Sodium Orthovanadate Inhibits Proliferation and Triggers Apoptosis in Oral Squamous Cell Carcinoma in vitro
A. A. Khalil1,2* and M. J. Jameson1*
1University of Virginia Health System, Division of Head and Neck Oncologic and Microvascular Surgery, Department of Otolaryngology, Head and Neck Surgery, 1215 Charlottesville, Virginia, USA; E-mail: mark.jameson@virginia.edu2Menoufiya University, National Liver Institute, Department of Biochemistry, Egypt; E-mail: ashkalil2010@gmail.com
* To whom correspondence should be addressed.
Received July 25, 2016; Revision received September 8, 2016
Sodium orthovanadate (SOV) is a general inhibitor of tyrosine phosphatases, a large family of enzymes that catalyze the removal of phosphate groups from tyrosine residues. SOV is commonly used in the laboratory to preserve the protein tyrosyl phosphorylation state of proteins under study. It has shown promising antineoplastic activity in some human cancer cell lines; this effect has not been fully investigated in head and neck squamous cell carcinoma. In this study, the effect of SOV on cell growth, proliferation, viability, and apoptosis was assessed in Cal27 cells, an oral squamous cell carcinoma (OSCC) cell line. SOV exhibited dose-dependent inhibition of cell growth and decrease in cell viability and colony formation. The IC50 values for treatment lasting 72 h and 7 days were 25 and 10 µM, respectively. The cytotoxic effect of the drug was associated with poly(ADP-ribose)polymerase cleavage detected by immunoblot. Flow cytometry of Cal27 cells stained with annexin V-FITC and propidium iodide showed a dose-dependent increase in apoptosis that reached approximately 40% at 25 µM SOV. These findings demonstrate that SOV has in vitro antiproliferative and proapoptotic effect on OSCC cells.
KEY WORDS: sodium orthovanadate, oral cavity cancer, squamous cell carcinomaDOI: 10.1134/S0006297917020067
Abbreviations: OSCC, oral squamous cell carcinoma; PARP, poly(ADP-ribose)polymerase; PI, propidium iodide; SOV, sodium orthovanadate.
Cytoplasmic protein tyrosine phosphokinases and phosphatases regulate
the tyrosine phosphorylation status of proteins crucial to multiple
signal transduction pathways. Activation of phosphokinases or
inactivation of phosphatases leads to accumulation of phosphorylated
proteins resulting in regulation of cell cycle, growth, and
differentiation, DNA damage response, apoptosis, and other critical
cellular behaviors. The balance between tyrosine kinases and tyrosine
phosphatases establishes whether a cell will survive or undergo
apoptosis [1].
Sodium orthovanadate (SOV) is a phosphate analog that functions as a general inhibitor for protein tyrosine phosphatases, alkaline phosphatase, and ATPase, resulting in accumulation of phosphoproteins [1, 2]. It is commonly used in research settings to prevent tyrosine dephosphorylation in assays dependent on protein tyrosyl phosphorylation.
SOV has been found to affect apoptosis, but it has been shown to both suppress [3, 4] and induce [5]. This could be due to differences in the genetic background of the cell lines studied or in the apoptotic stimuli used [6]. Induction of apoptosis by SOV involves generation of reactive oxygen species in the cytosol, or in mitochondria [7] with the release of cytochrome c and activation of caspases [8]. Activated caspases such as caspase-3 and caspase-8 provoke further mitochondrial damage and activate cellular substrates such as poly(ADP-ribose)polymerase (PARP), leading to apoptosis [9, 10]. In this study, we explored the effect of SOV on the growth and proliferation of Cal27 oral squamous cell carcinoma (OSCC) cells in vitro.
MATERIALS AND METHODS
Tissue culture and reagents. Cal27 cells were obtained from the American Type Culture Collection and were grown in DMEM/F12 medium supplemented with 5% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2. SOV (NaVO3) was obtained from Sigma (USA); alamarBlue and trypan blue were obtained from Invitrogen (USA); anti-PARP, anti-β-actin, annexin V-fluorescein isothiocyanate (FITC), and propidium iodide (PI) were obtained from Millipore (USA).
