Cells Resistant to Toxic Concentrations of Manganese Have Increased Ability to Repair DNA
K. A. Zakharcheva, L. V. Gening, K. Yu. Kazachenko, and V. Z. Tarantul*
Institute of Molecular Genetics, Russian Academy of Sciences, 123182 Moscow, Russia; E-mail: tarantul@img.ras.ru* To whom correspondence should be addressed.
Received July 8, 2016; Revision received September 5, 2016
Manganese (Mn) is crucially important for vital activity of cells and has many biological functions. Nevertheless, high doses of Mn taken up by an organism over a long period may cause neurodegenerative diseases such as manganism and Parkinsonism. The molecular mechanisms of this Mn toxicity are still poorly studied. It is now believed that Mn-induced pathophysiological neural processes are multifaceted and affect several metabolic pathways. In particular, Mn ions might affect the processes of DNA replication and repair. To test this possibility, we obtained an SKOV-3 cell line resistant to the toxic action of Mn ions. We found that these cells are characterized by the activation of poly(ADP-ribose)polymerase, which leads to increased ability to repair DNA. Thus, the model used here supports the suggestion that at least one cause of Mn cytotoxicity might be disorders of the processes involved in DNA replication and repair.
KEY WORDS: Mn toxicity, manganism, Parkinsonism, DNA repairDOI: 10.1134/S0006297917010047
Abbreviations: DTT, dithiothreitol; misGvA, misincorporation of G versus A; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PAR, poly(ADP-ribose); PARP-1, poly(ADP-ribose)polymerase-1; Pol ι, DNA polymerase iota.
Manganese performs many diverse biological functions and is a trace
element necessary for the normal vital activity of mammals. Manganese
ions (Mn2+) are involved in many physiological processes:
protein, carbohydrate, and fat metabolism; energy metabolism;
connective tissue formation; and bone tissue mineralization. Manganese
ions also act as an activator and cofactor of many enzymes [1].
However, long-term uptake of high doses of these ions can lead to the development of diseases such as manganism and Parkinsonism. According to modern concepts, these diseases with similar symptoms are most likely caused by common molecular mechanisms [2]. A single dose or not very frequent uptake of even high doses of manganese has actually no effect on the human organism. However, it is difficult to control the content of manganese because it is extremely widespread in foods and in the environment, in contrast to other toxic and carcinogenic metals. It can be assumed that almost each individual suffers to some extent from its excessive uptake. For example, according to data of the United States Environmental Protection Agency, the reference dose of manganese is 0.14 mg per kg weight per day (9.8 mg per 70 kg) [3]. The Food and Nutrition Board of the National Research Council (United States) has established that 1.4 to 9 mg of manganese can enter the human organism daily only from food [4]. At the same time, the safe upper limit of chronic uptake of manganese is 11 mg per day.
The molecular mechanisms of the toxic effect of Mn2+ on the nervous system are still not fully known. One of several existing hypotheses associates manganese-induced toxicity with disorders of mitochondrial metabolism [5]; it is also supposed that Mn2+ are involved in production of reactive oxygen species [6]. In contrast, there is a hypothesis suggesting that divalent manganese ions increasing the level of inclusion of incorrect nucleotides by some DNA polymerases in the DNA-polymerase reaction in vitro are able thereby to cause enhanced level of mutagenesis and lead to cell death [7]. In eukaryotes, the best candidate for this function may be DNA polymerase iota (Pol ι), with its incorrect activity increasing hundreds of times in the presence of Mn2+ [8].
Another hypothesis associates manganese toxicity with inhibition of the enzyme poly(ADP-ribose)polymerase-1 (PARP-1) [9].
One approach for elucidation of the mechanisms of toxicity of Mn2+ is to study cells resistant to these ions. With this object in view, in this work we selected clones of SCOV-3 cells that are able to survive in the presence of toxic concentrations of Mn2+ and studied the molecular mechanisms of cell adaptation to such concentrations of these metal ions. The results indicate that enhanced cell resistance to Mn2+ is associated with higher DNA repair ability of the cells.
