Impact of Changes in Neurotrophic Supplementation on Development of Alzheimer’s Disease-Like Pathology in Oxys Rats
E. A. Rudnitskaya, N. G. Kolosova, and N. A. Stefanova*
Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia; E-mail: stefanovan@bionet.nsc.ru* To whom correspondence should be addressed.
Received October 9, 2016; Revision received November 29, 2016
Alzheimer’s disease (AD) is the most common type of age-related dementia. The development of neurodegeneration in AD is closely related to alterations in neurotrophic supplementation of the brain, which may be caused either by disorder of neurotrophin metabolism or by modification of its availability due to changes in the microenvironment of neurons. The underlying mechanisms are not fully understood. In this work, we used senescence-accelerated OXYS rats as a unique model of the sporadic form of AD to examine the relationship of development of AD signs and changes in neurotrophic supplementation of the cortex. Based on comparative analysis of the transcriptome of the frontal cerebral cortex of OXYS and Wistar (control) rats, genes of a neurotrophin signaling pathway with different mRNA levels in the period prior to the development of AD-like pathology in OXYS rats (20 days) and in the period of its active manifestation (5 months) and progression (18 months) were identified. The most significant changes in mRNA levels in the cortex of OXYS rats occurred in the period from 5 to 18 months of age. These genes were associated with neurogenesis, neuronal differentiation, synaptic plasticity, and immune response. The results were compared to changes in the levels of brain-derived neurotrophic factor (BDNF), its receptors TrkB and p75NTR, as well as with patterns of their colocalization, which reveal the balance of proneurotrophins and mature neurotrophins and their receptors. We found that alterations in neurotrophic balance indicating increased apoptosis precede the development of AD-like pathology in OXYS rats. Manifestation of AD-like pathology occurs against a background of activation of compensatory and regenerative processes including increased neurotrophic supplementation. Active progression of AD-like pathology in OXYS rats is accompanied by the suppression of activity of the neurotrophin system. Thus, the results confirm the importance of the neurotrophin system as a potential target for development of new approaches to slow age-related alterations in brain and AD development.
KEY WORDS: neurotrophic factors, Alzheimer’s disease, OXYS ratsDOI: 10.1134/S0006297917030105
Abbreviations: Aβ, β-amyloid; AβPP, Aβ precursor protein; AD, Alzheimer’s disease; BDNF, brain-derived neurotrophic factor; mBDNF, mature form of BDNF protein; phosphoTrkB(Y817), phosphorylated form of TrkB; proBDNF, immature form of BDNF protein; RNA-seq, method of massive parallel sequencing; TrkB, tropomyosin tyrosine kinase receptor B.
Alzheimer’s disease (AD) is a neurodegenerative disease that
causes senile dementia due to brain atrophy. The key signs of the
disease in brain include accumulation of the neurotoxic forms of
amyloid beta peptide (Aβ) causing formation of amyloid plaques,
hyperphosphorylation of tau-protein, inflammation, mitochondrial
dysfunction, oxidative stress, synaptic deficiency, and neuronal death
[1]. The mechanisms of these processes are closely
related to the disruption of neurotrophic supplementation in the brain
including changes in the content of brain-derived neurotrophic factor
(BDNF) and its receptors TrkB and p75NTR [2, 3]. During ontogenesis of the
nervous system, the mature form of the protein (mBDNF) regulates
division of neurons, their migration, differentiation, and establishing
of intercellular contacts via interacting with the high-affinity TrkB
receptor, while its precursor (proBDNF) regulates initiation of
apoptosis via interaction with the p75NTR receptor [4], which occurs even in the early stages of
development, when neuronal death plays an essential role in the
structural and functional development of the brain [5]. Decrease in BDNF content with aging and during the
development of AD against a background of disbalance in the contents of
the precursor and mature forms of the neurotrophin results in the loss
of control over neuron proliferation and differentiation, disruption of
neurotransmission, and development of neurodegeneration [6]. It was demonstrated that accumulation of the
C-terminal fragment of p75NTR protein on the cell membrane,
causing cell death during AD, could be mediated by an increase in the
content of Aβ precursor protein (AβPP) [3]. The Aβ can directly contact the extracellular
domain of p75NTR receptor, which results in activation of
the intracellular domain of this receptor (death domain), initiation of
the caspase 3 and caspase 8 cascades, and death of neurons [7, 8]. Progression of
neurodegenerative processes in AD is inevitably accompanied by the
disruption of trans-synaptic transmission and reduction of interneuron
connections [9]. Considering that retrograde
transport of Trk receptors activated by neurotrophins plays a crucial
role in initiation of signaling cascades of neuronal survival, its
dysfunction results in the disruption of response to neurotrophins and,
ultimately, to neuronal death [10]. The mechanisms
underlying the disruption of the neurotrophin system during the
development of AD are poorly understood. This is because it is
impossible to investigate these issues in humans, especially during
preclinical stages of AD, as well as because of the lack of adequate
biological models of the disease. The predominant existing models of AD
are monogenic, such as transgenic animals or animals with knockout
genes or with certain mutations. The use of these models could provide
better understanding of the contribution of a particular gene to the
development of hereditary form of the disease, but these models do not
reproduce all phenotypic manifestations of its sporadic form, such as
complex diseases of polygene nature, which accounts for approximately
95% of all AD cases.
The senescence-accelerated OXYS rat line exhibiting all key symptoms is a unique AD model [11]. The sequence of manifestation of signs – dysfunction of mitochondria, hyperphosphorylation of tau-protein, destructive changes in neurons and synapses, and their progression on a background of increasing level of AβPP, enhanced accumulation of Aβ, and formation of amyloid plaques in the brain – corresponds to the modern notions on pathogenesis of the sporadic form of AD in humans. The lack of mutations in the App, Psen1, and Psen2 genes in the genome of OXYS rats, which are characteristic for the familial form of the disease [12], suggests the OXYS rat line as a promising model of the sporadic form of AD. Mechanisms of development of the processes characteristic for this disease in OXYS rats remain unclear, but the observed neurodegenerative changes in the brain structures responsible for cognition and memory already at young age [13-15] suggest that they can be associated with changes in the neurotrophin system. This is corroborated by our recent data: lack of differences in the level of BDNF in hippocampus between Wistar and OXYS rats in the period when the phenotypic signs of AD are not yet revealed in OXYS rats, and its increase in the period of active manifestation of these signs followed by a decrease in the period of enhanced accumulation of Aβ in the brain [16].