Immunoblot. Treated cells in 6-cm dishes were washed with ice-cold PBS containing 2 mM SOV, collected, resuspended in lysis buffer, and processed as previously described [11]. Briefly, cell lysate was vortexed and incubated at 4°C for 30 min, then centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was collected, and sample buffer containing 0.1 M dithiothreitol (DTT) was added. Proteins were resolved by 12% SDS-PAGE and then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore). Proteins were visualized using infrared-emitting conjugated secondary antibodies – anti-mouse 680 Alexa (Molecular Probes, USA) or anti-rabbit IRDYE 800 (Rockland Immunochemicals, USA) – and the Odyssey imaging system (LI-COR Biosciences, USA).
AlamarBlue proliferation assay. Cells were plated at 5000 cells per well in 96-well plates and allowed to grow for 24 h prior to treatment with SOV, which was dissolved in PBS and added directly into the media. Control cells were treated with PBS only. All experiments were performed in triplicate. Plates were incubated for 72 h. AlamarBlue was added to each well according to the manufacturer’s protocol. Cells were incubated for 3 h at 37°C, and the fluorescence at 540 nm was recorded.
Trypan blue cell count. Cells were plated at 50,000 cells per well in a 12-well plate and allowed to grow for 24 h. The cells were treated with SOV dissolved in PBS and added directly into the media. Control cells were treated with PBS only. All treatments were studied in triplicate. The plates were incubated for 72 h. The medium from each well was collected, and the wells were trypsinized; anchored and floating cells were combined and centrifuged at 3000 rpm for 3 min at 4°C. The cells were resuspended in 1 ml PBS, and 100 µl of the suspension was stained with 100 µl of trypan blue. Living and dead (blue) cells were counted using a TC20 Automated Cell Counter (Bio-Rad, USA).
Colony formation assay. Cal27 cells were seeded at 250 cells per well in a 6-well plate and incubated overnight at 37°C. The cells were treated with SOV dissolved in PBS and added directly into the media. Control wells were treated with PBS alone. All treatments were carried out in triplicate wells. The plates were incubated for 14 days; during this period the medium was changed twice weekly with the appropriate concentration of SOV. The plates were washed with ice-cold PBS; the colonies were fixed with methanol for 15 min, stained with 2% crystal violet, and counted. Colonies consisting of ≥50 cells were scored.
Flow cytometry. Cal27 cells were grown in 6-cm dishes to 70% confluence and then incubated with SOV or PBS for 72 h. The medium from each well was collected; the cell monolayers were trypsinized, resuspended in the corresponding medium, centrifuged at 3000 rpm for 3 min at 4°C, washed once with cold PBS, and resuspended in annexin V-FITC and PI according to the manufacturer’s recommendations (Millipore). Flow cytometry was conducted in the UVA Flow Cytometry Core Facility. With annexin V/PI staining, cells that are viable are annexin and PI negative, cells that are in early apoptosis are annexin positive and PI negative, and cells that are in late apoptosis are annexin and PI positive. Necrotic and dead cells are annexin V-FITC negative and PI positive [12, 13].
Statistics. All statistical analyses were performed using Prism 3 (GraphPad Software, USA). Comparisons to untreated controls were performed using Student’s t-test. Findings were considered significance when p < 0.05.
RESULTS
SOV inhibits Cal27 cell proliferation. Cal27 cells were treated with 2.5, 5, 15, 25, and 50 µM SOV, and proliferation was assessed at 72 h and 7 days using alamarBlue. Figure 1a demonstrates dose-dependent inhibition of proliferation with both short and long-term SOV treatment; a greater growth inhibitory effect was observed with the longer exposure. After 72 h of treatment with 2.5 and 5 µM SOV, growth was inhibited by 2.5 and 10%, respectively; after 7 days, growth was inhibited by 17.5 and 33%, respectively. The IC50 was 25 µM for 72 h treatment and 10 µM for 7 day treatment. At 50 µM, SOV inhibited proliferation by 95% (72 h) and 99% (7 days).