MATERIALS AND METHODS
Cell culture. The mechanisms of the toxicity of Mn2+ were investigated using the SKOV-3 (ovarian adenocarcinoma) cell line. Previously, in cell extracts of this line we had found high Pol ι activity at manganese ion concentrations of 50-250 µM and T-stop overcoming (continuation of DNA synthesis after incorrect insertion of noncomplementary guanine opposite thymine of the template) [10].
The cells were grown at 37°C in 5% CO2 atmosphere in RPMI-1640 medium containing 10% newborn calf serum and penicillin at concentration 100 U/ml.
Study of cytotoxicity of Mn2+. The cytotoxicity of Mn2+ was studied by staining living cells with the vital dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), which is incorporated into the respiratory chain of living cells and inhibits mitochondrial dehydrogenases, interacting with oxygen with the formation of water-insoluble purple formazan crystals.
The cells were inoculated in cell culture microplates (50,000 cells per well) and, after 24 h, MnCl2 solution (25 to 200 µM) was added to the incubation medium. Cells grown without manganese were used as a control. The cells were incubated with the MnCl2 solution for 4 days. After that, the dye MTT was added to the medium up to final concentration 0.5 mg/ml. The staining process lasted for 3 h at 37°C in 5% CO2 atmosphere; then the incubation medium was sampled, and formazan crystals were dissolved in lysis buffer (sodium acetate, 410 mg; sodium dodecyl sulfate, 40 mg; acetic acid, 4 ml; dimethylformamide, 100 ml; and water, 100 ml).
The optical density of formazan crystals dissolved in the lysis buffer was measured with a spectrophotometer at 570 nm. Nonspecific absorbance was measured at 650 nm. The intensity of MTT staining (Dn) was calculated as the difference of optical densities at 570 and 650 nm. The MTT staining of the control cells (D0) was taken as 100%. The portion of living cells (F) was calculated by the formula: F = Dn/D0 × 100%. The results were analyzed in Origin 8.1.
Obtaining cell clones resistant to toxic concentrations of Mn2+. The SKOV-3 cells were incubated for 4 days with 100 µM MnCl2 in RPMI-1640 medium containing 10% serum. Then the medium was changed. The surviving cells were grown up to a monolayer in medium without Mn2+ and again incubated for 4 days in medium with 150 µM MnCl2. Then this procedure was repeated with 200 µM Mn2+. For obtaining a pure cell line resistant to the toxic concentrations of Mn2+, the resulting cells were inoculated in a 96-well microplate (3 cells per well) and grown up to a monolayer, followed by sampling of clones morphologically different from cells of the original SKOV-3 line. Twelve clones of cells resistant to enhanced concentrations of Mn2+ were obtained. The clone designated as cKZ-5 was used in further work.
The probability of generation of cells resistant to the toxic concentrations of Mn2+ was studied in the original SKOV-3 culture inoculated in a 24-well microplate (100,000 cells per well). The following day, Mn2+ was added to the cells at concentration 200 µM, and the cells were incubated for 4 days; then the medium was changed, and the ratio of surviving cells to the control (the cell number before the addition of Mn2+) was determined in a Goryaev chamber after 24 h.
DNA Pol ι activity measurement. The activity of Pol ι in cell extracts was measured by the misGvA method based on the difference in electrophoretic mobility between the primer extended by the Pol ι and the primer extended by other DNA polymerases in the reaction mixture [11].
The extracts were prepared as follows: the SKOV-3 cells and the cells resistant to the toxic manganese concentrations were grown up to a monolayer in 6-cm Petri dishes, trypsinized, and homogenized on ice with a Teflon homogenizer in 1× PBS solution, pH 7.4, in 50-µl volume. The resulting homogenate was centrifuged at 4°C for 10 min at 14,000g, and the supernatant was taken for further reaction.
The two complementary deoxyribooligonucleotides, a 17-membered primer 5′-GGAAGAAGAAGTATGTT-3′ and a 30-membered template 5′-CCTTCTTCATTCTAACATACTTCTTCTTCC-3′, forming a duplex with a protruding 5′-end during hybridization, were used in the reaction as substrate 1. The primer was labeled at the 5′-end with phage T4 polynucleotide kinase (10 units) and [γ_32P]ATP (2 MBq) for 30 min at 37°C in 70 mM Tris-HCl buffer, pH 7.6, containing 10 mM MgCl2 and 5 mM dithiothreitol (DTT). Then the enzyme was inactivated at 70°C for 10 min, and the labeled primer (10 pmol) was annealed from 15 pmol of the template in 200 µl with the addition of NaCl to 100 mM at 73°C for 3 min, followed by cooling to room temperature.