The objective of this study was to investigate the relationship between the development of AD symptoms in OXYS rats and the state of neurotrophic supplementation in the cerebral cortex. For this purpose, we analyzed the results of investigation of the prefrontal cortex transcriptome in OXYS and Wistar rats with the method of massive parallel sequencing (RNA-seq) [17]. We identified genes associated with the neurotrophin signaling pathway, the level of mRNA of which changed with age and was different in OXYS and Wistar (control) rats, and made functional annotation of these genes. These results were correlated with the age-related changes in the content of BDNF and its receptors TrkB and p75NTR in the prefrontal cortex of OXYS and Wistar rats, as well as their colocalization, which reflected the balance between proneurotrophins and mature neurotrophins as well as their receptors.
MATERIALS AND METHODS
Animals. This work was performed with the male rats of OXYS and Wistar lines in the Common Use Center for Gene pools of laboratory animals, Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, in accordance with the “Requirements for conducting experiments with laboratory animals” (86/609/EEC). Starting with age of four weeks, rats were kept in groups of five in 57 × 36 × 20 cm cages at 22 ± 2°C under conditions of fixed illumination regime (12 h light/12 h dark) with freely available water and food (granulated food for laboratory animals; Chara, Assortiment-Agro, Russia).
The RNA-seq method was used for determination of mRNA content of genes associated with the neurotrophin signaling pathway in the prefrontal cortex of OXYS and Wistar rats at the age of 20 days and of 5 and 18 months (three animals per group). The levels of TrkB receptor and its phosphorylated form (phosphoTrkB(Y817)) were determined in prefrontal cortex of the right cerebral hemisphere of the OXYS and Wistar rats at the age of 20 days and of 3 and 18 months (six animals per group) using Western blot analysis. The contents and colocalization of TrkB and phosphoTrkB(Y817), mBDNF and TrkB, proBDNF and p75NTR were determined in the prefrontal cortex of the left cerebral hemisphere of the same animals using immunohistochemistry.
Analysis of data using RNA-seq. To create groups of differentially expressed genes of the neurotrophin signaling pathway, the considered genes were corrected according to the Benjamini–Hochberg multiple testing procedure with p < 0.01. These groups were functionally annotated using bioinformatics databases DAVID (Database for Annotation, Visualization and Integrated Discovery) and KEGG (Kyoto Encyclopedia of Genes and Genomes) with threshold of enrichment significance (EASE) p < 0.05.
Western blot analysis. Brain samples were isolated on ice and stored at –80°C. Protein was isolated using lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 mM EDTA, protease inhibitors (Sigma-Aldrich, USA)) at ratio 1 : 4. Protein concentration was determined with the Bradford assay (Bio-Rad, USA). Proteins were separated with electrophoresis in 12% polyacrylamide gel in Tris-glycine buffer (25 mM Tris, 190 mM glycine, 0.1% SDS) with operating power of 120 V and transferred onto a nitrocellulose membrane (GE Healthcare, USA). Following blocking with 5% BSA (Sigma-Aldrich) in sodium phosphate buffer solution (PBS) with 0.1% Tween-20 at room temperature (1 h), the membranes were incubated with primary antibodies against TrkB, phosphoTrkB(Y817), and β-actin (Abcam, USA) at 4°C (16 h) and with secondary antibodies at room temperature (Abcam; 1 h). Emission intensity was evaluated using the ImageJ program.
Immunohistochemical analysis. The left hemisphere of the brain was fixed in 4% paraformaldehyde in PBS, pH 7.4 (48 h), and then in 30% sucrose solution in PBS (~48 h) at 4°C and stored at −80°C. Sagittal brain sections (20 µm) were prepared on a Microm HM-505N cryostat (Microm, Germany) at –20°C. The sections were mounted on Polysine® microscope slides (Menzel-Glaser, Germany), incubated in 5% BSA in PBS at room temperature (1 h) with primary antibodies against mBDNF (Millipore, USA) and TrkB, TrkB and phosphoTrkB(Y817), proBDNF and p75NTR (Abcam) at 4°C (16 h) and with secondary antibodies at room temperature (Abcam; 1 h), and washed in PBS; then mounting medium with DAPI (Amersham, USA) was applied to stain nuclei. The samples were analyzed with an Axioplan 2 light microscope (Zeiss, Germany).
Statistical analysis of results was conducted using the Statistica 6.0 program. Factorial dispersion analysis (ANOVA) with post-hoc comparison of group average (Newman–Keuls criterion) was used. Genotype and age were considered as independent factors. Data are presented as mean ± SEM. The results were considered statistically significant at p < 0.05.
