Functioning of Yeast Pma1 H+-ATPase under Changing Charge: Role of Asp739 and Arg811 Residues
V. V. Petrov
Skryabin Institute of Biochemistry and Physiology of Microorganisms, 142290 Pushchino, Moscow Region, Russia; E-mail: vpetrov07@gmail.com
Received June 17, 2016; Revision received August 24, 2016
The plasma membrane Pma1 H+-ATPase of the yeast Saccharomyces cerevisiae contains conserved residue Asp739 located at the interface of transmembrane segment M6 and the cytosol. Its replacement by Asn or Val (Petrov et al. (2000) J. Biol. Chem., 275, 15709-15716) or by Ala (Miranda et al. (2011) Biochim. Biophys. Acta, 1808, 1781-1789) caused complete blockage of biogenesis of the enzyme, which did not reach secretory vesicles. It was proposed that a strong ionic bond (salt bridge) could be formed between this residue and positively charged residue(s) in close proximity, and the replacement D739A disrupted this bond. Based on a 3D homology model of the enzyme, it was suggested that the conserved Arg811 located in close proximity to Asp739 could be such stabilizing residue. To test this suggestion, single mutants with substituted Asp739 (D739V, D739N, D739A, and D739R) and Arg811 (R811L, R811M, R811A, and R811D) as well as double mutants carrying charge-neutralizing (D739A/R811A) or charge-swapping (D739R/R811D) substitutions were used. Expression of ATPases with single substitutions R811A and R811D were 38-63%, and their activities were 29-30% of the wild type level; ATP hydrolysis and H+ transport in these enzymes were essentially uncoupled. For the other substitutions including the double mutations, the biogenesis of the enzyme was practically blocked. These data confirm the important role of Asp739 and Arg811 residues for the biogenesis and function of the enzyme, suggesting their importance for defining H+ transport determinants but ruling out, however, the existence of a strong ionic bond (salt bridge) between these two residues and/or importance of such bridge for structure–function relationships in Pma1 H+-ATPase.
KEY WORDS: yeast, plasma membrane, secretory vesicles, Pma1 H+-ATPase, transmembrane segments, H+ transport, site-directed mutagenesisDOI: 10.1134/S0006297917010059
In memory of C. W. Slayman
Plasma membrane H+-ATPase of yeast and fungi (Pma1), encoded by gene PMA1 [1], belongs to a widely distributed and physiologically important group of P2-ATPases [2], which also includes plant plasma membrane H+-ATPase and mammalian H+,K+-, Na+,K+-, and Ca2+-ATPases. Being both phosphohydrolases and ion pumps, these enzymes couple energy from ATP hydrolysis to transport of different mono- and divalent cations across cellular and subcellular membranes [2, 3]. Pma1 H+-ATPase is a vital enzyme creating electrochemical proton gradient (ΔµH+) across the plasma membrane, which provides energy for functioning of secondary solute transport systems, maintaining ion homeostasis and intracellular pH, and probably indirectly regulating cell metabolism [4]. Knockout of the PMA1 gene is lethal for the cell [1].
Despite the physiological differences of P2-ATPases, their structures are similar: they have one major catalytic subunit with molecular weight of ~100 kDa anchored in the lipid bilayer by four hydrophobic transmembrane segments at the N-terminal and by six at the C-terminal ends of the enzyme; all these are α-helices of different length and inclination. These segments form the enzyme membrane domain, in which amino acid residues involved in forming the cation binding and transporting cites are located. P2-ATPases have a common catalytic mechanism in which ATP binds to a conserved aspartic residue in the cytosolic catalytic center to form covalent β-aspartyl phosphate [5], the energy of which is used to transport cations across the membrane. However, these ion pumps differ in both ion specificity and stoichiometry of transport. Site-directed mutagenesis data for Ca2+- [6-9], Na+,K+- [9-12], and H+,K+-ATPases [13-18] of animal, H+-ATPase of plant [19, 20], and Ca2+,Mn2+-ATPase of yeast cells [21, 22] point out that determinants of cation specificity and stoichiometry lie in transmembrane segments M4, M5, M6, and M8 of these enzymes; in the same segments [23-30] and the L5-6 loop connecting M5 and M6 [31-33], mutagenesis of the yeast Pma1 H+-ATPase revealed residues whose substitution affect the normal functioning and/or biogenesis of the enzyme. Involvement of M4, M5, M6, and M8 segments in formation of the cation binding and transport sites was confirmed by crystallographic analysis of animal Ca2+- [34-39] and Na+,K+-ATPases [40-44] and plant H+-ATPase [45]. Segments M6 and M8 seem to be the most interesting; the first is probably responsible for the ion transport specificity, while the latter for its stoichiometry [25, 26].
P2-ATPases undergo significant changes during their reaction cycle, reflecting the reversible transition of the enzyme between conformations E1 and E2. During the cycle, membrane segments M1-M6 (especially, M4, M5 and M6, which form transport cites) bend, change inclination, partly unwind, and shift from the membrane, while segments M7-M10 are less mobile [35-38]. In segment M6 of the Saccharomyces cerevisiae Pma1 ATPase, Asp730 appeared to be one of the most important residues, being responsible for one of the transport determinants, cation specificity. This residue participates in forming two sites of cation binding and translocation [30, 33]. In segment M8, Glu803 appeared to be the most important residue, being responsible for transport stoichiometry [25, 26]. There are also other residues whose roles seem to be important for structure–function organization of the Pma1 ATPase [25, 26, 30]. In particular, replacement of Asp739 amino acid residue located at the border of membranous and cytosolic phases by Ala completely blocked the biogenesis of yeast Pma1 ATPase [30]. It was suggested that Asp739 could play an important role in the biogenesis and functioning of the enzyme, particularly due to forming a strong ionic bond with a nearby positively charged amino acid residue. Formation of such a salt bridge was earlier postulated for Asp730 in segment M6 and Arg695 in segment M5 [46]. Located at the border of segment M8 and the cytosol, Arg811 could be the positively charged residue that is able to form a bond with Asp739. Its replacement by Ala led to threefold decrease in the enzyme expression and activity and twofold increase in uncoupling of H+ transport and ATP hydrolysis, which points to its involvement in maintaining the functioning of the ATPase [26].
The results presented here further extend the systematic study of the structure–function relations in the yeast plasma membrane Pma1 H+-ATPase by focusing on residues Asp739 and Arg811 with the goal of finding whether these residues form a strong ionic bond (salt bridge), which may be essential for maintaining the structure and function of the enzyme.
MATERIALS AND METHODS
Yeast strain. The S. cerevisiae strain SY4 (MATa; ura3-52; leu2-3, 112; his4-619; sec6-4; GAL; pma1::YIpGAL-PMA1) was used throughout the work; it contained both chromosomal (PMA1) and plasmid (pma1) copies of the gene encoding Pma1 H+-ATPase; they were under control of different promoters: the chromosomal copy of the wild-type ATPase gene was placed under the control of the GAL1 promoter (PGAL-PMA1), and the plasmid allele (on the centromeric plasmid YCp2HSE) was under the heat shock-inducible HSE promoter (PHSE-pma1) [47]. The plasmid pma1 gene was of either the wild type or mutant. The SY4 strain also carries a temperature-sensitive mutation in the SEC6 gene, which blocks the fusion of secretory vesicles to the plasma membrane under heat shock and leads to accumulation of secretory vesicles.
Site-directed mutagenesis. To introduce mutations into the BglII-SalI fragment (519 bp) of the pma1 gene that has been previously subcloned into a modified version of the Bluescript plasmid (Stratagene, USA), mutant oligonucleotides were synthesized and used to introduce mutations by means of a kit for oligonucleotide-directed mutagenesis (Amersham, USA) or polymerase chain reaction [25, 26, 47, 48]. Upon mutagenesis, each fragment was sequenced to verify the presence of the mutation and the absence of unwanted base changes. Then, the fragments were used to replace corresponding parts in the plasmid pPMA1.2 [47] carrying the entire coding sequence of the pma1 gene by means of restriction endonucleases BglII, HindIII, SacI, and SalI and T4-DNA-ligase. To express the Pma1 H+-ATPase in secretory vesicles, a HindIII-SacI piece (3.77 kb) of pPMA1.2 containing the entire coding sequence of the gene by means of restriction endonucleases HindIII and SacI and T4-DNA-ligase was transferred into centromeric plasmid YCp2HSE under the control of the HSE promoter (PHSE-pma1); this plasmid was then used to transform the SY4 cells [47]. The plasmid DNA (YCp2HSE-PMA1) from the new strains was extracted and sequenced again to confirm the presence of the mutations.