Fig. 1. Dose response of Cal27 cell proliferation (a) and viability (b) after SOV treatment. Assays were performed in triplicate as described in “Materials and Methods” using alamarBlue ((a) relative fluorescence at 590 nm) and trypan blue ((b) number of live cells as percent of control). Results are shown as mean ± standard error of the mean (SEM).
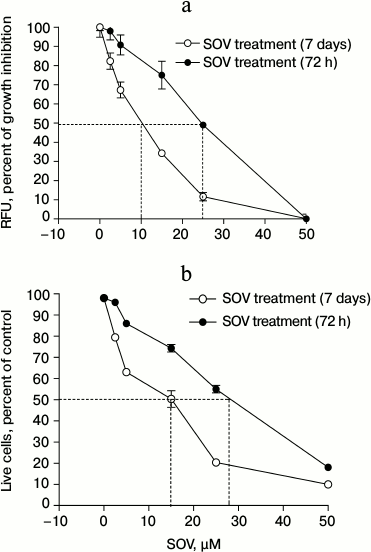
SOV decreases Cal27 cell viability. Similar experiments were performed to assess cell viability using trypan blue. Nonviable cells were differentiated from viable cells by their blue staining. SOV cytotoxicity in Cal27 cells was dose-dependent (Fig. 1b) with IC50 of 28 and 15 µM after 72 h and 7 day treatment, respectively.
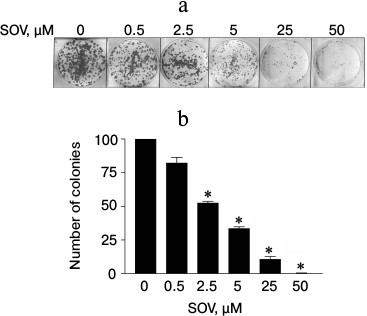
SOV inhibits clonogenic survival of Cal27 cells. The effect of chronic exposure (14 days) to SOV on clonogenic survival was evaluated using a colony formation assay. A dose-dependent decrease in the number of colonies was observed (Fig. 2); 50 µM SOV resulted in a 95% reduction in colony formation compared to untreated cells (p < 0.05).
Fig. 2. Dose response of Cal27 colony formation after SOV treatment. The colony formation assay was performed in triplicate as described in “Materials and Methods”. a) Representative stained colonies. b) Mean colonies ± standard error of the mean (SEM) at each SOV concentration. * p < 0.05 compared to control untreated cells.
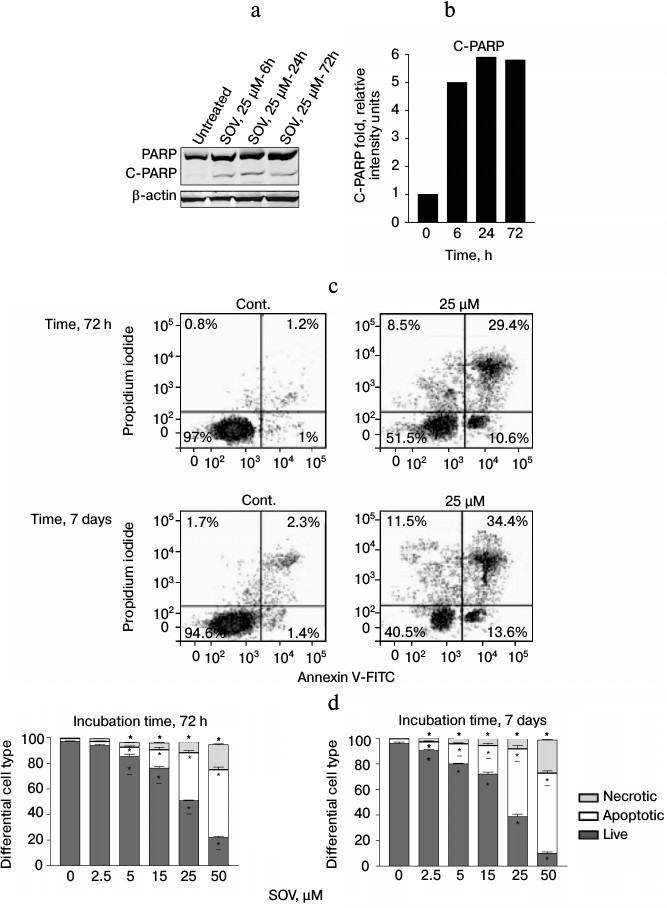
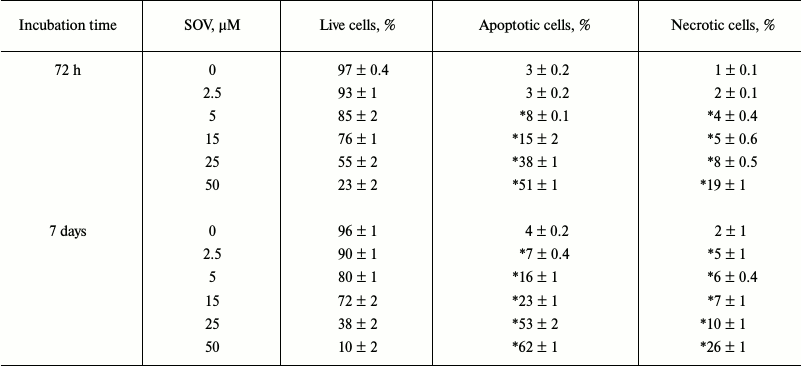
SOV induces apoptosis in Cal27 cells. PARP cleavage was assessed by immunoblot in Cal27 cells as a marker of apoptosis after SOV treatment (Fig. 3, a and b). Cal27 cells were treated with 25 µM SOV and collected at 6, 24, and 72 h. SOV induced a 5-fold increase in cleaved PARP (C-PARP) within 6 h of treatment. C-PARP level was maximal (6-fold increase) at 24 h, and this was sustained through 72 h. SOV-induced apoptosis was further examined by flow cytometry of Cal27 