RESULTS
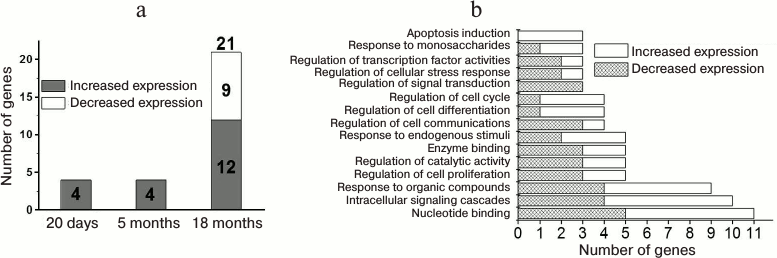
Age-related changes in expression of genes of neurotrophin signaling pathway in prefrontal cerebral cortex of OXYS and Wistar rats. Analysis of the results (Fig. 1a) showed that the level of mRNA of four genes (Ngf, Mapk14, Bax, and Nras) in 20-day-old OXYS and Wister rats was different, and at the age of 5 months another group of four genes associated with the neurotrophin signaling pathway (Ntf3, Camk2d, Shc4, and Prkcd) (according to the KEGG pathway database) exhibited different expression. The expression of these genes in OXYS rats was higher (p < 0.01). At 18 months of age, the number of differentially expressed genes in the cortex of OXYS rats compared to Wistar rats reached 21 (Fig. 1a). Among those, 12 genes displayed higher expression (Ngf, Mapk14, Shc4, Prkcd, Mapk3, Pik3r2, Akt1, Sh2b1, Nfkbie, Rps6ka2, Maged1, and Camk2b) and nine genes showed decreased expression (Rps6ka6, Calm2, Calm1, Frs2, Rap1a, Ripk2, Akt3, Cdc42, and Kras). Functional annotation showed that the changes in expression of neurotrophin signaling pathway genes in the brain of 18-month-old OXYS rats were associated with intracellular signaling cascades (Akt1, Cdc42, Kras, and others), induction of apoptosis (Maged1, Prkcd, and Ngf), binding of nucleotides (Akt1, Ripk2, Camk2b, and others) and enzymes (Akt1, Cdc42, Rap1a, and others), regulation of activity of transcription factors (Kras, Mapk3, and Ripk2), cell cycle (Akt1, Cdc42, Mapk14, and Ngf), cell proliferation (Maged1, Kras, Ngf, and others) and differentiation (Akt1, Mapk14, Ripk2, and Ngf), cellular stress response (Akt1, Cdc42, and Ripk2), and others (Fig. 1b). Hence, significant changes in the expression of neurotrophin signaling pathway genes in the cortex of OXYS rats were observed at the age of 18 months; furthermore, the decrease in mRNA levels of genes was associated with neuronal signal transmission, and increase was associated with apoptosis.
Fig. 1. Age-related changes in expression of the neurotrophin signaling pathway genes in prefrontal cortex of OXYS and Wistar rats. a) Number of differentially expressed genes of the neurotrophin signaling pathway in the cortex of OXYS rats in comparison with Wistar rats at the age of 20 days and of 5 and 18 months. b) Significant gene ontology terms combining inter-line differences in expression of the neurotrophin signaling pathway genes in prefrontal cortex of OXYS and Wistar rats at the age of 18 months (p < 0.05).
From the age of 20 days to 5 months, the expression of 51 genes of the neurotrophin signaling pathway changed in the cortex of Wistar rats: mRNA level decreased for 17 genes and increased for 34 genes. In the case of OXYS rats, the expression of 61 genes changed: mRNA level of 24 genes decreased, and of 37 genes increased. Moreover, the expression of 45 genes changed in the same direction for Wistar and OXYS rats: mRNA level of 15 genes decreased and of 30 genes increased. The changes in expression of genes encoding products associated with cell proliferation (Psen1, Map2k1, Bax, and others), mitochondrial (Psen1, Bax, Bcl2, and Mapk9) and nuclear-cytoplasmic transport (Mapk1, Camk4, Gsk3b, and Mapk3), regulation of ion transport (Bax, Bcl2, Akt3, and Akt2) and synaptic transmission (Ntf3, Psen1, Gsk3b, and others), phosphorylation (Bcl2, Camk2d, Camk2b, and others), nucleotide binding (Pdk1, Braf, Map2k1, and others), and apoptosis induction (Psen1, Gsk3b, Bax, Psen2, and others) and its regulation (Ntf3, Ywhab, Foxo3, Psen1, and others) were common for OXYS and Wistar rats.
From the age of 20 days to 5 months, the expression of 15 genes changed in OXYS rats but not in Wistar rats, in particular, the mRNA level of eight genes decreased (Mapk12, Ngf, Nfkbib, Mapkapk2, Tp53, Csk, Raf1, and Rapgef1) and of seven genes increased (Rap1b, Rap1a, Rhoa, Cdc42, Map3k1, Prkcd, and Sos2). The genes, whose expression increased in the cortex of OXYS rats, were associated with regulation of signal transduction (Cdc42, Rhoa, and Prkcd) and polymerization of actin filaments (Map3k1 and Rhoa), organization of cellular components (Cdc42, Map3k1, and Rhoa), binding of enzymes (Cdc42, Map3k1, and others), etc., while the expression of genes associated with regulation of the cell cycle (Mapk12, Tp53, and Ngf) decreased (Fig. 2a).
Fig. 2. Functional annotation of neurotrophin signaling pathway genes, which showed age-related changes of expression in the cortex of OXYS rats. Significant gene ontology terms combining age-related differences in expression of the neurotrophin signaling pathway genes in prefrontal cortex of OXYS versus Wistar rats at the age from 20 days to 5 months (a) and from 5 to 18 months (b); p < 0.05.