Isolation of secretory vesicles. To obtain secretory vesicles, the SY4 strain cells were grown for 16 h at 23°C in 800 ml of liquid medium containing 0.67 g/liter YNB (Difco, USA), 20 mg/liter histidine, and 2% galactose under continuous agitation until the mid-exponential phase (A600 ~ 0.7-1.0). Then, the cells were washed free from galactose and transferred into 400 ml of the same medium containing 2% glucose. After 3 h incubation with glucose, the yeast cells were subjected to a heat shock by increasing the temperature to 39°C and incubated for 2 h. Ten minutes before the end of the heat shock, 4 ml of 1 M NaN3, which blocks metabolism, was added to prevent fusion of vesicles to the plasma membrane [47], and the cell suspension was chilled in ice water. Cells were sedimented by centrifugation, washed with 10 mM NaN3, and resuspended in 1.4 M sorbitol, 10 mM NaN3, and 50 mM KH2PO4-NaOH, pH 7.5. Zymolyase 20T (ICN, USA) was added, and the cells were incubated at 39°C for 30-40 min to obtain spheroplasts. After spheroplasts were obtained, they were washed and resuspended in the same buffer and treated with concanavalin A to make plasma membranes heavier. The spheroplasts were mechanically lysed in hypotonic medium (1 mM EDTA, 10 mM triethanolamine-acetic acid, pH 7.2, 1 mM diisopropylfluorophosphate, 2 µg/ml chymostatin, and leupeptin, pepstatin, and aprotinin, 1 µg/ml each) containing 0.4 M sorbitol (purification by gel filtration) or 12.5% sucrose (purification by sucrose density gradient) as described earlier [25, 47, 48]. After preparation of the spheroplasts, all procedures were performed at 0-4°C. The homogenate was centrifuged at low speed, the pellet was discarded, and secretory vesicles containing newly synthesized ATPase were isolated from the supernatant by differential centrifugation and purification by either gel filtration [25, 47, 48] or centrifugation in a sucrose density gradient [25, 26]. They were suspended in 0.8 M sorbitol, 1 mM EDTA, 10 mM triethanolamine-acetic acid, pH 7.2, with the above-mentioned protease inhibitors (except for diisopropylfluorophosphate) as described earlier [25, 48].
Quantitation of expressed ATPase. The amount of ATPase in secretory vesicles was determined by SDS-PAGE and Western blotting as described earlier [25, 47, 48]. Blots were treated with polyclonal antibody against Pma1 H+-ATPase and then with 125I-labeled protein A (ICN, USA). The mutant Pma1 ATPase expression in the secretory vesicles was assayed using a Phosphorimager with ImageQuant software (Molecular Dynamics, USA) and calculated relative to a wild-type control run in parallel on the day of preparation [48].
ATP hydrolysis was measured for 10-30 min at 30°C in 0.5 ml of incubation mixture containing 10 mM MgSO4, 5 mM Na2ATP, 50 mM MES-Tris, pH 5.7, 5 mM KN3, and an ATP-regenerating system (5 mM phosphoenolpyruvate and 50 µg/ml pyruvate kinase (40 U/mg protein; Sigma-Aldrich, USA)) in the absence or presence of 100 µM sodium orthovanadate. To obtain Km, the true concentration of MgATP was calculated as described previously [49]. Inorganic phosphate was measured according to Fiske and Subbarow [50].
ATP-dependent H+ transport in secretory vesicles was registered at 29°C by fluorescence quenching of the pH-sensitive dye acridine orange using a Hitachi F2000 spectrofluorimeter (excitation, 430 nm; emission, 530 nm) equipped with F2000 Intracellular Cation Measurement System software (Hitachi, Japan) [47, 48]. Secretory vesicles (50-100 µg protein) were suspended in 1.5 ml of 0.6 M sorbitol, 100 mM KCl, 20 mM KNO3, 5.0 mM Na2ATP, 2 µM acridine orange, 20 mM HEPES-KOH, pH 6.7; after stabilization of the baseline fluorescence (100-150 s), the reaction was started by adding 10 mM MgCl2.
Coupling between H+ transport and ATP hydrolysis. To estimate the coupling ratio between H+ transport and ATP hydrolysis, the initial rates of ATP hydrolysis were assayed over a range of MgATP concentrations in parallel with measuring the initial rates of H+ transport under the same concentrations: in 0.1 ml (ATP hydrolysis) or 1.5 ml (H+ transport) of 0.6 M sorbitol, 20 mM HEPES-KOH, pH 6.7, 100 mM KCl, 20 mM KNO3, containing 0.2-3.0 mM Na2ATP and 5.2-8.0 mM MgCl2 at 29°C [25, 26]. The ATP hydrolysis reaction was started by adding secretory vesicle suspension; fluorescence quenching was initiated by addition of 5.2-8.0 mM MgCl2. The reaction mixture for assaying H+ transport additionally contained 2 µM acridine orange and 50-100 µg of secretory vesicle protein. The reaction of ATP hydrolysis was carried out for 30-40 min in the presence or absence of 0.1 mM Na3VO4 and stopped by addition of 1 ml of 1.25% trichloroacetic acid; inorganic phosphate was measured as above [50].
Protein assay. Protein was determined by a modified Lowry method [51] with bovine serum albumin as a standard. Aliquots of the secretory vesicle-resuspending buffer were added to the standard to compensate for absorbance changes.
Homology modeling. Three-dimensional homology models of the yeast Pma1 H+-ATPase were constructed using amino acid sequence alignment obtained by the ClustalX algorithm [52] and cryoelectronic structures of the animal SERCA1a Ca2+-ATPase and plant AHA2 H+-ATPase as described earlier [26, 30].
RESULTS
Choice of amino acid residues to be studied. Alanine-scanning mutagenesis of amino acid residues in membrane segments M6 [27, 30] and M8 [26, 29] was used previously to study structure–function relations in the yeast Pma1 H+-ATPase. It was found that more than half of the replacements (10 out of 19) in M6 [30] caused serious impairment of the enzyme functioning and/or biogenesis (Table 1). There were much fewer such residues (6 out of 21) in M8 [26]; the primary reason for this was block in biogenesis (Table 1). The main result of this study was the conclusion that negatively charged residues Asp730 (in M6) and Glu803 (in M8) are involved in forming the H+ (H3O+) ion transport sites [25-27, 29, 30, 33].
Table 1. Replacements by alanine of amino
acid residues in membrane segments M6 and M8 that led to loss of ATPase
activity and/or expression of Pma1 ATPase in secretory vesicles
(%)1
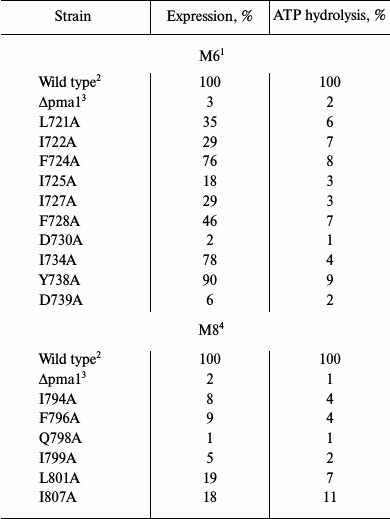
1 Specific expression (as Pma1 ATPase amount in secretory
vesicles) of 100-kDa ATPase subunit was determined by quantitative
immunoblotting as described in “Materials and Methods” and
is represented as percent of wild-type enzyme expression run in
parallel on the same day. ATP hydrolysis was assayed at pH 5.7.
2 Data are from [30]; 100% corresponded
to 4.53 ± 0.26 µmol Pi/min (U) per mg
protein. Data are average of 31 experiments for the wild type and 2-5
for mutants.
3 Data are from [26]; 100% corresponded
to 5.58 ± 0.21 U per mg of protein. Data are average of 31
experiments for the wild type and 2-5 for mutants.