cells. Annexin V labeled with a fluorescent tag (FITC) was used in conjunction with a live/dead dye (PI) to identify and quantify the apoptotic population. The characteristic pattern differentiates early apoptosis (PI negative, annexin positive) from either late apoptosis (PI positive, annexin positive) or dead cells (PI positive, annexin negative). Cal27 cells were incubated with 0, 2.5, 5, 15, 25, and 50 µM SOV for either 72 h or 7 days and subsequently stained with annexin V-FITC and PI as described above. Figure 3c shows a representative flow cytometry result for treatment with 25 µM SOV at 72 h and 7 days. The table and Fig. 3d summarize triplicate experiments across the entire dose range and show a dose dependent increased in apoptosis and necrosis with SOV treatment, greater at 7 days than at 72 h. The apoptotic population increased from 3% in untreated control cells to 51% after 72 h of treatment with 50 µM SOV (p < 0.05). The necrotic population similarly increased from 1% in untreated control cells to 19% in cells exposed to 50 µM SOV for 72 h (p < 0.05). After 7 days of treatment, apoptotic and necrotic populations reached a maximum of 62 and 26%, respectively (p < 0.05).
Fig. 3. Cal27 apoptosis after treatment with SOV. a) Cal27 cells were treated with 25 µM SOV, and the cells were collected at 6, 24, and 72 h. Immunoblot was performed for intact and cleaved PARP (PARP, C-PARP) with β-actin as loading control. b) Fold change in C-PARP based on densitometry of immunoblot normalized to β-actin band. c) Cal27 cells were treated with various concentrations of SOV, and cells were collected at 72 h and 7 days. Flow cytometry was performed after staining with annexin V-FITC and PI; a representative result is shown for 25 µM SOV. d) Average relative populations of necrotic, apoptotic, and living cells at 72 h and 7 days based on triplicate flow cytometry studies for the entire concentration range. * p < 0.05 compared to control untreated cells.