During the reaction, 20 µl of the reaction mixture containing the substrate (50 nM), the cell extracts (4 µl), the dATP and dGTP mixture (0.5 mM), Tris-HCl (50 mM), pH 8.0, and MnCl2 (0.2 mM) was incubated for 20 min at 37°C. The reaction was stopped by cooling on ice, followed by the addition of 20 µl of mixture containing 95% formaldehyde, 0.05% xylene cyanol, and 0.05% bromophenol blue. The samples were applied to 18% polyacrylamide gel with 7 M urea in Tris-borate buffer, pH 8.0. Electrophoresis was carried out at current intensity 15 mA prior to the exit of bromophenol blue from the gel. Then the gel was fixed for 10 min in solution containing 10% acetic acid and 30% ethyl alcohol, dried, and radioautographed for 24 h. The radioautograph was scanned with a Storm 840 phosphorimager (Amersham Biosciences, USA). The results were analyzed using ImageQuant 5.2.
Measurement of level of poly(ADP-ribosyl)ation. The level of poly(ADP-ribosyl)ation was measured by immunofluorescence assay using highly specific anti-PAR mouse monoclonal antibodies (clone 10H; Santa Cruz, USA) and Alexa 488-conjugated secondary antibodies. The cells were grown to a monolayer in a four-well microplate with 15-mm wells and then incubated with MnCl2 (100 µM) for 24 h. For inducing poly(ADP-ribosyl)ation, the cells were treated with 100 µM hydrogen peroxide for 5 min. Then the cells were washed with PBS solution and fixed on ice with methanol for 20 min, followed by twofold washing with the PBS solution. After washing, the cells were incubated for 30 min in PBS buffer containing 5% fetal bovine serum and 0.25% Triton X-100. Then the cells were stained with monoclonal antibodies 10H (5 mg/ml) in buffer containing 5% fetal bovine serum and 0.25% Triton X-100, at 4°C for 24 h. After being washed with PBS solution, the cells were stained with Alexa 488-conjugated secondary antibodies (1 : 1000 dilution) for 1 h at room temperature in the dark, followed by washing with PBS solution and nuclear chromatin staining with propidium iodide (PI) solution (1 mg/ml) in PBS (1 : 3000) for 5 min at room temperature. After staining, the cells were washed with the PBS solution, dried slightly, and placed on to glass slides. The cell preparations were examined under a fluorescence microscope (Axio Zeiss Imager Z1, Switzerland). The results were analyzed with Image J.
Analysis of correcting exonuclease activity in cell extracts. The correcting exonuclease activity in cell extracts was studied using the same 30-membered template as for the analysis of the Pol ι activity and the 18-membered primer 5′-GGAAGAAGAAGTATGTTG-3′. Oligonucleotide substrate 2 obtained after their annealing contained an unpaired nucleotide at the 3′-end of the 18-membered primer, which was identical to the aberrant product synthesized by Pol ι on substrate 1. At the same time, all manipulations with substrate 2 were performed exactly as in the study of Pol ι activity described above.
Determination of AP endonuclease activity in cell extracts. The AP endonuclease activity was determined using a duplex of 23-membered oligonucleotides, where oligonucleotide A was 5′-CTCTCCCTTCUCTCCTTTCCTCT-3′ containing a uracil residue at position 11 and labeled with P32 at the 5′-end. Oligonucleotide B was 3′-GAGAGGGAAGAGAGGAAAGGAGA-5′ and was annealed with nucleotide A without any changes. A duplex containing the AP region was obtained by treating the initially obtained duplex with E. coli uracil glycosylase in buffer containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM DTT, and 1 mM EDTA for 10 min at 37°C. The AP endonuclease activity was analyzed as described in [12].