4 Secretory vesicles were isolated from cells carrying
expression plasmid YCp2HSE with pma1 gene (positive control).
5 Secretory vesicles were isolated from cells carrying
expression plasmid YCp2HSE without pma1 gene (negative
control).
Since charged residues are more likely to be involved in formation of sites of binding and translocation of transported cations, our attention was focused on data for one more negatively charged residue. This residue, Asp739, located at the border of segment M6 and cytosol (Fig. 1) appeared to be extremely important for the biogenesis and functioning of the enzyme: when replaced by Ala, the newly synthesized mutant enzyme D739A did not reach secretory vesicles (Table 1), similarly to the D730A enzyme [30]. Likewise, the analogous results were obtained for earlier constructed substitutions of the Asp739 residue by neutral Val and very similar stereochemically but charge-free Asn [25]. It was suggested that one of the reasons for such dramatic effect of the Asp739 replacements could be its involvement in creating a strong ionic bond (salt bridge) with proximate positively charged amino acid residues [30], one of which could be Arg811 (Fig. 2). The replacement of this residue by Ala, even without blocking the enzyme biogenesis, essentially decreased its expression and activity (Table 1) as well as its coupling of H+ transport to ATP hydrolysis [26]. However, earlier reported substitutions of Arg811 by close in size Met or Leu almost completely blocked the enzyme biogenesis and functioning [25]. The suggestion that Asp739 and Arg811 residues form a salt bridge in 3D structure seemed to be very appealing, since the presence of such a bridge was earlier postulated for Arg695 located in M5 and the above-mentioned Asp730 in M6 [46].
Fig. 1. Amino acid residues in membrane segment M6 of yeast Pma1 ATPase that are important for biogenesis and functioning of the enzyme. The N-terminal end of M6 is highlighted in light gray. The highlighted in bold Asp739 residue is in cytosol under the M8 segment. Modified version from [30].
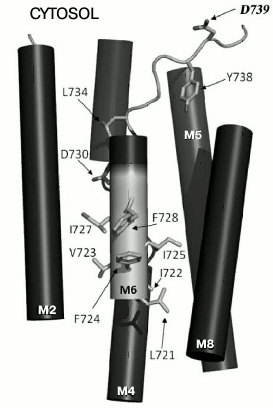
Fig. 2. Amino acid residues in membrane segment M8 of yeast Pma1 ATPase that are important for the biogenesis and functioning of the enzyme. The bold-highlighted Arg811 residue is in the cytosol under the M8 segment and extension of the M6 segment. Modified version from [26].
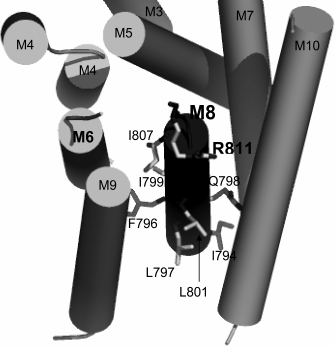
Indeed, the homology model of the central part of the yeast Pma1 H+-ATPase membrane domain was built using amino acid sequences and cryoelectronic structures of the animal SERCA1 Ca2+-ATPase [35-37] and plant AHA2 H+-ATPase [45], and it appeared that Asp739 and Arg811 are in close proximity and could interact with each other (Figs. 1 and 2). Therefore, we decided to study the roles of the Asp739 and Arg811 residues in greater detail and to explore whether forming of a salt bridge between them is possible.
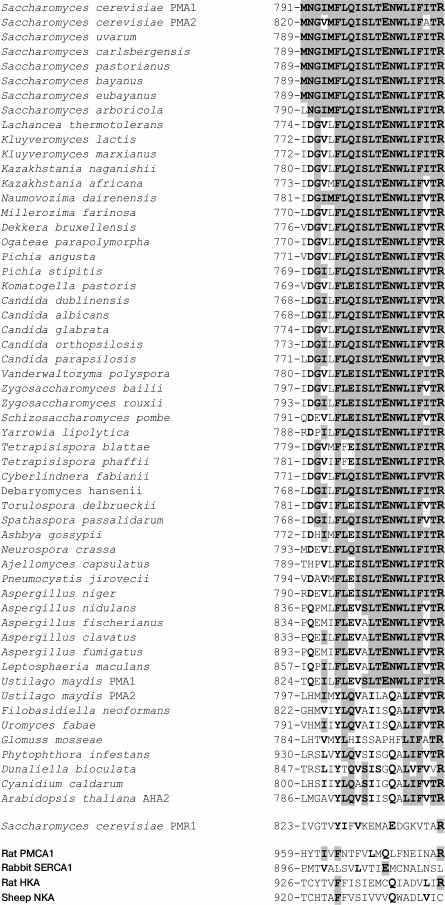
Conservation of the studied amino acid residues. As previously shown [29, 32, 33], conservation of an amino acid residue in yeast Pma1 ATPase could be of great importance, but this is not a strict rule. Conservation (identity) of the M6 residues in the ATPases of ascomycetes (from S. cerevisiae to Leptosphaeria maculans; Fig. 3) and the basidiomycete Ustilago maydis Pma1 is 74-100%. However, for the U. maydis Pma2 and the rest of fungi, algae, and plants, it dropped to 21-47%. In animal ATPases, only an aspartyl residue that corresponds to Asp730 of the S. cerevisiae Pma1 ATPase is strictly conserved; there are either polar Asn (SERCA1) or charged Glu (PMCA1, NKA, HKA; Fig. 3) shifted by one position towards the C-end in place of the S. cerevisiae Pma1 Asp739. Homology of the M6 residues is higher: 100% for ascomycetes and U. maydis Pma1 and 37-53% for the rest of fungi, algae, and plants. Homology of the M6 residues in animal ATPases ranges from 25 (PMCA1) to 40% (HKA; Fig. 3).
Conservation of M8 residues is in general lower than that in segment M6: among H+-ATPases, it is 62-100% for ascomycetes and U. maydis Pma1 and 19-38% for the rest of the cases (Fig. 4). Homology of M8 residues is similar to that in M6 for ascomycetes and U. maydis Pma1; for the rest of the cases it was 62%. M8 residue homology in animal cation pumps was significantly lower compared to M6; from 14 (SERCA1) to 24% (PMCA1). In segment M8, the residue corresponding to Glu803 was found in all H+-ATPases of ascomycetes and Pma1 of U. maydis; shifted by one position towards the C-end, a Gln residue was found in the rest of H+-ATPases (Fig. 4). It is worth mentioning that Arg811 is found in all published sequences of H+-ATPases of fungi, algae, and plants; it is also present in the yeast Ca2+,Mn2+-ATPase and in PMCA1 Ca2+- and H+,K+-ATPases, but not in the SERCA1 Ca2+- and Na+,K+-ATPases of animal cells (Fig. 4).
Fig. 3. Sequence alignment of amino acid residues in transmembrane segment M6 of ATPases from fungi, algae, plants, and animals. Identical and homologous residues are highlighted in bold, and identical residues are highlighted in gray. The Asp730 and Asp739 residues from S. cerevisiae Pma1 H+-ATPase and those identical or homologous residues in other P2-ATPases are highlighted. Transported cations are shown. The amino acid sequences were aligned using the ClustalX algorithm [52].

Fig. 4. Sequence alignment of amino acid residues in transmembrane segment M8 of ATPases from fungi, algae, plants, and animals. Identical and homologous residues are highlighted in bold, and identical residues are highlighted in gray. The Glu803 and Arg811 residues from S. cerevisiae Pma1 H+-ATPase and those identical or homologous residues in other P2-ATPases are highlighted. For details, see legend to Fig. 3.
Constructing and expressing mutants in secretory vesicles. To obtain substitutions, oligonucleotides carrying replacements of Asp739 and Arg811 residues were synthesized using the mostly frequently found codons in the yeast plasma membrane H+-ATPase [53]. To obtain double substitutions, parental DNA was annealed with two oligonucleotides carrying the mutations: for D739A/R811A, with oligonucleotides oVP39 and oVP41; for D739R/R811D, with oligonucleotides oVP38 and oVP40 (Table 2). Then, by means of site-directed mutagenesis, BglII-SalI fragments carrying mutations were obtained and cloned into the pma1 gene on the YCp2HSE plasmid under control of the heat shock-induced promoter HSE. After the S. cerevisiae cells were transformed with the plasmid, the yeast contained two copies of the gene: the chromosomal PMA1 gene was under the control of GAL1 and the plasmid pma1 copy, under the HSE promoter [47]. This allowed obtaining even inactive enzymes (Table 1).