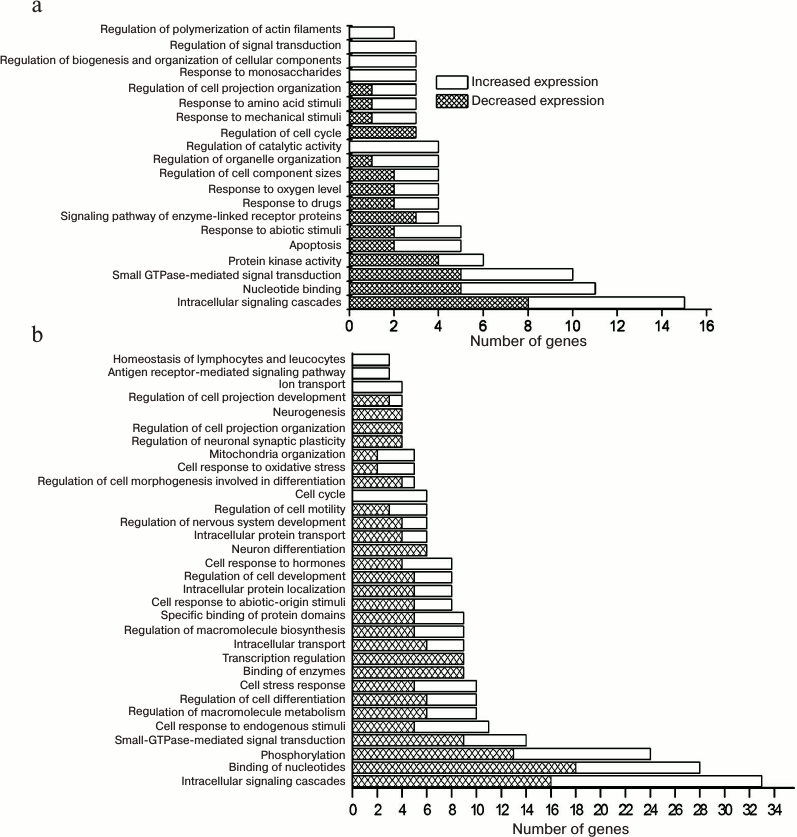
The expression of five genes changed in Wistar rats from the age of 5 to 18 months (mRNA level of Camk4, Ntrk3, and Mapk14 decreased, and that of Rps6ka1 and Shc4 – increased), while the mRNA level of 54 genes changed in OXYS rats: the level of mRNA of 26 genes decreased and that of 28 genes increased. In particular, the expression of the genes associated with nervous system regulation (Ntrk3, Bdnf, Ywhah, and others), phosphorylation (Gsk3b, Mapk3, Mapk9, and others), mitochondria organization (Akt1, Bax, Map3k1, Mapk9, and Tp73), intracellular localization (Akt1, Cdc42, Ywhah, and others) and transport of proteins (Akt1, Ywhah, Gsk3b, and others), regulation of macromolecule biosynthesis (Akt1, Cdc42, Ywhah, Mapk3, and others) and metabolism (Akt1, Cdc42, Ywhah, Mapk9, and others), cell stress response (Gsk3b, Bax, Mapk9, Mapk8, and others), apoptosis (Akt1, Bax, Psen2, Bad, Tp73, and others), etc. changed in OXYS rats with the progression of AD signs (Fig. 2b).
It is significant that the expression of only three genes of the neurotrophin signaling pathway changed in the OXYS rat cortex in the same direction from the age of 20 days to 18 months: the mRNA level of Ptpn11, Crkl, and Mapk3 genes increased. At the same time, the expression of 30 genes of the neurotrophin signaling pathway in the cortex of OXYS rats changed in different direction depending on the age: the mRNA level of 20 genes (Ywhah, Gsk3b, Calm1, Calm2, Camk4, and others) increased from the age of 20 days to 5 months and decreased from 5 to 18 months, while the mRNA level of 10 genes (Ikbkb, Nfkbib, Psen2, Camk2b, Rapgef1, Mapkapk2, and others) decreased from the age of 20 days to 5 months and increased from 5 to 18 months.
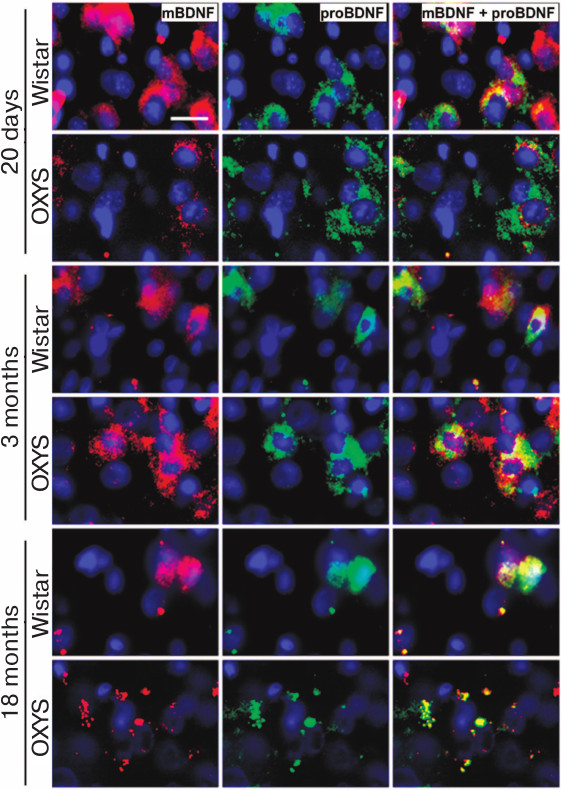
Age-related change in mBDNF and proBDNF content in prefrontal cerebral cortex of OXYS and Wistar rats. The analysis of the results of immunohistochemical staining of the brain sections showed that the mature form of BDNF was predominant in the prefrontal cortex of 20-day-old Wistar rats, while the proBDNF was predominant in OXYS rats (Fig. 3). The content of both protein forms in the prefrontal cortex of OXYS rats increased by the age of 3 months and became higher than in the Wistar rats (Fig. 3). By the age of 18 months, the content both mBDNF and proBDNF decreased in the cerebral cortex of both rat lines.
Fig. 3. Content of mBDNF and proBDNF in the prefrontal cerebral cortex of OXYS and Wistar rats of different age. By the age of 20 days, the intensity of the mBDNF signal is lower and of the proBDNF signal is higher in OXYS rats than in Wistar rats. By the age of 3 months, the intensity of signals from both forms of protein increases in OXYS rats. By the age of 18 months, the intensity of mBDNF and proBDNF signals decreases in the rats of both lines. Brain sections are stained with specific antibodies against mBDNF (red) and proBDNF (green); nuclei stained with DAPI (blue). Layer of pyramidal neurons in the prefrontal cerebral cortex. Scale bar: 20 µm.