Cal27 apoptosis and necrosis after 72 h and 7 days of treatment with SOV
Note: Assays were performed in triplicate using flow cytometry as
described in “Materials and Methods”. Results are shown as
mean ± standard error of the mean (SEM); * p < 0.05
compared to control untreated cells.
DISCUSSION
The Cal27 OSCC cell line was established in 1982 from a tumor of a 56-year-old Caucasian male with poorly differentiated squamous cell carcinoma of the central tongue. Since then, Cal27 cells have been widely used as a model representative OSCC cell line for in vitro and in vivo studies [14]. SOV is a nonspecific tyrosine phosphatase inhibitor affecting tyrosine phosphorylation status of many signaling molecules including those that regulate cell cycle, growth, and differentiation, DNA damage response, apoptosis, and other critical cellular behaviors [1, 15, 16]. Prior work with SOV highlights its potential usefulness as an antiproliferative agent in cancer. The present study demonstrates that short- and long-term treatment with SOV reduces Cal27 growth rate and cell viability. The dose response curves using alamarBlue and trypan blue were similar, suggesting that the antiproliferative effect of SOV was accompanied by decreased cell viability. Prolonged exposure time of the cells to the drug increased its toxicity with resultant reduced IC50. Chronic exposure of Cal27 to SOV for 2 weeks reduced colony formation; this effect reached 90% at 25 µM. Similar studies with different cell lines have shown comparable antiproliferative effects of SOV but with different sensitivity depending on the cells under study. The present in vitro results recapitulate the observations of Klein et al. who showed that SOV at 50 µM inhibited the growth of human epithelial cancer cell lines from lung (A549), kidney (HTB4), and prostate (DU145) [17]. Yang et al. also showed a similar effect at 50 µM on the growth and proliferation of esophageal squamous carcinoma cell line EC109 [18]. On the other hand, Goncalves et al. demonstrated a dual effect of SOV on the growth of papillary thyroid carcinoma (PTC) cells, with some stimulation of proliferation at low concentrations but inhibitory effect on cell growth at concentrations greater than 10 µM, with an IC50 of about 50 µM [19]. Delwar et al. studied SOV treatment of A549 cells and demonstrated an IC50 100 µM after 72 h that was reduced to 17.5 µM after 7 days of treatment [20].
Existing evidence links SOV cytotoxicity with activation of the apoptotic pathway. In the present study, cleavage (implying activation) of PARP was observed by immunoblot analysis as early as 6 h after treatment; this effect persisted beyond 72 h, suggesting that the long-term growth inhibitory effect of SOV in Cal27 cells may be largely due to apoptosis. Differential cell analysis using flow cytometry showed that the number of apoptotic cells (early and late apoptosis) increased significantly with SOV treatment for 72 h or 7 days. At the IC50 concentration, the apoptotic population increased to about 40% after 72 h and 55% after 7 days of treatment. The corresponding levels of necrotic cells were 8 and 10%, respectively, suggesting that SOV causes predominantly apoptosis rather than necrosis. The present study did not evaluate the mechanism(s) of induction of apoptosis by SOV, but Gunther et al. have demonstrated that SOV increases reactive oxygen species (ROS) in tumor cells that inhibit proliferation and trigger apoptosis [21]. Zhang et al. showed that SOV causes cell cycle arrest at G2/M phase in A529 cells via a mechanism that generates hydrogen peroxide [22]. Ray et al. and Yang et al. have shown vanadium in vivo and in vitro causes suppression of cell proliferation, induction of apoptosis, and cell cycle arrest [18, 23]. Despite the complexity of the signaling pathways, which include many key proteins, there is considerable evidence that inorganic salts of vanadium activate phosphotyrosine phosphorylases, specifically the MAPKs [24]. MAPKs include extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SARK), and p38. MAPKs are central regulatory proteins through which diverse extracellular signals are transduced into intracellular events mainly throughout modulation of the downstream molecule NF-κB. JNK/SARK, and ERK signaling pathways have opposite effects on apoptosis with JNK/SARK and p38 are mainly proapoptotic signals, while ERK signals are mainly survival and antiapoptotic [25, 26]. Data in the literature suggested that vanadium compounds could modulate NF-κB activation in normal and cancer cells via MAPKs signaling pathway, leading to cell apoptosis. These proapoptotic effects of vanadium compounds in relation to protein tyrosine phosphorylation and NF-κB depend on the type of cells and the dose of vanadium compound [27-29]. p53 protein is another critical molecule in modulating the response to vanadate. In studies with tumor cells defective in p53 or p53-knockout cells, vanadium compounds were found to inhibit the cell cycle and induce apoptosis [22]. Moreover, activation of NF-κB by ROS generated by vanadium compounds enhances the apoptotic effect in these p53 defective cells [30, 31]. On the other hand, in tumor cells with functional p53, vanadium compounds were found to stimulate the cell cycle, thus inhibiting apoptosis [22, 32].