Table 2. Oligonucleotides synthesized to
replace amino acid residues Asp739 for Ala, Arg, Val, and Asn and
Arg811 for Ala and Asp
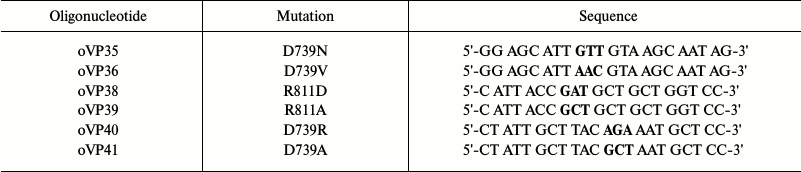
Note: Triplets encoding replacements are in bold.
The yeast was grown at 23°C in galactose medium until mid-exponential phase; the wild-type enzyme was synthesized. Then the cells were transferred into glucose medium and the synthesis from the PMA1 gene was interrupted; when the temperature was elevated to 39°C, the synthesis started from the plasmid pma1 gene, encoding either the wild-type or mutant ATPase. Strain SY4 also carried temperature-sensitive mutation sec6-4, which blocked the fusion of secretory vesicles to plasma membrane; therefore, elevation of temperature to 39°C led to accumulation of secretory vesicles with enzyme synthesized de novo from the plasmid pma1 gene [47]. Secretory vesicles isolated from the strain carrying plasmid YCp2HSE without pma1 gene (Δpma1; Table 3) was used as control for the presence of plasma membranes. The secretory vesicles were virtually free from plasma membrane contamination (4%) and were used to measure the enzyme expression (as the Pma1 ATPase amount in secretory vesicles), and phosphohydrolyzing activity determined by ATP hydrolysis sensitivity to the P-ATPase specific inhibitor orthovanadate [5]. Proton transport into secretory vesicles was registered by fluorescence quenching of the pH-sensitive dye acridine orange [25, 26, 47, 48]. To estimate coupling ratio of ATP hydrolysis to H+ transport, ATPase activity and fluorescence quenching were measured in parallel under the same conditions over a range of MgATP concentrations.
For comparison, previously obtained strains carrying substitutions R811M and R811L [25] were also used; they were studied as described above (see below).
Expression of mutant enzymes with single substitutions and measurement of ATP hydrolysis. Pma1 H+-ATPase is synthesized in endoplasmic reticulum and during maturation reaches secretory vesicles, which deliver the enzyme to the plasma membrane by fusion. There are at least two points of quality control at these steps: enzyme with serious defects does not reach secretory vesicles [54-56].
Of the mutant enzymes presented here, the majority was retained at early stages of biogenesis: from 1% (D739V and D739N) to 11% (R811M) of the mutant enzymes reached secretory vesicles (Table 3). Considering the amount of plasma membrane in the secretory vesicles preparation (4%, Δpma1; Table 3), we concluded that substitutions D739V (1%), D739N (1%), D739A (3%), and D739R (4%) led to complete, and R811L (7%) and R811M (11%) to an almost complete block in the newly synthesized ATPase trafficking, thus preventing mutant enzymes from reaching secretory vesicles (Table 3). Such behavior is typical for protein with serious defects of folding, which cause retention of the defective protein in endoplasmic reticulum [55, 56].
Table 3. Effect of different replacements of
Asp739 and Arg811 amino acid residues on expression of Pma1 ATPase in
secretory vesicles, ATP hydrolysis, and H+ transport
(%)1
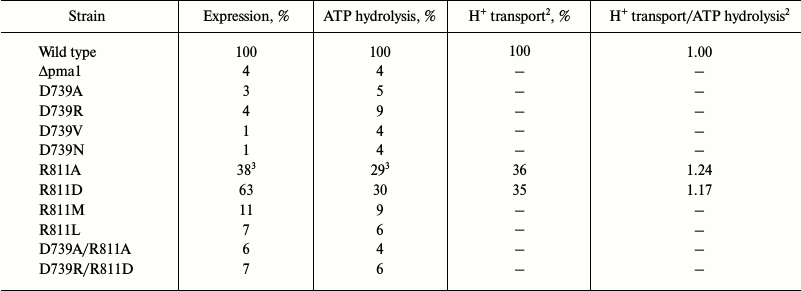
1 For details, see legend to Table 1 and “Materials and Methods”; 100%
corresponded to 4.37 ± 0.58 U per mg protein (ATP
hydrolysis). H+ transport was registered by fluorescence
quenching of acridine orange expressed as percent (% F) as described
under “Materials and Methods”; 100% corresponded to 651
± 67 % F per mg protein (H+ transport). Data are
averages of 11-18 experiments for the wild type and 2-7 for the
mutants.
2 Sensitivity of the method did not allow registering
H+ transport when ATPase activity was below 13-15% of the
wild-type level.
3 Data are from [25].
To detect synthesis of mutant ATPases that were unable to reach secretory vesicles, the SY4 cells were transferred from galactose into glucose medium as described above and metabolically labeled with [35S]methionine [32, 33, 57]. Then total membrane fraction was isolated and treated with trypsin at trypsin/protein ratio 1 : 20, subjected to SDS-PAGE and immunoblotting, and treated as described previously [25, 57]. Impairment of folding could be judged by sensitivity of protein to trypsin. Such experiment was done for the D739V mutant. By contrast to the wild type, whose 100-kDa subunit remained stable as a 97-kDa band after the cleavage of a small fragment by 10 min [32, 33], the mutant D739V ATPase was completely hydrolyzed by 0.5 min, pointing to serious folding defects that led to changes in exposure of trypsin cleavage sites. Both the 100- and 97-kDa bands of the mutant enzyme were completely hydrolyzed (not shown). Obviously, the same result should be expected for the other mutant enzymes with blocked biogenesis (compare to trypsinolysis of mutant ATPases with Asp714 residue replacements – D714A, D714C, D714V, and D714E [33].
Of the eight studied mutants with single replacements only two, R811A and R811D, were significantly expressed (38 and 63%, respectively; Table 3) and possessed reasonable activity (29-30%), which allowed their study in greater detail, including kinetics of ATP hydrolysis and proton transport (see below and Tables 3 and 4). The kinetics of ATP hydrolysis by the active mutant enzymes differed from that of the wild type only for Ki – sensitivity for the specific inhibitor orthovanadate decreased by 2.5-3.5-fold (Table 4), which could be explained by a shift in equilibrium between conformational states E1 and E2 that have different affinity for the reaction substrates and inhibitors [54]. However, these changes were rather insignificant to suggest a significant shift in the enzyme conformational state.
Proton transport and its coupling to ATP hydrolysis. Since the normal enzyme functioning is tightly bound to the ability of the pump to couple ATP hydrolysis to ATP-dependent H+ translocation [32, 33], it was of special interest to determine whether the mutants with significant enzymatic activity influence H+ transport and its coupling to ATP hydrolysis. Two enzymatically active mutants, R811A and R811D, were used to determine their ability to maintain ATP-dependent H+ transport. To do so, ATP-dependent fluorescence quenching of acridine orange was used and expressed as percent of fluorescence quenching per mg protein (H+ gradient extent; Table 3). It appeared that the H+ transport level by these mutants was the same or even somewhat higher (by 17-24%) than the ATP hydrolysis level (H+ transport/ATP hydrolysis ratio; Table 3).