Level of TrkB-receptor and its phosphorylated form in prefrontal cerebral cortex of OXYS and Wistar rats of different age. As shown with dispersion analysis, the level of TrkB in prefrontal cortex was independent of genotype and age of the animals (F1,30 = 0.7, p = 0.41 and F2,30 = 2.3, p = 0.11, respectively; Fig. 4a). The level of phosphoTrkB(Y817) also was independent of the animal genotype (F1,29 = 0.5, p = 0.49), but decreased with age (F2,29 = 6.3, p < 0.005) (Fig. 4b). Comparison of group average showed that significant difference was demonstrated only for OXYS rats: at the age of 18 months they had level of phosphoTrkB(Y817) receptor lower than at the age of 20 days (p < 0.005). The phosphoTrkB(Y817)/TrkB ratio indicates the degree of activation of TrkB receptor. This indicator for the prefrontal cortex was not dependent on genotype or on age of the animals (Fig. 4c) (F1,29 = 0.001, p = 0.97 and F2,29 = 0.08, p = 0.92, respectively).
Fig. 4. Age-related changes in the level of TrkB receptor, phosphoTrkB(Y817) receptor, and their ratio in the prefrontal cerebral cortex of OXYS and Wistar rats. a) The level of TrkB receptor did not change significantly with age in both rat lines. b) The level of phosphoTrkB(Y817) receptor decreased moderately with age in the OXYS rats. c) The phosphoTrkB(Y817)/TrkB ratio did not change with age (data of Western blot analysis); (^) age differences, p < 0.05. d) Polyacrylamide gel bands of TrkB, phosphoTrkB(Y817) and β-actin proteins from 20-day- and 3- and 18-month-old OXYS (O) and Wistar (W) rats. e) Immunohistochemical staining of brain sections of 20-day-old and of 3- and 18-month-old OXYS and Wistar rats with specific antibodies against TrkB receptor and phosphoTrkB(Y817) receptor; nuclei stained with DAPI. Layer of pyramidal neurons in the prefrontal cerebral cortex. Scale bar: 10 µm.
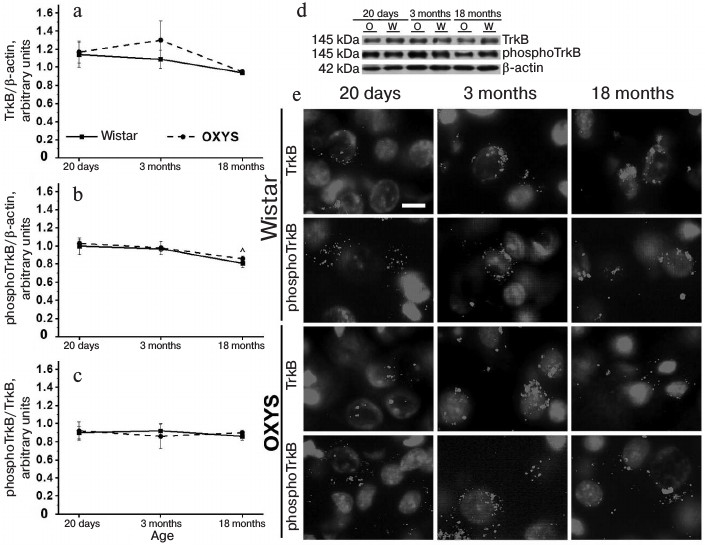
Immunohistochemical analysis did not reveal any differences in the intensity of signals from TrkB receptors and phosphoTrkB(Y817) receptors in the prefrontal cerebral cortex of 20-day-old and 3-month-old Wistar and OXYS rats. The content of TrkB receptor and its phosphorylated form in cerebral cortex of both rat lines decreased with age, but the decrease was more significant in OXYS rats. As a result, the intensity of TrkB receptor and phosphoTrkB(Y817) receptor signals in the prefrontal cortex of 18-month-old OXYS rats was lower than in the case of Wistar rats (Fig. 4d).
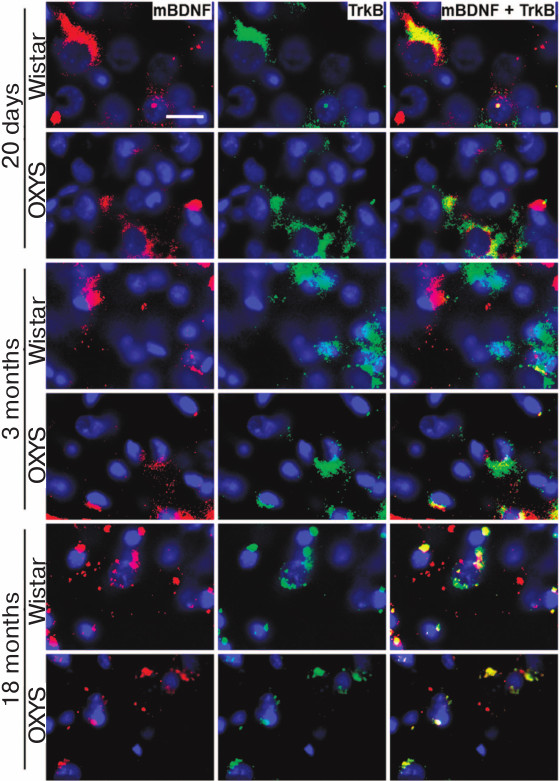
Content of BDNF and its receptors TrkB and p75NTR in prefrontal cerebral cortex of OXYS and Wistar rats of different age. Immunohistochemical analysis did not reveal any differences between the rat lines in the intensity of the TrkB receptor signal, but it demonstrated high frequency of its colocalization with mBDNF in the prefrontal cortex of the 20-day-old rats of both lines (Fig. 5). By the age of 3 months, the pattern changed: despite the absence of inter-line differences in the intensity of the TrkB receptor signals, the frequency of its colocalization with mBDNF in the prefrontal cortex of OXYS rats was higher than in the case of Wistar rats (Fig. 5). By the age of 18 months, the frequency of colocalization of mBDNF and TrkB receptor decreased in the cerebral cortex of both rat lines (Fig. 5).