Another important observation: studies had shown vanadium compounds cause differential DNA damage in tumor cells compared to nontumor cells when present at the same dose [33]. Vanadium compounds cause extensive DNA damage with subsequent apoptosis in tumor cells, while a less intensive damage in nontumor cells, which may stimulate DNA repair enzymes and cell cycle arrest, thus protecting cells from apoptosis [33, 34]. It is clear that the mechanisms underlying the antitumor properties of vanadium are quite complex and impose signal modulation and cross talking among different signaling phosphoproteins.
In conclusion, this study provides in vitro data demonstrating that SOV is capable of inhibiting cell proliferation and inducing apoptosis of human OSCC cells. These data raise the possibility that SOV could have a role in OSCC therapy; however, additional studies using in vivo models are required to further investigate this possibility.
REFERENCES
1.Alonso, A., Sasin, J., Bottini, N., Friedberg, I.,
Friedberg, I., Osterman, A., Godzik, A., Hunter, T., Dixon, J., and
Mustelin, T. (2004) Protein tyrosine phosphatases in the human genome,
Cell, 117, 699-711.
2.Chen, Y. Y., Chu, H. M., Pan, K. T., Teng, C. H.,
Wang, D. L., Wang, A. H., Khoo, K. H., and Meng, T. C. (2008) Cysteine
S-nitrosylation protects protein-tyrosine phosphatase 1B against
oxidation-induced permanent inactivation, J. Biol. Chem.,
283, 35265-35272.
3.Lawson, A. E., Bao, H., Wickrema, A.,
Jacobs-Helber, S. M., and Sawyer, S. T. (2000) Phosphatase inhibition
promotes antiapoptotic but not proliferative signaling pathways in
erythropoietin-dependent HCD57 cells, Blood, 96,
2084-2092.
4.Chin, L. S., Murray, S. F., Harter, D. H., Doherty,
P. F., and Singh, S. K. (1999) Sodium vanadate inhibits apoptosis in
malignant glioma cells: a role for Akt/PKB, J. Biomed. Sci.,
6, 213-218.
5.Gamero, A. M., and Larner, A. C. (2001) Vanadate
facilitates interferon alpha-mediated apoptosis that is dependent on
the Jak/Stat pathway, J. Biol. Chem., 276,
13547-13553.
6.Figiel, I., and Kaczmarek, L. (1997) Orthovanadate
induces cell death in rat dentate gyrus primary culture,
Neuroreport, 8, 2465-2470.
7.Khalil, A., Morgan, R. N., Adams, B. R., Golding,
S. E., Dever, S. M., Rosenberg, E., Povirk, L. F., and Valerie, K.
(2011) ATM-dependent ERK signaling via AKT in response to DNA
double-strand breaks, Cell Cycle, 10, 481-491.