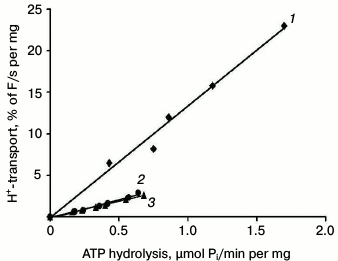
Nevertheless, the formal ratio between H+ transport and ATP hydrolysis does not reflect the true mechanism of H+-ATPase functioning [32]. To study this in greater detail and to estimate the coupling level of H+ transport to ATP hydrolysis by the enzyme, fluorescence quenching was measured over a range of different MgATP concentrations (for details, see [25, 26, 32, 33]) as percent of quenching per mg protein per second during the initial linear period (initial rate; Table 4 and Fig. 5); the rate of ATP hydrolysis by ATPase was measured in parallel at the same concentrations and conditions. Since increase in MgATP concentrations led to loss of linearity of fluorescence quenching [33], the ATP concentrations during initial rate measurements did not exceed 2-3 mM. Then the initial rate of H+ transport was plotted as a function of the ATP hydrolysis initial rates over a range of MgATP concentrations (Fig. 5). In every case, there was a roughly linear relationship between the rates of quenching and hydrolysis, as could be expected if the stoichiometry of proton transport remained unchanged over the entire range of pump velocities. The coupling ratio of the wild-type enzyme was 1.00 (Table 4). For mutants R811D and R811A, this ratio decreased twofold (0.45 and 0.52, respectively) compared to the wild type (Table 4). It should be noted that as in the case of L5-6 extracytosolic loop mutants [32, 33], the formal ratio of ATP-hydrolyzing and H+-translocating activities of the ATPase gives incorrect value (small increase in coupling ratio; Table 3), while the direct measurement of the coupling ratio reveals its twofold decrease (Table 4). In other words, these values differ by 2.5-fold. Thus, the coupling ratio reflects real mechanisms of the enzyme functioning that is in good agreement with our previous conclusions [32, 33].
Table 4. Effect of Arg811 replacements on
kinetics of ATP hydrolysis and its coupling to H+
transport

Note: Km was measured in mM ATP,
Ki in µM of sodium orthovanadate. Coupling
ratio was calculated by H+ transport rate as function of ATP
hydrolysis rate. Data are averages of 3-6 experiments. Details are
given in “Materials and Methods” and legends to Tables 1 and 3.
Fig. 5. Coupling of H+ transport and ATP hydrolysis by the wild type (1) and mutant enzymes with replacements of the Arg811 residue – R811A (2) and R811D (3). Initial rates of H+ transport were measured at ATP concentrations of 0.2-3.0 mM; then initial rates of H+ transport were plotted as a function of ATP hydrolysis rates by the Pma1 ATPase measured under the same conditions. Percent of acridine orange fluorescence quenching (% F) was measured by dividing the fluorescence quenching (ΔF) by its maximal level (Fmax). Average of the wild-type ATPase activity in secretory vesicles used for this series of experiments was 5.86 U per mg protein under standard conditions, H+-transport – 912% F per mg protein (H+ gradient extent at ATP concentration of 5 mM) and 33% F/mg per s (initial rate of H+-transport at 3 mM ATP). For details, see “Materials and Methods”.
Characterization of mutant enzymes with double substitutions. Results of substituting Asp730 and Asp739 for Val or Asn [25] and for Ala [27, 30] were the starting point of this study: biogenesis of mutant enzymes with these substitutions was completely blocked (Tables 1 and 3), which suggested the extreme importance of these aspartyl residues for the Pma1 H+-ATPase structure and function. In the case of Asp730, the suggestion that this residue forms a salt bridge with positively charged Arg695 in M5 segment was an attempt to explain the result of substitutions of this residue. It was also suggested that this residue is not involved in formation of the proton binding and translocation site [46]. The role of Asp739 was not specially discussed. However, it was later proposed that this residue could also form a strong ionic bond with positively charged residues nearby [30]; one such residue could be Arg811. It should be noted that all replacements of Asp739 (D739N, D739V, D739A, D739R) led to a complete block in the enzyme biogenesis (Tables 1 and 3) caused by the retention of the mutant enzyme at early stages of biogenesis [25, 54].
In the case of the Arg811 residue substitutions, R811L and R811M caused virtually complete block in biogenesis, while the replacement of Arg811 by Ala led to almost threefold decrease in expression and activity and twofold uncoupling of the enzyme (Tables 3 and 4). The R811D substitution was expressed 1.7-fold more than R811A, but the activity of this mutant enzyme and its transport and kinetics characteristics were the same as those of the R811A mutant. This led to the suggestion that this residue is important for structure–function relationships in the ATPase as is and/or in interacting with other residues. In the latter case, the consequences of replacement by neutral Ala or Leu and polar Met or countercharge Asp could be explained by destruction of the salt bridge between Asp739 and Arg811. Indeed, computer homology modeling showed that these residues are close to each other (Figs. 1 and 2). To test the suggestion whether residues Asp739 and Arg811 form a salt bridge, double mutants carrying replacements either neutralizing (D739A/R811A) or swapping (D739R/R811D) charges of the residues were constructed.
The secretory vesicle isolation scheme allows preparation of mutant forms of the enzyme that lost activity (Table 1). Despite this, folding of both double mutants was so impaired that these mutant enzymes were retained at early stages of biogenesis; therefore, their expression in secretory vesicles (taking into account contamination by plasma membranes) was not higher than 2-3% (Table 3). Thus, they were not different from the mutants with single substitutions that changed charge proportion in the enzyme molecule. However, if single substitutions of Arg811 by Val and Met blocked the mutant enzyme biogenesis, replacements for Ala and even Asp allowed significant expression but with significant decrease in activity and impairment of coupling (Tables 3 and 4).
It is worth noting that both in the case of removing (D739A/R811A) or swapping charges (D739R/R811D), the results were very similar. If a salt bridge was formed and/or its presence should be necessary for maintaining the Pma1 ATPase structure and functioning, the D739A/R811A mutant should be inactive (regardless of the influence of these substitutions on biogenesis), while the D739R/R811D mutant should have reasonable expression and activity. By contrast, both mutants were inactive and did not reach secretory vesicles, which was evidence that there is no salt bridge formed between Asp739 and Arg811 residues and/or this bridge does not influence the enzyme structure, biogenesis, and functioning.
DISCUSSION
Salt bridges and their role in protein structure. Salt bridges are noncovalent interactions and appear relatively often in various proteins, being responsible for conformational specificity and molecular recognition and catalysis [58-60]. They are formed most often between a carboxylic group (RCOO–) of aspartic or glutamic acid and an amine (RNH3+) or guanidine (RNHC(NH2)2+) group of lysine or arginine, respectively [58]. Salt bridges appeared to be a combination of two type of noncovalent interactions – hydrogen bonds and electrostatic interactions; amino acids with ionized side chains such as His, Tyr, and Ser could also be involved in formation of these bonds. Both hydrogen bonds and electrostatic interactions and their combination could be important for stabilization of a protein and its conformers [61-65]. On the other hand, it was suggested that salt bridges could destabilize structure while being on the protein surface [62]. Salt bridges could also be essential for protein thermostability by rigidifying its structure at high temperature [66, 67]. In particular, although thermophilic and mesophilic proteins have similar hydrophobicities, oligomeric states, and hydrogen bonds, they differ in the number of salt bridges: their number in thermophilic proteins is 20% higher than in mesophilic ones [67].
Among transport proteins, existence of salt bridges was proposed earlier for Escherichia coli sucrose [68] and lactose LacY [69-71] carriers and the S. cerevisiae Pma1 H+-ATPase [46] and HKA H+,K+-ATPase of animals [16, 72, 73]. Thus, results of biochemical, biophysical, and crystallographic studies led to the conclusion that several salt bridges could be formed between oppositely charged residues in the LacY transmembrane α-helices IV (Glu126), V (Arg144), VII (Asp237, Asp240), VIII (Glu269), X (Lys319), and XI (Lys358) [69-71]. When substrate binds, the salt bridge between Glu126 and Arg144 breaks, allowing the guanidine group of Arg144 to form a hydrogen bond both with oxygen atoms of galactopyranosyl and Glu269 [70].
A similar picture was seen in the HKA H+,K+-ATPase [16, 72, 73]. Based on homology modeling and mutagenesis data, the presence of a salt bridge was suggested between residues Glu820 (in M6) and Lys791 (in M5); this salt bridge was specific for the E2 conformation [72], while the polar Asn792 residue (M5) was involved in formation of hydrogen bonding with residues Lys791 (M5), Ala339, and Val341 (M4) [73].