Fig. 5. Content of mBDNF and TrkB receptor in the prefrontal cerebral cortex of OXYS and Wistar rats of different age. OXYS rats at the age of 3 months demonstrate higher frequency of colocalization of mBDNF and TrkB receptor than the Wistar rats. The intensity of mBDNF and TrkB receptor signals decreases in OXYS rats by the age of 18 months. Immunohistochemical staining of brain sections with specific antibodies against mBDNF (red) and TrkB receptor (green); nuclei stained with DAPI (blue). Layer of pyramidal neurons in the prefrontal cerebral cortex. Scale bar: 20 µm.
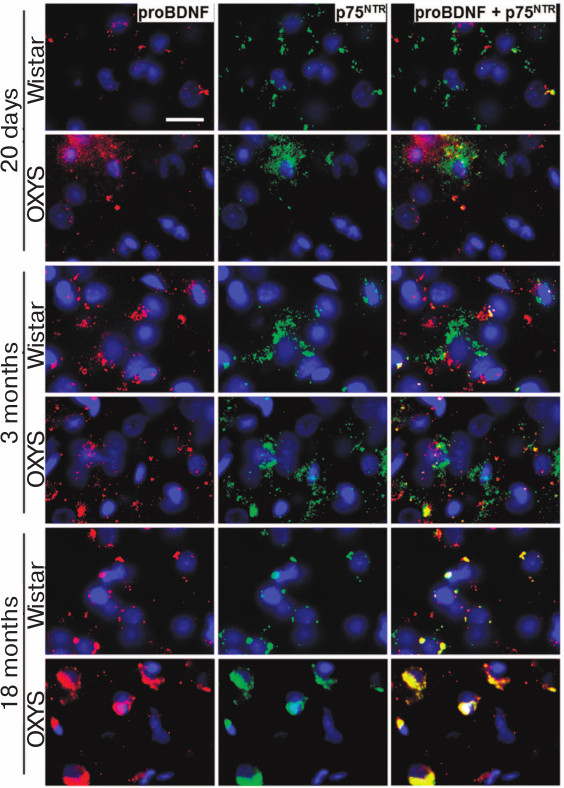
The analysis of proBDNF and p75NTR receptor content showed that the intensity of signals from both proteins in the prefrontal cortex of 20-day-old OXYS rats was higher than in the Wistar rats, while their colocalization was low in the rats of both lines (Fig. 6). The same pattern was observed in the 3-month-old Wistar rats, while the frequency of colocalization of proBDNF with p75NTR receptor in the cerebral cortex of OXYS rats increased. By the age of 18 months, the intensity of p75NTR receptor signal in the prefrontal cortex of Wistar rats decreased (Fig. 6), and the frequency of its colocalization with proBDNF and the level of free proBDNF increased. The content of p75NTR receptor in the cerebral cortex of OXYS rats increased from the age of 20 days to 3 months, and it remained high at the age of 18 months. Thus, the intensity of proBDNF and p75NTR receptor signals for the 18-month-old OXYS rats and the frequency of their colocalization in the prefrontal cortex were significantly higher than in Wistar rats (Fig. 6), which along with the cerebral cortex transcriptome analysis indicated activation of apoptosis.
Fig. 6. Content of proBDNF and p75NTR receptor in the prefrontal cerebral cortex of OXYS and Wistar rats of different age. In comparison with Wistar rats, the intensity of proBDNF and p75NTR receptor signals in the 20-day-old OXYS rats was higher, as well as the frequency of their colocalization at the age of 3 months and the signal intensity and frequency of colocalization at the age of 18 months. Immunohistochemical staining of brain sections with specific antibodies against proBDNF (red) and p75NTR receptor (green); nuclei stained with DAPI (blue). Layer of pyramidal neurons in the prefrontal cerebral cortex. Scale bar: 20 µm.
DISCUSSION
The development of age-related and AD-associated neurodegenerative processes is strongly linked to the changes in neurotrophic supplementation of the brain, which occurs due to either disruption of neurotrophin metabolism or modification of its accessibility due to change in the neuron microenvironment. The mechanisms initiating these processes as well as the differences between the changes in neurotrophic supplementation due to aging and during the development of AD remain poorly understood. Information on the dynamics of disruptions in the brain neurotrophin system during AD development is lacking. We have evaluated changes in neurotrophic supplementation in the prefrontal cerebral cortex of Wistar rats occurring with aging, and of OXYS rats in the period preceding the development of AD signs (20 days) and in the stages of their active manifestation (3-5 months) and progression (18 months). It is remarkable that the levels of mRNA of four genes of neurotrophin signaling increase in OXYS rats already at the age of 20 days, with three of them (Ngf, Bax, and Nras) associated with apoptosis. The shift of the proBDNF/mBDNF balance toward the immature form of the protein that was observed in the cerebral cortex of OXYS rats in this period favored activation of apoptosis. The period of postnatal development of rat brain associated with correction of errors and removal of “transitional” cellular population required for optimization of the neuronal network via apoptosis of a significant number of neurons is completed by this age [18]. It is likely that the increased activity of apoptosis in OXYS rats in comparison with Wistar rats is related to hypoxia, the signs of adaptation to which we have observed earlier in the brain of 20-day-old OXYS rats during investigation of energy metabolism [19]. Oxidative stress indirectly manifested by enhanced accumulation of a large deletion of mitochondrial DNA (4834 bp) in the hippocampus of OXYS rats in the early postnatal period can be either the cause or result of activation of apoptosis [14].
