, M., Saggese, U., Rotilio, G. C.,
Antonini, E., and Wyman, J. (1971) Redox equilibrium of sperm-whale
myoglobin, Aplysia myoglobin, and Chironomus thummi
hemoglobin, Biochemistry, 10, 1604-1609.
60.Zhang, B. J., Andrew, C. R., Tomkinson, N. P.,
and Sykes, A. G. (1992) Reactivity patterns for redox reactions of
monomer forms of myoglobin, hemocyanin and hemerythrin, Biochim.
Biophys. Acta, 1102, 245-252.
61.Colotti, G., Verzili, D., Boffi, A., and
Chiancone, E. (1994) Identification of the site of ferrocyanide binding
involved in the intramolecular electron transfer process to oxidized
heme in Scapharca dimeric hemoglobin, Arch. Biochem.
Biophys., 311, 103-106.
62.Egyed, A., May, A., and Jacobs, A. (1980)
Transferrin-bipyridine iron transfer mediated by hemoproteins,
Biochim. Biophys. Acta, 629, 391-398.
63.Eguchi, L. A., and Saltman, P. (1987) Kinetics
and mechanisms of metal reduction by hemoglobin. 1. Reduction of
iron(III) complexes, Inorg. Chem., 26, 3665-3669.
64.Harrington, J. P., and Hicks, R. L. (1994)
Spectral analysis of Fe(III) complex reduction by hemoglobin: possible
mechanisms of interaction, Int. J. Biochem., 26,
1111-1117.
65.Cassatt, J. C., Marini, C. P., and Bender, J. W.
(1975) The reversible reduction of horse metmyoglobin by the iron(II)
complex of
trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetate,
Biochemistry, 14, 5470-5475.
66.Yamada, T., Marini, C. P., and Cassatt, J. C.
(1978) Oxidation-reduction reactions of hemoglobin A, hemoglobin M
Iwate, and hemoglobin M Hyde Park, Biochemistry, 17,
231-236.
67.Lim, A. R., and Mauk, A. G. (1985) Kinetic
analysis of metsulphmyoglobin and metmyoglobin reduction by
Fe(EDTA)2–, Biochem. J., 229,
765-769.
68.Shekhovtsova, E. A., Goraev, E. V., Sivozhelezov,
V. S., and Postnikova, G. B. (2005) The oxidation of sperm whale,
horse, and pig oxymyoglobins catalyzed by ferrocyanide ions: kinetics
and mechanism, Biophysics (Moscow), 50, 33-42.
69.Shekhovtsova, E. A., Goraev, E. V., Sivozhelezov,
V. S., and Postnikova, G. B. (2005) The mechanism of oxymyoglobin
oxidation catalyzed by ferrocyanide ions: chemically modified and
mutant sperm whale myoglobins, Biophysics (Moscow), 50,
552-561.
70.Postnikova, G. B., Moiseeva, S. A., Goraev, E.
V., and Shekhovtsova, E. A. (2007) Ferrocyanide – a novel
catalyst for oxymyoglobin oxidation by molecular oxygen, FEBS
J., 274, 5360-5369.
71.Postnikova, G. B., Tselikova, S. V., and
Sivozhelezov, V. S. (1992) Study of electron transport in heme
proteins. X. Effect of pH, ionic strength, and zinc ions and the rate
of ferricytochrome c reduction by oxymyoglobin from swine heart,
Mol. Biol. (Moscow), 26, 880-890.
72.Cher, M., and Davidson, N. (1955) The kinetics of
the oxygenation of ferrous iron in phosphoric acid solution, J.
Amer. Chem. Soc., 77, 793-798.
73.Stadtman, E. R., and Oliver, C. N. (1991)
Metal-catalyzed oxidation of proteins, J. Biol. Chem.,
266, 2005-2008.
74.Gao, X., Liu, Y., and Song, Zh. (2007) Catalytic
effect of ferricyanide between myoglobin and luminol and effect of
temperature, Luminescence, 22, 88-91.
75.Song, Zh., Wang, L., and Hou, S. (2004) A study
of the chemiluminescence behavior of myoglobin with luminol and its
analytical application, Anal. Bioanal. Chem., 378,
529-535.
76.Goucher, C. R., and Taylor, J. F. (1964)
Compounds of ferric iron with adenosine triphosphate and other
nucleoside phosphates, J. Biol. Chem., 239,
2251-2255.