As for the Pma1 H+-ATPase, the presence of a salt bridge was postulated for the Arg695 (M5) and Asp730 (M6) residues [46]. It appeared later that this interpretation was in disagreement with results obtained for the animal H+,K+- [18], Na+,K+- [74], and Ca2+-ATPases [35-38], yeast Ca2+,Mn2+-ATPase [21, 22], and yeast [25, 30, 33] and plant [19, 20] plasma membrane H+-ATPases. According to these data, Asp730 (and corresponding aspartyl residues in other P2-ATPases) is a residue that forms binding sites for transported cations; in Ca2+-ATPase, this residue is part of both transport sites [35-38], as well as in the yeast and fungal Pma1 H+-ATPase [30, 33]. It was also found that when the plasmid DNA encoding the pma1 gene was extracted from strains that should be carrying double substitutions D730A/R695A and D730R/R695D and sequenced, the sequence of the wild-type enzyme was found in both cases (K. E. Allen, unpublished data), confirming that a salt bridge between Asp730 and Arg695 is not formed. It is worth noting that a sulfhydryl bridge, whose presence was suggested for the Neurospora crassa Pma1 ATPase [75], was not found in S. cerevisiae Pma1 ATPase as well [48].
Role of charged residues Asp739 and Arg811. Although the aim of this work was not detailed study of the role of the Asp739 and Arg811 residues, but mainly their involvement in forming a salt bridge, the following should be mentioned. It looks possible that the role of these residues is similar but not identical to that known for Asp730 and Glu803, respectively. The first residue is involved in binding and coordinating the hydrated H+ (H3O+) and possibly defines the cation specificity [21, 22, 30-33]. The latter is involved in regulating (modulating) the ion transport stoichiometry [25, 26]. As mentioned above, the residues, which correspond to Asp739, are shifted by one position towards the C-terminal end in the P2-ATPases that transport cations other than H+; there is Asn (SERCA1) or Glu (PMR1, PMCA1, HKA, NKA; Fig. 3) in this position in these ATPases. The Asp739 residue might also be involved in, if not direct binding of H3O+, then in its coordination. During binding of the transported cations, unwinding of α-helices (including M6) occurs near the binding site where Asp730 is located [34, 35]; at the same time, Asp739 is already located in the unwound part of M6 stretching into the cytosol (Fig. 1).
Arg811 residue is also located in the unwound part of M8 stretching into the cytosol (Fig. 2). Since the active substitutions of Arg811 (R811A and R811D) significantly decrease coupling of ATP hydrolysis to H+ transport by the mutant enzymes, we suggest that this residue participates in H+ transport stoichiometry modulation similarly to the Glu803 residue and could be directly involved in H3O+ binding. Interestingly, there is also Arg in the position of this residue in the animal P2-ATPases that also transport H+ (PMCA and HKA; Fig. 4); it may be possible that the Arg811 residue participates in determining cation specificity.
It should also be noted that many organisms have more than one isoform of P2-ATPase; animals have several such isoforms and they are tissue-specific. Fungi have two isoforms: Pma1 and Pma2, the usually being less active and dormant. The fungus U. maydis, a biotrophic corn pathogen, also has two copies of the PMA gene. However, by contrast to other fungi, amino acid sequences of the U. maydis Pma1 and Pma2 ATPases differ significantly (Figs. 3 and 4), and the Pma2 isoform expression is even higher than that of Pma1 [76] compared to such isoforms in S. cerevisiae, where the Pma2 isoform is much less active, dormant, and insignificantly expressed [77, 78]. In U. maydis, the ascomycetous Pma1 ATPase is encoded by the PMA1 gene, while the plant type Pma2 ATPase similar to the Arabidopsis thaliana AHA2 is encoded by the PMA2 gene. Despite this, the residues corresponding to Asp739 and Arg811 are in “their places” in both isoforms of the enzyme, which points to their importance for the biogenesis and functioning of the H+-ATPase and their possible involvement in defining the proton transport determinants. Evidently, the cation transport determinants are defined not only by individual residues, but by their arrangement. Thus, corresponding to the Asp730 residue (Asp778) occupies “its position” in the Pmr1 Ca2+,Mn2+-ATPase of the endoplasmic reticulum, while the Asp739 residue is replaced by Glu788, which is shifted by one position towards the C-end of the enzyme similar to PMCA1 ATPase (Fig. 3). In the Pmr1 ATPase M8 segment, the residue corresponding to Arg811 (Arg843; Fig. 4) is in “its place”, while a glutamyl residue that corresponds to Glu803 is shifted by one position towards the C-end.
Thus, the results of the substitutions, especially the double substitution with swapped charges (D739R/R811D), suggest that a functional salt bridge is not formed between the Asp739 and Arg811 residues. Pma1 ATPase is not a thermophilic protein, while the animal P2-ATPases and E. coli LacY permease are on the border between mesophilic and thermophilic proteins, which could be a reason for difference in the occurrence of salt bridges in these enzymes [67]. It is established that determinants for cation specificity and stoichiometry of transport are defined by the presence of several amino acid residues in transmembrane segments M4, M5, M6, and M8 [6-45]. We can also suggest that the Asp739 (M6) and Arg811 (M8) could be among these residues, while transport determinants are defined by the arrangement of single amino acid residues that are involved in cation transport.
Acknowledgements
The author is grateful to Prof. C. W. Slayman (Yale University School of Medicine) who was a scientific adviser for this project, to Dr. J. P. Pardo for computer modeling, and to K. E. Allen for expert technical assistance.
REFERENCES
1.Serrano, R., Kielland-Brandt, M. C., and Fink, G.
R. (1986) Yeast plasma membrane ATPase is essential for growth and has
homology with (Na+,K+)K+- and
Ca2+-ATPases, Nature, 319, 689-693.
2.Lutsenko, S., and Kaplan, J. H. (1995) Organization
of P-type ATPases: significance of structural diversity,
Biochemistry, 34, 15607-15613.
3.Axelsen, K. B., and Palmgren, M. G. (1998)
Evolution of substrate specificities in the P-type ATPase superfamily,
J. Mol. Evol., 46, 84-101.
4.Petrov, V. V., and Okorokov, L. A. (1992)
Energization of yeast plasmalemma is necessary for activation of its
Н+-ATРase by glucose, Biokhimiya
(Moscow), 57, 1705-1711.
5.Goffeau, A., and Slayman, C. W. (1981) The
proton-translocating ATPase of the fungal plasma membrane, Biochim.
Biophys. Acta, 639, 197-223.
6.Andersen, J. P., and Vilsen, B. (1994) Amino acids
Asn796 and Thr799 of the Ca2+-ATPase of sarcoplasmic
reticulum bind Ca2+ at different sites, J. Biol.
Chem., 269, 15931-15936.
7.Rice, W. J., and MacLennan, D. H. (1996) Scanning
mutagenesis reveals a similar pattern of mutation sensitivity in
transmembrane sequences M4, M5, and M6, but not in M8, of the
Ca2+-ATPase of sarcoplasmic reticulum (SERCA1a), J. Biol.
Chem., 271, 31412-31419.
8.Zhang, Z., Lewis, D., Strock, C., Inesi, G.,
Nakasako, M., Nomura, H., and Toyoshima, C. (2000) Detailed
characterization of the cooperative mechanism of Ca2+
binding and catalytic activation in the Ca2+ transport
(SERCA) ATPase, Biochemistry, 39, 8758-8767.
9.Vilsen, B., and Andersen, J. P. (1998) Mutation to
the glutamate in the fourth membrane segment of
Na+,K+-ATPase and Ca2+-ATPase affects
cation binding from both sides of the membrane and destabilizes the
occluded enzyme forms, Biochemistry, 37, 10961-10971.
10.Jewell-Motz, E. A., and Lingrel, J. B. (1993)
Site-directed mutagenesis of the Na,K-ATPase: consequences of
substitutions of negatively-charged amino acids localized in the
transmembrane domains, Biochemistry, 32, 13523-13530.
11.Kuntzweiler, T. A., Arguello, J. M., and Lingrel,
J. B. (1996) Asp804 and Asp808 in the transmembrane domain of the
Na,K-ATPase α subunit are cation coordinating residues, J.
Biol. Chem., 271, 29682-29687.