RESULTS
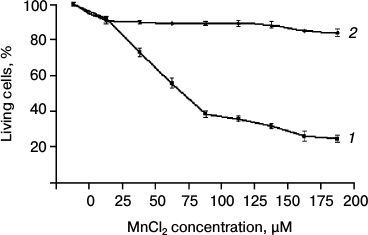
Cytotoxicity of Mn2+ in SKOV-3 cell culture. The cytotoxicity of Mn2+ was measured by the MTT test. The SKOV-3 cells were incubated in a 96-well microplate with MnCl2 at concentrations from 0 to 200 µM for 4 days (Fig. 1a). The cytotoxic effect of Mn2+ began to be observed only on day three. The optical density of MTT-stained samples was used to calculate the percentage of surviving cells relative to the control (without MnCl2). The presented diagram (Fig. 1) shows that more than half of the original cells die at Mn2+ concentrations above 75 µM. At 200 µM Mn2+, cell death reached more than 80%.
Fig. 1. Cytotoxicity of Mn2+ measured by the MTT test: 1) original SKOV-3 cells; 2) cKZ-5 cells resistant to toxic concentrations of Mn2+.
Obtaining cell clones resistant to toxic concentrations of Mn2+. The cells obtained by incubation of SKOV-3 cells with different concentrations of Mn2+ (see “Materials and Methods”) tolerated up to 200 µM Mn2+ and differed from the initial SKOV-3 cell culture in lower proliferation rate and altered morphology. The shape of these cells was more elongated, untypical of the initial SKOV-3 cells (Fig. 2, a and b).
Fig. 2. Cell morphology: a) original SKOV-3 cell culture; b) clone cKZ-5 cells resistant to toxic manganese concentrations; c) concentric ring-like colonies of clones resistant to Mn2+.

Then the cells were inoculated into a 96-well microplate at 3 cells per well to obtain a pure cell culture containing only the cells resistant to the toxic manganese concentrations. As a result, 11 clones were found that grew with the form of concentric ring-like colonies (Fig. 2c) untypical of the original SKOV-3 cells and other clones. These clones were samples and tested for survival in the presence of toxic concentrations of Mn2+. The cells were inoculated into a 24-well microplate and grown to a monolayer, and then they were incubated with Mn2+ at concentrations from 0 to 200 µM for 4 days; the percentage of surviving cells was determined by MTT staining (Figs. 1 and 2). The survival rate of selected cells in the presence of Mn2+ concentrations toxic for the original SKOV-3 cells increased considerably. The incubation of resistant cells with 200 µM of Mn2+ resulted in about 20% cell death on average, i.e. three times less than for the original SKOV-3 cell line. The resulting cells were designated as cKZ-5.
The probability of transformation of a separate SKOV-3 cell during the selection into a cell resistant to toxic concentrations of Mn2+ was 3.6·10–5.
DNA Pol ι activity in manganese-resistant clones. The range of manganese concentrations toxic for SKOV-3 cells is approximately the same (from 50 to 100 µM Mn2+) as when enhanced activity of Pol ι is observed [10]. Consequently, it may be supposed that this DNA polymerase causes cell death by inducing enhanced mutagenesis. For elucidating this question, the Pol ι activity was studied in the cells resistant to the toxic concentrations of Mn2+. The comparison of DNA Pol ι activity in the resistant clones with the activity in the original SKOV-3 cell culture by the misGvA method showed that the product of DNA Pol ι activity (18G) is present both in the resistant clones and in the original SKOV-3 cell line (Fig. 3). At the same time, its amount is approximately the same in all of the analyzed cells. Thus, we cannot see any noticeable relationship between Pol ι activity and cell resistance to the high concentrations of Mn2+.
Fig. 3. Pol ι activity in SKOV-3 cells and in clones resistant to toxic concentrations of Mn2+ measured by the misGvA method. The number of nucleotides in the DNA polymerase reaction products and in the initial primer is indicated on the electrophoregram on the right. Band 18G is a product of Pol ι. Band 18A and other bands are products of other DNA polymerase extracts.
