MicroRNA-294 Promotes Cellular Proliferation and Motility through the PI3K/AKT and JAK/STAT Pathways by Upregulation of NRAS in Bladder Cancer
Yongwei Li1#, Zhengfei Shan1#, Chu Liu1#, Diandong Yang1*, Jitao Wu1*, Changping Men1, and Yankai Xu2
1Department of Urology, The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai 264000, China; E-mail: jtwu1006@163.com2Department of Urology, Yantai Affiliated Hospital of Binzhou Medical University, Yantai 264100, China
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received October 10, 2016; Revision received November 18, 2016
In our study we examined the role of microRNA-294 (miR-294) in bladder cancer and related mechanisms. Real-time polymerase chain reaction (RT-PCR) was performed to determine the expression level of miR-294. Western blot was used to determine the expression of NRAS, mainly factors in the PI3K/AKT and JAK/STAT pathways. Cell counting kit-8 assay, clonogenic assay, wound-healing assay, transwell and flow cytometry were used to explore, respectively, cell proliferation, survival, migration, invasion, and apoptosis of bladder cancer cell line T24. The expressions of miR-294 in bladder cancer cells including J82, HT1376, T24, and SW780 were significantly increased compared to those in human bladder epithelium cells (both HCV29 and SV-HUC-1). The proliferation rate, surviving fraction, migration, and invasion of T24 cells in miR-294 mimetic transfected group were significantly increased, while they were significantly decreased by miR-294 inhibitor transfection. Moreover, miR-294 suppression could increase the apoptotic rate of T24 cells. In addition, drug resistance of T24 cells to cisplatin was increased in miR-294 mimetic-treated group, while it was decreased by miR-294 inhibitor compared to empty control. Overexpression of miR-294 could upregulate NRAS expression in T24 cells and activate PI3K/AKT and JAK/STAT pathways. We found that miR-294 expression was positively related with proliferation and motility of T24 cells. Moreover, miR-294 suppression could promote the sensitivity of T24 cells to cisplatin. We also found miR-294 could upregulate NRAS and activate the PI3K/AKT and JAK/STAT pathways in T24 cells.
KEY WORDS: microRNA-294, NRAS, PI3K/AKT and JAK/STAT pathways, bladder cancerDOI: 10.1134/S0006297917040095
Bladder cancer is the ninth most common type of cancer in the world and the most common urinary tract malignancy in China. Of these, bladder urothelial carcinoma accounted for almost 90% [1]. The incidence of bladder cancer is on a rising trend in recent decades. It is 3-4-fold more common in males than in females. The morbidity of bladder cancer increases with age and has a peak between 50-70 years, especially 65 years. Bladder cancer is responsible for over 130,000 annual deaths worldwide each year. Moreover, bladder cancer has the highest lifetime treatment cost in all cancers, which has brought serious economic burden to patients. Bladder cancer has become a public health problem across the world.
The pathological type of bladder cancer generally includes transitional cell carcinoma (TCC, also known as urothelial cancer), squamous cell carcinoma, and adenocarcinoma. TCC constitutes more than 90% of these bladder cancers [2, 3]. About 70-85% of TCCs are superficial, including Ta, T1, and carcinoma in situ, which is also termed non-muscle invasive bladder cancer (NMIBC) [4, 5]. The remaining 15-30% of TCCs is muscle invasive bladder cancer (T2-T4), which is less favorable for patients, and nearly 50% of patients will die within five years of diagnosis [5].
In clinical practice, transurethral resection of bladder tumor (TURBT) is the standard treatment for non-muscle invasive bladder cancers. However, the recurrence of tumors was approximately 50-80%, and 10-25% of recurrent tumors would progress to muscle-invasive bladder cancers [6-8]. Recurrence and tumor progression after TURBT are the main causes of death in bladder cancer patients. The mechanisms of carcinogenesis of bladder cancer have not been fully investigated. Therefore, better understanding of pathogenesis is essential for development of novel, effective therapies for bladder cancer.
MicroRNAs (miRNAs) are a class of small noncoding RNAs, widely present in eukaryotes, about 22 nucleotides in length, and usually regulate gene expression via translational repression and mRNA degradation [9, 10]. Recent studies have shown that miRNAs are important regulators that play a critical role in cancer, and half of the miRNAs are located in cancer-associated genomic regions as tumor suppressor or oncogenes depending on their targets [11]. Until now, many human miRNAs have been shown to be dysregulated in bladder cancer, such as miR-19a [12], miR-34a [13], miR-129 [14], and miR-133a [15], which contribute to the development and progression of bladder cancer.
It has been reported that the miR-290 family was highly expressed in undifferentiated mouse embryonic stem (ES) cells and was rapidly downregulated upon differentiation [16, 17]. Recent research showed that these ES cell-specific miRNAs, including miR-291a-3p, miR-294, and miR-295, could promote ES cell proliferation or at least partly promoted the transition of cells from G1 to S phase [18]. In the present study, we sought to evaluate the role of miR-294 in bladder cancer, as well as its mechanism, and hoped to provide theoretical basis and novel method for bladder cancer treatment.
MATERIALS AND METHODS
Cell culture. Human bladder cancer cell lines (T24, HT1376, J82, and SW780) and immortalized human bladder epithelium cell lines (HCV29 and SV-HUC-1) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) in a humidified atmosphere incubator with 5% CO2 at 37°C.
Generation of stably engineered cell lines. For miRNA transfection, cells were transfected with miR-294 mimetic, miR-294 inhibitor, or scramble control. Stably transfected cells were treated with 1.5 µg/ml puromycin for 10 days beginning 48 h after infection. The expression level of stably engineered cell lines was detected by real-time PCR. Cell transfections were performed using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s protocol. The miR-294 mimetic, miR-294 inhibitor, and scramble control were synthesized by GenePharma (China). After 48 h of transfection, cells were collected for further investigation.