12.Nielsen, J. M., Pedersen, P. A., Karlish, S. J.
D., and Jorgensen, P. L. (1998) Importance of intramembrane carboxylic
acids for occlusion of K+ ions at equilibrium in renal
Na,K-ATPase, Biochemistry, 37, 1961-1968.
13.Swarts, H. G., Klaassen, C. H., De Boer, M.,
Fransen, J. A., and De Pont, J. J. (1996) Role of negatively charged
residues in the fifth and sixth transmembrane domains of the catalytic
subunit of gastric H+,K+-ATPase, J. Biol.
Chem., 271, 29764-29772.
14.Hermsen, H. P., Koenderink, J. B., Swarts, H. G.,
and De Pont, J. J. (1998) The negative charge of glutamic acid-795 is
essential for gastric H+,K+-ATPase activity,
Biochemistry, 39, 1330-1337.
15.Hermsen, H. P., Swarts, H. G., Koenderink, J. B.,
and De Pont, J. J. (2000) The carbonyl group of glutamic acid-820 in
the gastric H+,K+-ATPase alpha-subunit is
essential for K+ activation of the enzyme activity,
Biochem. J., 331, 465-472.
16.Swarts, H. G. P., Koenderink, J. B., Willems, P.
H., Krieger, E., and De Pont, J. J. (2005) Asn792 participates in the
hydrogen bond network around the K+-binding pocket of
gastric H,K-ATPase, J. Biol. Chem., 280, 11488-11494.
17.Asano, S., Io, T., Kimura, T., Sakamoto, S., and
Takeguchi, N. (2001) Alanine-scanning mutagenesis of the sixth
transmembrane segment of gastric H+,K+-ATPase
alpha-subunit, J. Biol. Chem., 276, 31265-31273.
18.Asano, S., Morii, M., and Takeguchi, N. (2004)
Molecular and cellular regulation of the gastric pump, Biol. Pharm.
Bull., 27, 1-12.
19.Buch-Pedersen, M. J., Venema, K., Serrano, R.,
and Palmgren, M. G. (2000) Abolishment of proton pumping and
accumulation in the E1P conformational state of a plant plasma membrane
H+-ATPase by substitution of a conserved aspartyl residue in
transmembrane segment 6, J. Biol. Chem., 275,
39167-39173.
20.Buch-Pedersen, M. J., and Palmgren, M. G. (2003)
Conserved Asp684 in transmembrane segment M6 of the plant plasma
membrane P-type proton pump AHA2 is molecular determinant of proton
translocation, J. Biol. Chem., 278, 17845-17851.
21.Wei, Y., Chen, J., Rosas, G., Tompkins, D. A.,
Holt, P. A., and Rao, R. (2000) Phenotypic screening of mutations in
Pmr1, the yeast secretory pathway
Ca2+/Mn2+-ATPase, reveals residues critical for
ion selectivity and transport, J. Biol. Chem., 275,
23927-23932.
22.Mandal, D., Woolf, T. B., and Rao, R. (2000)
Manganese selectivity of pmr1, the yeast secretory pathway ion pump, is
defined by residue Gln783 in transmembrane segment 6. Residue Asp778 is
essential for cation transport, J. Biol. Chem., 275,
23933-23938.
23.Ambesi, A., Pan, R. L., and Slayman, C. W. (1996)
Alanine-scanning mutagenesis along membrane segment 4 of the yeast
plasma membrane H+-ATPase. Effects on structure and
function, J. Biol. Chem., 271, 22999-23005.
24.Dutra, M. B., Ambesi, A., and Slayman, C. W.
(1998) Structure-function relationships in membrane segment 5 of the
yeast Pma1 H+-ATPase, J. Biol. Chem., 273,
17411-17417.
25.Petrov, V. V., Padmanabha, K. P., Nakamoto, R.
K., Allen, K. E., and Slayman, C. W. (2000) Functional role of charged
residues in the transmembrane segments of the yeast plasma membrane
H+-ATPase, J. Biol. Chem., 275,
15709-15716.
26.Guerra, G., Petrov, V. V., Allen, K. E., Miranda,
M., Pardo, J. P., and Slayman, C. W. (2007) Role of transmembrane
segment M8 in the biogenesis and function of yeast plasma-membrane
H+-ATPase, Biochim. Biophys. Acta, 1768,
2383-2392.
27.Miranda-Arango, M., Pardo, J. P., and Petrov, V.
V. (2009) Role of transmembrane segment M6 in the biogenesis and
function of the yeast Pma1 H+-ATPase, J. Biomol. Struct.
Dyn., 26, 866-868.
28.Petrov, V. V. (2009) Heat shock affects
functioning of the yeast Pma1 H+-ATPase, J. Biomol.
Struct. Dyn., 26, 857-858.
29.Petrov, V. V. (2010) Point mutations in Pma1
H+-ATPase of Saccharomyces cerevisiae: influence on
its expression and activity, Biochemistry (Moscow), 75,
1055-1064.
30.Miranda, M., Pardo, J. P., and Petrov, V. V.
(2011) Structure-function relationships in membrane segment 6 of the
yeast plasma membrane Pma1 H+-ATPase, Biochim. Biophys.
Acta, 1808, 1781-1789.
31.Petrov, V. V. (2011) Role of M5-M6 loop in the
biogenesis and function of the yeast Pma1 H+-ATPase, J.
Biomol. Struct. Dyn., 28, 1024-1025.
32.Petrov, V. V. (2015) Point mutations in the
extracytosolic loop between transmembrane segments M5 and M6 of the
yeast Pma1 H+-ATPase: alanine-scanning mutagenesis, J.
Biomol. Struct. Dyn., 33, 70-84.
33.Petrov, V. V. (2015) Role of loop L5-6 connecting
transmembrane segments M5 and M6 in biogenesis and functioning of yeast
Pma1 H+-ATPase, Biochemistry (Moscow), 80,
31-44.
34.Toyosima, C., Nakasako, M., Nomura, H., and
Ogawa, H. (2000) Crystal structure of the calcium pump of sarcoplasmic
reticulum at 2.6 Å resolution, Nature, 405,
647-655.
35.Toyosima, C., and Nomura, H. (2002) Structural
changes in the calcium pump accompanying the dissociation of calcium,
Nature, 418, 605-611.
36.Takahashi, M., Kondou, Y., and Toyoshima, C.
(2007) Interdomain communication in calcium pump as revealed in the
crystal structures with transmembrane inhibitors, Proc. Natl. Acad.
Sci. USA, 104, 5800-5805.
37.Toyoshima, C., Norimatsu, Y., Iwasawa, S., Tsuda,
T., and Ogawa, H. (2007) How processing of aspartyl phosphate is
coupled to lumenal gating of the ion pathway in the calcium pump,
Proc. Natl. Acad. Sci. USA, 104, 19831-19836.
38.Toyoshima, C. (2008) Structural aspects of ion
pumping by Ca2+-ATPase of sarcoplasmic reticulum, Arch.
Biochem. Biophys., 476, 3-11.
39.Toyoshima, C., Iwasawa, S., Ogawa, H., Hirata,
A., Tsueda, J., and Inesi, G. (2013) Crystal structures of the calcium
pump and sarcolipin in the Mg2+-bound E1 state,
Nature, 495, 260-264.
40.Morth, J. P., Pedersen, B. P., Toustrup-Jensen,
M. S., Sorensen, T. L., Petersen, J., Andersen, J. P., Vilsen, B., and
Nissen, P. (2007) Crystal structure of the sodium-potassium pump,
Nature, 450, 1043-1049.
41.Shinoda, T., Ogawa, H., Cornelius, F., and
Toyosima, C. (2009) Crystal structure of the sodium-potassium pump at
2.4 Å resolution, Nature, 459, 446-450.
42.Ogawa, H., Shinoda, T., Cornelius, F., and
Toyoshima, C. (2009) Crystal structure of the sodium-potassium pump
(Na+,K+-ATPase) with bound potassium and ouabain,
Proc. Natl. Acad. Sci. USA, 106, 13742-13747.
43.Nyblom, M., Poulsen, H., Gourdon, P., Reinhard,
L., Andersson, M., Lindahl, E., Fedosova, N., and Nissen, P. (2013)
Crystal structure of Na+, K+-ATPase in the
Na+-bound state, Science, 342, 123-127.