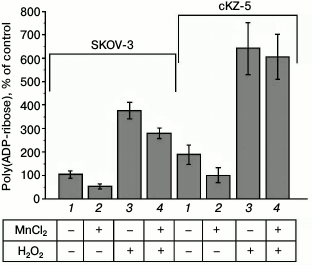
PARP activity in SKOV-3 cells and in cell clones resistant to Mn2+. The poly(ADP-ribosyl)ation level in the cells was determined by immunological detection of poly(ADP-ribose) using highly specific monoclonal antibodies and Alexa 488-conjugated secondary antibodies. Enhanced poly(ADP-ribosyl)ation was caused by inducing errors in the DNA structure by 5-min hydrogen peroxide treatment of the cells. Figure 4 shows that poly(ADP-ribosyl)ation in the cells resistant to Mn2+ is almost 2-fold higher than in the original SKOV-3 line both at the basal level and under the influence of hydrogen peroxide, which is indicative of enhnaced PARP activity in the cells resistant to Mn2+. Previously, it was shown that nontoxic concentrations of Mn2+ after 24-h incubation inhibit poly(ADP-ribosyl)ation in the cells [9]. According to our data, the level of poly(ADP-ribosyl)ation in the original SKOV-3 cell line decreased by 50% during the incubation with 100 µM MnCl2 for 24 h and by 33% after the treatment with hydrogen peroxide for 5 min. In the cells resistant to Mn2+, the level of poly(ADP-ribosyl)ation also decreased by 50% during the incubation with 100 µM MnCl2 for 24 h and remained essentially unvaried (decreased by no more than 5%) after the teatment with hydrogen peroxide.
Fig. 4. Level of poly(ADP-ribosyl)ation in original SKOV-3 cells and in cells resistant to Mn2+ (cKZ-5 clone): 1) control; 2) poly(ADP-ribosyl)ation in cells incubated with MnCl2 for 24 h; 3) poly(ADP-ribosyl)ation in cells treated with H2O2; 4) poly(ADP-ribosyl)ation in cells incubated with MnCl2 and H2O2.
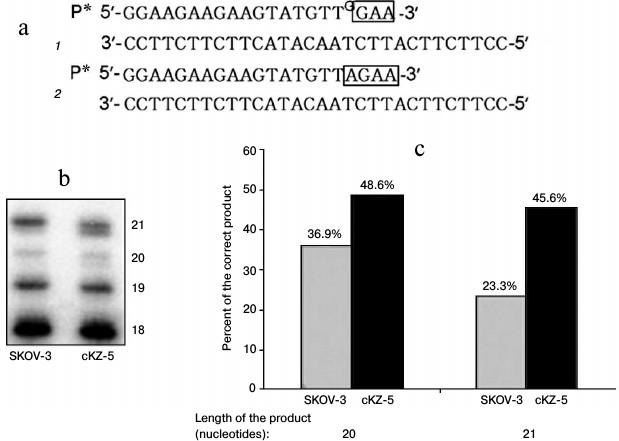
Analysis of correcting exonuclease activity in cell extracts. Repair activity in cells was analyzed using an oligonucleotide substrate that we used previously to study the activity of Pol ι, with the only difference being in this case, at the 3′-end of the 32P-labeled primer there was an additional nucleotide G noncomplementary to T of the template. This substrate is actually identical to the product of Pol ι. Since, in this case the primer contains an unpaired nucleotide, the following two variants of DNA synthesis are possible under the conditions of DNA polymerase reaction in the presence of dATP and dTTP (Fig. 5): 1) synthesis continues after unpaired G, with formation of less mobile products (Fig. 5, bands 20 and 21 mer); 2) unpaired G is removed by enzymes of the cell extract, and DNA synthesis continues after the inserted complementary nucleotide A, with formation of more mobile reaction products (Fig. 5, bands 20 and 21 mer). Thus, Fig. 5 shows that the extracts of cells resistant to Mn2+ yield much more product with the correct nucleotide A.
Fig. 5. Analysis of repair activities of cell extracts: a) variants (1 and 2) of DNA synthesis in cell extracts using oligonucleotide substrate 2 (see “Materials and Methods”); b) electrophoregram of DNA polymerase reaction products (size in nucleotides is designated); c) percentage of product containing correct nucleotide of total reaction products, 20- and 21-membered, formed by SKOV-3 and cKZ-5 cell extracts.