Comparison of changes in the transcriptome of prefrontal cerebral cortex of OXYS and Wistar rats from the age of 20 days to 5 months revealed unidirectional changes in the expression of 45 genes of the neurotrophic signaling pathway in both rat lines. Their increased expression is associated with the development and differentiation of neurons, growth of axons, and decreased with regulation of neurogenesis and axonogenesis and immune response. Previously, we showed that the first signs of neurodegenerative changes developed in the brain of OXYS rats by the age of 3-5 months: destructive changes in neurons and synapses [11], disruption of prolonged post-tetanic potentiation [20], hyperphosphorylation of tau-protein [11, 12], demyelination [21], and mitochondria dysfunction [11, 12]. Moreover, due to the plasticity of biological systems, compensatory reactions are initiated in the brain of OXYS rat in parallel with the development of neurodegenerative changes, directed to prevention of these processes: the density of neurons and synapses increases, as well as the BDNF level in hippocampus [15, 16]. The fact that the level of mRNA of neurotrophin signaling pathway genes changes in the brain of OXYS rats between the age of 20 days and 5 months while the same parameter in Wistar rats remains unchanged supports the hypothesis on initiation of compensatory mechanisms in the cerebral cortex of OXYS rats. The increase in their expression in the brain of OXYS rats is associated with regulation of signal transduction and biogenesis and organization of cellular components, which indicates enhancement of synaptogenesis and, hence, activation of the neurotrophin system in the stage of manifestation of AD signs. This activation could be the cause of either increase in neuron and synapse population or enhancement of neuronal death in the brain of young OXYS rats. Indeed, we have revealed the signs of functional stress in the neurotrophin system of 3-month-old OXYS rats: increase in mBDNF and proBDNF content in the cerebral cortex and higher frequency of their colocalization with TrkB receptors, indicating activation of neuroprotective mechanisms, and increase in p75NTR, which is an indicator of activation of axon retraction mechanisms, destruction of synapses, and apoptosis.
Hyperphosphorylation of tau-protein [22], which causes disruption and disorganization of the microtubule system, development of transport collapse, and blocking of neuronal transport pathways [23], could facilitate the changes in the neurotrophin system in the stage of manifestation of AD symptoms in OXYS rats. The disruption of exocytosis and intracellular transport of neurotrophin receptors becomes critical for the development of signaling cascades initiated by neurotrophins [24]. At the same time, the increase in expression of the neurotrophin signaling pathway genes associated with regulation of biogenesis, organization of cellular components, and polymerization of actin filament in the cerebral cortex of OXYS rats with age increase from 20 days to 5 months corroborates the notion on activation of compensatory processes aimed to restore intracellular transport and, as a result, signaling cascades induced by neurotrophins.
The compensatory activation of the neurotrophin system is followed by its exhaustion. The progression of AD signs in OXYS rats (from the age of 5 to 18 months) occurs on the background of the change in mRNA level of 54 neurotrophin signaling pathway genes in the cerebral cortex of OXYS rats, while the expression of only five genes changes in Wistar rats during this period. The decrease in mRNA level of genes in OXYS rats is associated with the processes of neurogenesis, neuron differentiation, regulation of cell projection organization, synaptic plasticity, and the increase is associated with immune system functions. It must be mentioned that the unidirectional change of expression of only three genes (Ptpn11, Crkl, and Mapk3) was observed in OXYS rats in the period from the age preceding the development of AD signs to the age when these signs are clearly pronounced (from the age of 20 days to 18 months), while the expression of 30 genes changed in different directions, for example, the Bdnf gene with the level of its mRNA increasing by the age of 5 months and decreasing by the age of 18 months.
We did not observe any age-related changes in the content of TrkB protein (mBDNF receptor) in the prefrontal cortex of either OXYS or Wistar rats; however, the level of the phosphorylated form of the TrkB receptor (phosphoTrkB(Y817)) decreased in OXYS rats. Following phosphorylation of tyrosine 817 in TrkB, the phospholipase Cγ is recruited to the signaling cascade [25], which results in formation of Ins(1,4,5)P3, diacylglycerol, and increase in intracellular Ca2+ concentration, which in turn activate calmodulin and protein kinase C. These signaling pathways control various aspects of cell functioning including survival, differentiation, growth, and synaptic plasticity.
Hence, changes in neurotrophin system balance indicating the enhancement of apoptosis precede the appearance of the AD signs in OXYS rats, and manifestation of the signs occurs on the background of activation of compensatory processes including the neurotrophin system, which is followed by the suppression of the activity of the neurotrophin system during the period of disease. In general, the results agree with the notion that the disruption of neurotrophic supplementation of brain significantly affects AD pathogenesis and demonstrate potential benefits of the use of the neurotrophin system as a target for the development of new approaches for correction of age-related changes in the brain and the development of AD.
Acknowledgements
This work was financially supported by the Russian Science Foundation (project No. 16-15-10005).
REFERENCES
1.Morley, J. E., Armbrecht, H. J., Farr, S. A., and
Kumar, V. B. (2012) The senescence accelerated mouse (SAMP8) as a model
for oxidative stress and Alzheimer’s disease, Biochim.
Biophys. Acta, 1822, 650-656.
2.Rohe, M., Synowitz, M., Glass, R., Paul, S. M.,
Nykjaer, A., and Willnow, T. E. (2009) Brain-derived neurotrophic
factor reduces amyloidogenic processing through control of SORLA gene
expression, J. Neurosci., 29, 15472-15478.
3.Sotthibundhu, A., Sykes, A. M., Fox, B., Underwood,
C. K., Thangnipon, W., and Coulson, E. J. (2008) β-Amyloid 1-42
induces neuronal death through the p75 neurotrophin receptor, J.
Neurosci., 28, 3941-3946.
4.Bothwell, M. (2014) NGF, BDNF, NT3 and NT4, in
Neurotrophic Factors (Lewin, G. R., and Carter, B. D., eds.)
Springer, Berlin, pp. 3-15.
5.Dekkers, M. P. J., Nikoletopoulou, V., and Barde,
Y. A. (2013) Death of developing neurons: new insights and implications
for connectivity, J. Cell Biol., 203, 385-393.