8.Krejsa, C. M., Nadler, S. G., Esselstyn, J. M.,
Kavanagh, T. J., Ledbetter, J. A., and Schieven, G. L. (1997) Role of
oxidative stress in the action of vanadium phosphotyrosine phosphatase
inhibitors. Redox independent activation of NF-kappaB, J. Biol.
Chem., 272, 11541-1149.
9.Burkle, A. (2000) Poly(ADP-ribosyl)ation, genomic
instability, and longevity, Ann. NY Acad. Sci., 908,
126-132.
10.Burkle, A. (2001) Poly(APD-ribosyl)ation, a DNA
damage-driven protein modification and regulator of genomic
instability, Cancer Lett., 163, 1-5.
11.Jameson, M. J., Beckler, A. D., Taniguchi, L. E.,
Allak, A., Vanwagner, L. B., Lee, N. G., Thomsen, W. C., Hubbard, M.
A., and Thomas, C. Y. (2010) Activation of the insulin-like growth
factor-1 receptor induces resistance to epidermal growth factor
receptor antagonism in head and neck squamous carcinoma cells, Mol.
Cancer Ther., 10, 2124-2134.
12.Rieger, A. M., Nelson, K. L., Konowalchuk, J. D.,
and Barreda, D. R. (2011) Modified annexin V/propidium iodide
apoptosis assay for accurate assessment of cell death, J. Vis.
Exp., doi: 10.3791/2597.
13.Vermes, I., Haanen, C., and Reutelingsperger, C.
(2000) Flow cytometry of apoptotic cell death, J. Immunol.
Methods, 243, 167-190.
14.Jiang, L., Ji, N., Zhou, Y., Li, J., Liu, X.,
Wang, Z., Chen, Q., and Zeng, X. (2009) CAL 27 is an oral adenosquamous
carcinoma cell line, Oral. Oncol., 45, e204-207.
15.Sakai, A. (1997) Orthovanadate, an inhibitor of
protein tyrosine phosphatases, acts more potently as a promoter than as
an initiator in the BALB/3T3 cell transformation,
Carcinogenesis, 18, 1395-1399.
16.Gordon, J. A. (1991) Use of vanadate as
protein-phosphotyrosine phosphatase inhibitor, Methods Enzymol.,
201, 477-482.
17.Klein, A., Holko, P., Ligeza, J., and Kordowiak,
A. M. (2008) Sodium orthovanadate affects growth of some human
epithelial cancer cells (A549, HTB44, DU145), Folia Biol.
(Krakow), 56, 115-121.
18.Yang, J., Zhang, Z., Jiang, S., Zhang, M., Lu,
J., Huang, L., Zhang, T., Gong, K., Yan, S., Yang, Z., and Shao, G.
(2016) Vanadate-induced antiproliferative and apoptotic response in
esophageal squamous carcinoma cell line EC109, J. Toxicol. Environ.
Health A, 79, 864-868.
19.Goncalves, A. P., Videira, A., Soares, P., and
Maximo, V. (2011) Orthovanadate-induced cell death in
RET/PTC1-harboring cancer cells involves the activation of caspases and
altered signaling through PI3K/Akt/mTOR, Life Sci., 89,
371-377.
20.Delwar, Z. M., Siden, A., Cruz, M. H., and
Yakisich, J. S. (2012) Menadione/sodium orthovanadate combination
eliminates and inhibits migration of detached cancer cells, ISRN
Pharmacol., 2012, 307102.
21.Gunther, T. M., Kviecinski, M. R., Baron, C. C.,
Felipe, K. B., Farias, M. S., Da Silva, F. O., Bucker, N. C., Pich, C.
T., Ferreira, E. A., Wilhelm Filho, D., Verrax, J., Calderon, P. B.,
and Pedrosa, R. C. (2013) Sodium orthovanadate associated with
pharmacological doses of ascorbate causes an increased generation of
ROS in tumor cells that inhibits proliferation and triggers apoptosis,
Biochem. Biophys. Res. Commun., 430, 883-888.
22.Zhang, Z., Chen, F., Huang, C., and Shi, X.