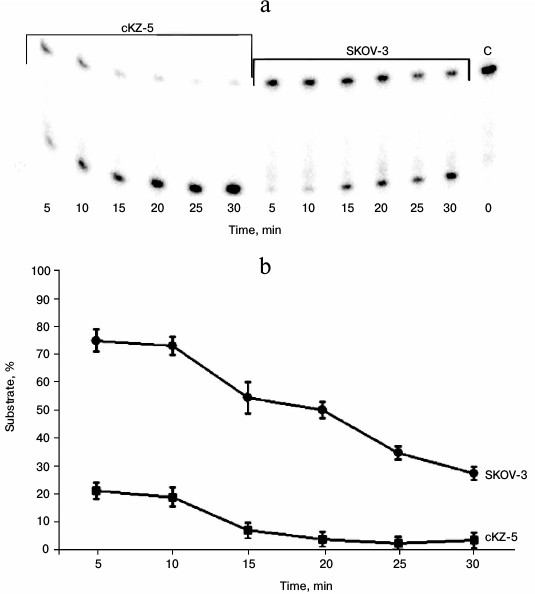
Study of AP endonuclease activity in cell extracts. The 5′ end-labeled 23-membered oligonucleotide substrate with a uracil residue at position 11 removed by uracil glycosylase was used in the experiments. Under the influence of apurinic endonucleases, this substrate is destroyed with the formation of 11- and 12-membered oligonucleotides. Figure 6 shows that the extracts of cells resistant to Mn2+ much more effectively destroy this substrate compared to the extracts of the original SKOV-3 cells.
Fig. 6. Analysis of AP endonuclease activity in cell extracts: a) electrophoregram of products formed by treatment of substrate containing a depyrimidinated region with cKZ-5 and SCOV-3 cell extracts. On right (extreme sample): C – control, i.e. substrate treated with E. coli uracil glycosylase; b) graphical representation of rates of substrate consumption in the reaction with cKZ-5 and SKOV-3 cell extracts.
DISCUSSION
The molecular mechanisms of the neurotoxic effect of manganese on the human organism in case of its chronic consumption are not quite clear yet. It is supposed that Mn2+ causes a disturbance in mitochondrial metabolism [5] and development of oxidative stress [6]. Manganese ions may cause changes in the level of neurotransmitters, as well as the activation of proteinases and apoptosis [13]. Other studies suggest that Mn2+ can have a mutagenic effect by directly interacting with DNA or influencing the activity of DNA polymerases [14, 15].
It is obvious that all of the destructive effects described can lead to cell death in the human organism, which in some cases is probably mediated by a genotoxic effect. In this work, we decided to use an alternative approach to solution of this problem. Accordingly, to understand the mechanisms of influence of Mn2+ on genome stability and to determine the factors that may be involved here, we obtained cells resistant to the effects of Mn2+ and studied their properties. The cells appeared with high frequency and had an altered morphology (Fig. 2). Probably, some of these properties can account for the causes of toxicity of Mn2+ and are associated with the development of manganism and Parkinsonism.
Among the great number of DNA polymerases present in cells, only Pol ι is strongly activated by Mn2+ compared to magnesium ions, which are the major activator of DNA replication in cells [8]. Increase in the activity of incorrect Pol ι in the presence of high Mn2+ concentrations may lead to an increase in errors during DNA synthesis and be the cause of toxicity of these metal ions [16]. If this is true, then the activity of Pol ι in the cells resistant to Mn2+ must be lower. However, our analysis of the Pol ι activity in all of the studied clones resistant to enhanced concentrations of Mn2+ showed that the activity of this enzyme is approximately the same as in the original SKOV-3 cells (Fig. 3). Thus, the survival of cells at enhanced concentrations of Mn2+ can be explained by the fact that they are able to more effectively eliminate DNA damage appearing due to the effect of faulty Pol ι.