44.Kanai, R., Ogawa, H., Vilsen, B., Cornelius, F.,
and Toyoshima, C. (2013) Crystal structure of a Na+-bound
Na+,K+-ATPase preceding the E1P state,
Nature, 502, 201-206.
45.Pedersen, B. P., Buch-Pedersen, M., Morth, J. J.
P., Palmgren, M. G., and Nissen, P. (2007) Crystal structure of the
plasma membrane proton pump, Nature, 450,
1111-1114.
46.Gupta, S. S., DeWitt, N. D., Allen, K. E., and
Slayman, C. W. (1998) Evidence for a salt bridge between transmembrane
segments 5 and 6 of the yeast plasma-membrane H+-ATPase,
J. Biol. Chem., 273, 34328-34334.
47.Nakamoto, R. K., Rao, R., and Slayman, C. W.
(1991) Expression of the yeast plasma membrane H+-ATPase in
secretory vesicles. A new strategy for directed mutagenesis, J.
Biol. Chem., 266, 7940-7949.
48.Petrov, V. V., and Slayman, C. W. (1995)
Site-directed mutagenesis of the yeast PMA1 H+-ATPase.
Structural and functional role of cysteine residues, J. Biol.
Chem., 270, 28535-28540.
49.Fabiato, A., and Fabiato, F. (1979) Calculator
programs for computing the composition of the solutions containing
multiple metals and ligands used for experiments in skinned muscle
cells, J. Physiol. (Paris), 75, 463-505.
50.Fiske, C. H., and Subbarow, Y. (1925) The
colorimetric determination of phosphorus, J. Biol. Chem.,
66, 375-400.
51.Bensadoun, A., and Weinstein, D. (1976) Assay of
proteins in the presence of interfering materials, Anal.
Biochem., 70, 241-250.
52.Thompson, J. D., Higgins, D. G., and Gibson, T.
J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple
sequence alignment through sequence weighting, position-specific gap
penalties and weight matrix choice, Nucleic Acids Res.,
22, 4673-4680.
53.Serrano, R. (1988) Structure and function of
proton translocating ATPase in plasma membranes of plants and fungi,
Biochim. Biophys. Acta, 947, 1-28.
54.Ambesi, A., Miranda, M., Petrov, V. V., and
Slayman, C. W. (2000) Biogenesis and function of the yeast
plasma-membrane H+-ATPase, J. Exp. Biol., 203,
156-160.
55.Ferreira, T., Mason, A. B., Pypaert, M., Allen,
K. E., and Slayman, C. W. (2002) Quality control in the yeast secretory
pathway: a misfolded PMA1 H+-ATPase reveals two checkpoints,
J. Biol. Chem., 277, 21027-21040.
56.Mason, A. B., Allen, K. E., and Slayman, C. W.
(2014) C-terminal truncations of the Saccharomyces
cerevisiae PMA1 H+-ATPase have major impacts on protein
conformation, trafficking, quality control, and function, Eukaryot.
Cell, 13, 43-52.
57.Nakamoto, R. K., Verjovski-Almeida, S., Allen, K.
E., Ambesi, A., Rao, R., and Slayman, C. W. (1998) Substitutions of
aspartate 378 in the phosphorylation domain of the yeast PMA1
H+-ATPase disrupt protein folding and biogenesis, J.
Biol. Chem., 273, 7338-7344.
58.Dougherty, D. A. (2006) Modern Physical
Organic Chemistry, University Science Books, Sausalito, CA.
59.Bairagya, H. R., Mukhopadhyay, B. P., and Bera,
A. K. (2011) Role of salt bridge dynamics in inter domain recognition
of human IMPDH isoforms: insight to inhibitor topology for isoform II,
J. Biomol. Struct. Dyn., 29, 441-462.
60.Bairagya, H. R., and Mukhopadhyay, B. P. (2013)
An insight to the dynamics of conserved water-mediated salt bridge
interaction and interdomain recognition in hIMPDH isoforms, J.
Biomol. Struct. Dyn., 31, 788-808.
61.Morozov, V. N., and Kallenbach, N. R. (1996)
Stabilization of helical peptides by mixed spaced salt bridges, J.
Biomol. Struct. Dyn., 14, 285-291.
62.Hendsch, Z. S., and Tidor, B. (1994) Do salt
bridges stabilize proteins? A continuum electrostatic analysis,
Protein Sci., 3, 211-226.
63.Sindelar, C. V., Hendsch, Z. S., and Tidor, B.
(1998) Effects of salt bridges on protein structure and design,
Protein Sci., 7, 1898-1914.
64.Strop, P., and Mayo, S. L. (2000) Contribution of
surface salt bridges to protein stability, Biochemistry,
39, 1251-1255.
65.Kumar, S., and Nussinov, R. (2002) Close-range
electrostatic interactions in proteins, ChemBioChem, 3,
604-617.
66.Kumar, S., Tsai, C.-J., Ma, B., and Nussinov, R.
(2000) Contribution of salt bridges toward protein thermostability,
J. Biomol. Struct. Dyn., 17, S1, 79-85.
67.Panja, A. S., Bandopadhyay, B., and Maiti, S.
(2015) Protein thermostability is owing to their preferences to
non-polar smaller volume amino acids, variations in residual
physico-chemical properties and more salt-bridges, PLoS, doi:
10.1371/journal.pone.0131495.
68.Frillingos, S., Sahin-Toth, M., Lengeler, J. W.,
and Kaback, H. R. (1995) Helix packing in the sucrose permease of
Escherichia coli: properties of engineered charge pairs between
helixes VII and XI, Biochemistry, 34, 9368-9373.
69.Frillingos, S., and Kaback, H. R. (1995) Chemical
rescue of Asp237>Ala and Lys358>Ala mutants in the lactose
permease of Escherichia coli, Biochemistry, 35,
13363-13367.
70.Weinglass, A., Whitelegge, J. P., Faull, K. F.,
and Kaback, H. R. (2004) Monitoring conformational rearrangements in
the substrate-binding site of a membrane transport protein by mass
spectrometry, J. Biol. Chem., 279, 41858-41865.
71.Guan, L., and Kaback, H. R. (2009) Properties of
a LacY efflux mutant, Biochemistry, 48, 9250-9255.
72.Koenderink, J. B., Swarts, H. G. P., Willems, P.
H., Krieger, E., and De Pont, J. J. (2004) A conformation-specific
interhelical salt bridge in the K+ binding site of gastric
H,K-ATPase, J. Biol. Chem., 279, 16417-16424.
73.Durr, K. L., Seuffert, I., and Friedrich, T.
(2010) Deceleration of the E1P-E2P transition and ion transport by
mutation of potentially salt bridge-forming residues Lys-791 and
Glu-820 in gastric H+/K+-ATPase, J. Biol.
Chem., 285, 39366-39379.
74.Jorgensen, P. L., Hakansson, K. O., and Karlish,
S. J. (2003) Structure and mechanism of Na,K-ATPase: functional sites
and their interactions, Annu. Rev. Physiol., 65,
817-849.
75.Rao, U. S., and Scarborough, G. A. (1990)
Chemical state of the cysteine residues in the Neurospora
crassa plasma membrane H+-ATPase, J. Biol.
Chem., 265, 7227-7235.
76.Roblez-Martinez, L., Pardo, J. P., Miranda, M.,
Mendez, T. L., Matus-Ortega, M. G., Mendoza-Hernandez, G., and
Guerra-Sanchez, G. (2013) The basidiomycete Ustilago maydis has
two plasma membrane H+-ATPases related to fungi and plants,
J. Bionerg. Biomembr., 45, 477-290.
77.Supply, P., Wach, A., Thines-Sempoux, D., and
Goffeau, A. (1993) Proliferation of intracellular structures upon
overexpression of the PMA2 ATPase in Saccharomyces cerevisiae,
J. Biol. Chem., 268, 19744-19752.
78.Supply, P., Wach, A., and Goffeau, A. (1993)
Enzymatic properties of the PMA2 plasma membrane-bound
H+-ATPase of Saccharomyces cerevisiae, J. Biol.
Chem., 268, 19753-19759.