(2002) Vanadate induces G2/M phase arrest in p53-deficient mouse embryo
fibroblasts, J. Environ. Pathol. Toxicol. Oncol., 21,
223-231.
23.Ray, R. S., Ghosh, B., Rana, A., and Chatterjee,
M. (2007) Suppression of cell proliferation, induction of apoptosis and
cell cycle arrest: chemopreventive activity of vanadium in vivo
and in vitro, Int. J. Cancer, 120, 13-23.
24.Choi, Y. J., Lim, S. Y., Woo, J. H., Kim, Y. H.,
Kwon, Y. K., Suh, S. I., Lee, S. H., Choi, W. Y., Kim, J. G., Lee, I.
S., Park, J. W., and Kwon, T. K. (2003) Sodium orthovanadate
potentiates EGCG-induced apoptosis that is dependent on the ERK
pathway, Biochem. Biophys. Res. Commun., 305,
176-185.
25.Tang, C., Liang, J., Qian, J., Jin, L., Du, M.,
Li, M., and Li, D. (2014) Opposing role of JNK-p38 kinase and ERK1/2 in
hydrogen peroxide-induced oxidative damage of human trophoblast-like
JEG-3 cells, Int. J. Clin. Exp. Pathol., 7, 959-968.
26.Xia, Z., Dickens, M., Raingeaud, J., Davis, R.
J., and Greenberg, M. E. (1995) Opposing effects of ERK and JNK-p38 MAP
kinases on apoptosis, Science, 270, 1326-1331.
27.Daum, G., Kalmes, A., Levkau, B., Wang, Y.,
Davies, M. G., and Clowes, A. W. (1998) Pervanadate inhibits
mitogen-activated protein kinase kinase-1 in a p38MAPK-dependent
manner, FEBS Lett., 427, 271-274.
28.Zhao, Z., Tan, Z., Diltz, C. D., You, M., and
Fischer, E. H. (1996) Activation of mitogen-activated protein (MAP)
kinase pathway by pervanadate, a potent inhibitor of tyrosine
phosphatases, J. Biol. Chem., 271, 22251-22255.
29.Leon, I. E., Porro, V., Di Virgilio, A. L., Naso,
L. G., Williams, P. A., Bollati-Fogolin, M., and Etcheverry, S. B.
(2014) Antiproliferative and apoptosis-inducing activity of an
oxidovanadium(IV) complex with the flavonoid silibinin against
osteosarcoma cells, J. Biol. Inorg. Chem., 19, 59-74.
30.Parrondo, R., De las Pozas, A., Reiner, T., Rai,
P., and Perez-Stable, C. (2010) NF-kappaB activation enhances cell
death by antimitotic drugs in human prostate cancer cells, Mol.
Cancer, 9, 182.
31.Markopoulou, S., Kontargiris, E., Batsi, C.,
Tzavaras, T., Trougakos, I., Boothman, D. A., Gonos, E. S., and
Kolettas, E. (2009) Vanadium-induced apoptosis of HaCaT cells is
mediated by c-fos and involves nuclear accumulation of clusterin,
FEBS J., 276, 3784-3799.
32.Morita, A., Yamamoto, S., Wang, B., Tanaka, K.,
Suzuki, N., Aoki, S., Ito, A., Nanao, T., Ohya, S., Yoshino, M., Zhu,
J., Enomoto, A., Matsumoto, Y., Funatsu, O., Hosoi, Y., and Ikekita, M.
(2010) Sodium orthovanadate inhibits p53-mediated apoptosis, Cancer
Res., 70, 257-265.
33.Wozniak, K., and Blasiak, J. (2004) Vanadyl
sulfate can differentially damage DNA in human lymphocytes and HeLa
cells, Arch. Toxicol., 78, 7-15.
34.Korbecki, J., Baranowska-Bosiacka, I., Gutowska,
I., and Chlubek, D. (2012) Biochemical and medical importance of
vanadium compounds, Acta Biochim. Pol., 59, 195-200.