Our analysis has shown that Mn2+-resistant cells demonstrate constitutively higher level of poly(ADP-ribosyl)ation compared to the original cells (Fig. 4). According to modern concepts, the main function of PARP in cells is to initiate the repair of damaged DNA [17]. Initiation is performed by the attachment of poly(ADP-ribose) (PAR), with the involvement of this enzyme, to particular protein factors that perform DNA repair and replication in cells. The cofactor in this reaction is NAD. At the same time, PARP attaches PAR also to its own molecule, which subsequently leads to its degradation. Thus, DNA damage results in the activation of PARP, which, in turn, initiates DNA repair and dies in its process, while normal existence of the cell continues after its DNA has been repaired. However, in case of repeated DNA damages, the content of NAD in the cell becomes exhausted, which has a negative effect on glycolysis processes and mitochondrial functions. In addition, the cells accumulate large amounts of poly(ADP-ribosyl)ation products, which results in cell death by the so-called parthanatos pathway [17].
Our data show that Mn2+-resistant cells demonstrate not only higher level of poly(ADP-ribosyl)ation, but also an increase in the correcting exonuclease activity (Figs. 5 and 6).
Our data correlate with the results of some other studies. Previously, it was shown that enhanced activity of PARP is associated with some neurodegenerative diseases. So, the brain of Alzheimer patients was shown to have higher content of protein polyadenylation products [18]. A similar tendency is observed in Parkinson’s disease. In particular, some authors have shown that the treatment of mice with the toxin MPTP (1-methyl-4-phenyl-1,2,4,6-tetrahydropyridine) causing the symptoms of Parkinson disease [19] increases PARP activity in their brains [20-22].
Together with constitutively high PARP activity, the Mn2+-resistant cells that we obtained demonstrate also increased level of apurinic endonuclease activity (Fig. 6). In addition, extracts of these cells are more efficient compared to the original cells at removing the incorrect nucleotide inserted by Pol ι and replacing it by the correct one, thereby reducing the potential mutagenic effect of Pol ι (Fig. 5). Taken together, our data indicate the intensification of repair processes in the cells resistant to Mn2+, which minimizes the negative genotoxic effects of Mn2+.
The influence of the toxic concentrations of Mn2+ on cells results in the appearance of cells resistant to these metal ions with a probability exceeding the appearance of conventional mutations. At the same time, most cells with enhanced resistance to these metal ions were less viable. Obviously, tissues consisting of cells incapable of division (e.g. brain cells) must be less resistant to such influence. Taking into consideration that manganism and Parkinsonism develop in humans over a very long period, it cannot be ruled out that less viable cells gradually accumulate in the brain under manganese excess, which may finally lead to these pathologies.
Acknowledgements
The authors are grateful to A. S. Efremova and S. I. Shram for their assistance in determination of PARP activity and to D. О. Zharkov for his assistance in the analysis of AP endonuclease activity.
This work was supported by grants from the Presidium of the Russian Academy of Sciences Program “Molecular and Cell Biology”. The work was carried out using equipment of the “Center of Genetic and Cellular Technologies” of the Federal State Budgetary Institution of Science, Institute of Molecular Genetics of the Russian Academy of Sciences.
REFERENCES
1.Santamaria, A. B., and Sulsky, S. I. (2010) Risk
assessment of an essential element: manganese, J. Toxicol. Environ.
Health, 73, 128-155.
2.Roth, J. A. (2014) Correlation between the
biochemical pathways altered by mutated Parkinson-related genes and
chronic exposure to manganese, Neurotoxicology, 44,
314-325.
3.Integrated Risk Information System (IRIS) (1996)
United States Environmental Protection Agency (USEPA).
4.Toxicological Profile for Manganese (2000) United
States Department of Health and Human Services, Agency for Toxic
Substances and Disease Registry (ATSDR).
5.Zhang, F., Xu Z., Gao, J., Xu, B., and Deng, Y.
(2008) In vitro effect of manganese chloride exposure on energy
metabolism and oxidative damage of mitochondria isolated from rat
brain, Environ. Toxicol. Pharmacol., 26, 232-236.
6.Zhang, S., Fu, J., and Zhou, Z. (2004) In
vitro effect of manganese chloride exposure on reactive oxygen
species generation and respiratory chain complexes activities of
mitochondria isolated from rat brain, Toxicol. in vitro,
18, 71-77.