6.Yoshii, A., and Constantine-Paton, M. (2010)
Post-synaptic BDNF-TrkB signaling in synapse maturation, plasticity and
disease, Dev. Neurobiol., 70, 304-322.
7.Perini, G., Della-Bianca, V., Politi, V., Della
Valle, G., Dal-Pra, I., Rossi, F., and Armato, U. (2002) Role of p75
neurotrophin receptor in the neurotoxicity by β-amyloid peptides
and synergistic effect of inflammatory cytokines, J. Exp. Med.,
195, 907-918.
8.Jakob-Roetne, R., and Jacobsen, H. (2009)
Alzheimer’s disease: from pathology to therapeutic approaches,
Angew. Chem. Int. Ed. Engl., 48, 3030-3059.
9.De Calignon, A., Polydoro, M., Suarez-Calvet, M.,
William, C., Adamowicz, D. H., Kopeikina, K. J., Pitstick, R., Sahara,
N., Ashe, K. H., Carlson, G. A., Spires-Jones, T. L., and Hyman, B. T.
(2012) Propagation of tau pathology in a model of early
Alzheimer’s disease, Neuron, 73, 685-697.
10.Ceni, C., Unsain, N., Zeinieh, M. P., and Barker,
P. A. (2014) Neurotrophins in the regulation of cellular survival and
death, in Neurotrophic Factors (Lewin, G. R., and Carter, B. D.,
eds.) Springer, Berlin, pp. 193-221.
11.Stefanova, N. A., Kozhevnikova, O. S., Vitovtov,
A. O., Maksimova, K. Y., Logvinov, S. V., Rudnitskaya, E. A.,
Korbolina, E. E., Muraleva, N. A., and Kolosova, N. G. (2014)
Senescence-accelerated OXYS rats: a model of age-related cognitive
decline with relevance to abnormalities in Alzheimer’s disease,
Cell Cycle, 13, 898-909.
12.Stefanova, N. A., Muraleva, N. A., Korbolina, E.
E., Kiseleva, E., Maksimova, K. Y., and Kolosova, N. G. (2015) Amyloid
accumulation is a late event in sporadic Alzheimer’s disease-like
pathology in nontransgenic rats, Oncotarget, 6,
1396-1413.
13.Stefanova, N. A., Maksimova, K. Y., Kiseleva, E.,
Rudnitskaya, E. A., Muraleva, N. A., and Kolosova, N. G. (2015)
Melatonin attenuates impairments of structural hippocampal
neuroplasticity in OXYS rats during active progression of
Alzheimer’s disease-like pathology, J. Pineal Res.,
59, 163-177.
14.Loshchenova, P. S., Sinitsyna, O. I., Fedoseeva,
L. A., Stefanova, N. A., and Kolosova, N. G. (2015) Influence of
antioxidant SkQ1 on accumulation of mitochondrial DNA deletions in the
hippocampus of senescence-accelerated OXYS rats, Biochemistry
(Moscow), 80, 596-603.
15.Maksimova, K. Yu., Logvinov, S. V., and
Stefanova, N. A. (2015) Morphological characterization of OXYS and
Wistar rat hippocampus in the aging process, Morfologiya,
147, 11-16.
16.Rudnitskaya, E. A., Maksimova, K. Y., Muraleva,
N. A., Logvinov, S. V., Yanshole, L. V., Kolosova, N. G., and
Stefanova, N. A. (2015) Beneficial effects of melatonin in a rat model
of sporadic Alzheimer’s disease, Biogerontology,
16, 303-316.
17.Stefanova, N. A., Korbolina, E. E., Ershov, N.
I., Rogaev, E. I., and Kolosova, N. G. (2015) Changes in the
transcriptome of the prefrontal cortex of OXYS rats as the signs of
Alzheimer’s disease development, Vavilov. Zh. Genet.
Selekt., 19, 445-454.
18.Kim, W. R., and Sun, W. (2011) Programmed cell
death during postnatal development of the rodent nervous system,
Dev. Growth Differ., 53, 225-235.
19.Sergeeva, S., Bagryanskaya, E., Korbolina, E.,
and Kolosova, N. (2006) Development of behavioral dysfunctions in
accelerated-senescence OXYS rats is associated with early postnatal
alterations in brain phosphate metabolism, Exp. Gerontol.,
41, 141-150.
20.Beregovoy, N. A., Sorokina, N. S., Starostina, M.
V., and Kolosova, N. G. (2011) Age-specific peculiarities of formation
long-term posttetanic potentiation on OXYS rats, Bull. Exp. Biol.
Med., 151, 71-73.
21.Kolosova, N. G., Akulov, A. E., Stefanova, N. A.,
Moshkin, M. P., Savelov, A. A., Koptyug, I. V., Panov, A. V., and
Vavilin, V. A. (2011) Effect of malate on development of
rotenone-induced brain changes in Wistar and OXYS rats: an MRI study,
Dokl. Biol. Sci., 437, 72-75.
22.Stefanova, N. A., Muraleva, N. A., Skulachev, V.
P., and Kolosova, N. G. (2014) Alzheimer’s disease-like pathology
in senescence-accelerated OXYS rats can be partially retarded with
mitochondria-targeted antioxidant SkQ1, J. Alzheimers Dis.,
38, 681-694.
23.Braak, H., and Tredici, K. D. (2016) Potential
pathways of abnormal tau and α-synuclein dissemination in
sporadic Alzheimer’s and Parkinson’s diseases, Cold
Spring Harb. Perspect. Biol., 8, 1-24.
24.Park, H., and Poo, M. M. (2013) Neurotrophin
regulation of neural circuit development and function, Nat. Rev.
Neurosci., 14, 7-23.
25.Cazorla, M., Jouvenceau, A., Rose, C., Guilloux,
J. P., Pilon, C., Dranovsky, A., and Premont, J. (2010) Cyclotraxin-B,
the first highly potent and selective TrkB inhibitor, has anxiolytic
properties in mice, PLoS One, 5, 1-17.