7.El-Deiry, W. S., Downey, K. M., and So, A. G.
(1984) Molecular mechanisms of manganese mutagenesis, Proc. Natl.
Acad. Sci. USA, 81, 7378-7382.
8.Frank, E. G., and Woodgate, R. (2007) Increased
catalytic activity and altered fidelity of human DNA polymerase iota in
the presence of manganese, J. Biol. Chem., 282, 24689-24696.
9.Bornhorst, J., Ebert, F., Hartwig, A., Michalke,
B., and Schwerdtle, T. (2010) Manganese inhibits poly(ADP-ribosyl)ation
in human cells: a possible mechanism behind manganese-induced toxicity,
J. Environ. Monitor., 12, 2062-2069.
10.Lakhin, A. V., Efremova, A. S., Makarova, I. V.,
Grishina, E. E., Shram, S. I., Tarantul, V. Z., and Gening, L. V.
(2013) Effect of Mn(II) on erroneous activity of DNA polymerase iota in
the extracts of normal and tumor human cells, Mol. Genet. Mikrobiol.
Virusol., 1, 14-20.
11.Makarova, A. V., Grabow, C., Gening, L. V.,
Tarantul, V. Z., Tahirov, T. H., Bessho, T., and Pavlov, Y. I. (2011)
Inaccurate DNA synthesis in cell extracts of yeast producing active
human DNA polymerase iota, PLoS One, 6, e16612.
12.Torgasheva, N. A., Menzorova, N. I., Sibirtsev,
Y. T., Rasskazov, V. A., Zharkov, D. O., and Nevinsky, G. A. (2016)
Base excision DNA repair in the embryonic development of the sea urchin
Strongylocentrotus intermedius, Mol. Biosyst., 12,
2247-2256.
13.Quintanar, L. (2008) Manganese neurotoxicity: a
bioinorganic chemist’s perspective, Inorg. Chim. Acta,
361, 875-884.
14.Beckman, R. A., Mildvan, A. S., and Loeb, L. A.
(1985) On the fidelity of DNA replication: manganese mutagenesis in
vitro, Biochemistry, 8, 5810-5817.
15.Bhanot, O. S., and Solomon, J. J. (1994) The role
of mutagenic metal ions in mediating in vitro mispairing by
alkylpyrimidines, Environ. Health Perspect., 102,
81-90.
16.Lakhin, A. V., Tarantul, V. Z., and Gening, L. V.
(2014) Manganese-induced incorrect DNA synthesis as a possible cause of
manganism, Mol. Genet. Mikrobiol. Virusol., 1, 15-21.
17.Martire, S., Mosca, L., and D’Erme, M.
(2015) PARP-1 involvement in neurodegeneration: a focus on
Alzheimer’s and Parkinson’s diseases, Mech. Ageing
Dev., 146, 53-64.
18.Love, S., Barber, R., and Wilcock, G. K. (1999)
Increased poly(ADP-ribosyl)ation of nuclear proteins in
Alzheimer’s disease, Brain J. Neurol., 122,
247-253.
19.Dawson, V. L., and Dawson, T. M. (1996) Nitric
oxide neurotoxicity, J. Chem. Neuroanat., 10,
179-190.
20.Cosi, C., Chopin, P., and Marien, M. (1996)
Benzamide, an inhibitor of poly(ADP-ribose)polymerase, attenuates
methamphetamine-induced dopamine neurotoxicity in the C57B1/6N mouse,
Brain Res., 735, 343-348.
21.Wang, H., Shimoji, M., Yu, S. W., Dawson, T. M.,
and Dawson, V. L. (2003) Apoptosis inducing factor and PARP-mediated
injury in the MPTP mouse model of Parkinson’s disease, Ann. N.
Y. Acad. Sci., 991, 132-139.
22.Wu, X. L., Wang, P., Liu, Y. H., and Xue, Y. X.
(2014) Effects of poly(ADP-ribose)polymerase inhibitor 3-aminobenzamide
on blood-brain barrier and dopaminergic neurons of rats with
lipopolysaccharide-induced Parkinson’s disease, J. Mol.
Neurosci., 53, 1-9.