RNA extraction, cDNA synthesis, and quantitative real-time PCR (qPCR) assays. Total cellular RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. RNA was quantified and cDNA with a stem-loop RT primer was synthesized using 2 µg of total RNA and M-MLV reverse transcriptase (Invitrogen). The qPCR was performed in CFX96 real-time PCR system (Bio-Rad, USA) using TaqMan probes (Applied Biosystems, USA) according to the manufacturer’s instructions. The PCR reaction consisted of an initial denaturation step (95°C for 30 s), then 40 cycles (95°C for 5 s and 60°C for 34 s). The data were normalized using the endogenous U6 snRNA as control and calculated according to the 2–ΔΔCT method.
Cisplatin treatment. The T24 cells were seeded in 96-well plates at 1·104 per well. Cells were transfected with miR-294 mimic or inhibitor and cultured for 36 h. Then 200 µl cisplatin was added into each well with concentrations of 5-50 µg/ml. The cells were cultured and selected for the cell viability assay.
Cell counting kit-8 assay. Cell viability was assessed using a Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, USA). Briefly, after transfection and stimulation, CCK-8 solution was added to the culture medium, and then cells were incubated for 1 h at 37°C in a humidified 5% CO2 atmosphere. Absorbance was measured at 450 nm using a microplate reader (Bio-Rad).
Clonogenic assay. T24 cells were trypsinized and diluted to a density of 1·103 cells/ml, then the cells were plated in 60-mm dishes. After 2-3 weeks of culture, cell clones were fixed with 4% paraformaldehyde solution and stained with 0.1% crystal violet. Stained cell clones on plates after different treatments were captured using a ChemiDoc XRS+ imaging system (Bio-Rad). The surviving fraction (SF) was calculated as a ratio of the number of colonies to the number of cells plated (plating efficiency) divided by the same ratio calculated for the nontreated group.
Wound healing assay. T24 cells were plated in 60-mm dishes and incubated until confluence. After 3 h of pretreatment with 50 µM mitomycin c, wounds were created by scratching the cell sheets with a sterile 200 µl pipette tip. Pictures of a specific position on the scratched areas were taken using an inverted microscope (Leica, Germany) every 24 h. The wound widths were measured, and the relative wound widths were calculated. Data are shown as mean ± SD from three independent experiments.
Invasion assay. T24 bladder cancer cells in serum-free medium were seeded on the upper compartment of an insert with 8-mm pores in 24-well tissue culture plates (Corning Costar, USA) with Matrigel (BD Bioscience, USA). Modified RPMI 1640 medium containing 10% FBS in the lower chamber was used as a chemoattractant. After 12 h of incubation at 37°C, non-transfected cells were carefully removed from the upper surface of the filter using a cotton swab. Cells adhering to the lower membrane were fixed with 20% methanol and stained with 0.1% crystal violet, and then counted under an IX71 inverted microscope (Olympus, Japan).
Apoptosis assay. Apoptosis analysis was performed to identify and quantify apoptotic cells using an Annexin V-FITC/PI apoptosis detection kit (Beijing Biosea Biotechnology, China). The cells (1·104 cells/well) were seeded in 6-well plates. After being treated, the cells were washed twice with cold PBS and resuspended in binding buffer containing 10 µl Annexin V-FITC and 5 µl of PI (propidium iodide). Adherent and floating cells were combined and treated according to the manufacturer’s instruction and measured with flow cytometer (Beckman Coulter, USA) to differentiate apoptotic cells (Annexin V-positive and PI-negative) from necrotic cells (Annexin V- and PI-positive).
Western blot. The protein of the treated cells used for Western blotting was extracted by RIPA lysis buffer (Beyotime Biotechnology, China) supplemented with protease inhibitors (Roche, China). Then the proteins were quantified using the BCA™ Protein Assay Kit (Pierce, USA). The Western blot system was established using a Bio-Rad Bis-Tris Gel system according to the manufacturer’s instructions. Equal amounts of protein were separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. Primary antibodies were prepared in 5% blocking buffer at a dilution of 1 : 1000 and incubated with the membranes at 4°C overnight, followed by washing and incubation with secondary antibody labeled with horseradish peroxidase for 1 h at room temperature. After rinsing, the membrane-carried blots and antibodies were transferred into the Bio-Rad ChemiDoc™ XRS system, and then 200 µl of Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA) was added to cover the membrane surface. Signals were captured, and the intensity of the bands was quantified using Image Lab™ Software (Bio-Rad, China).
Statistical analysis. All experiments were repeated three times. The data of multiple experiments are presented as mean ± SD. Statistical analyses were performed using GraphPad statistical software. The p values were calculated by one-way analysis of variance (ANOVA). A p value of <0.05 was considered to indicate a statistically significant result.
RESULTS
High expression of miR-294 in bladder cancer cells. As shown in Fig. 1, the expression of miR-294 in bladder cancer cells including J82, HT1376, T24, and SW780 cell lines, was significantly increased compared with that in HCV29 and SV-HUC-1 cell lines (p < 0.05). The expression of miR-294 was upregulated in bladder cancer cell.
Fig. 1. miR-294 was significantly upregulated in bladder cancer cell lines. Expression level of miR-294 in two immortalized human bladder epithelium cells (HCV29 and SV-HUC-1) and four bladder cancer cell lines (T24, HT1376, J82, and SW780) were detected by qRT-PCR. Error bars indicate mean ± SD and symbol (*) indicates significant difference compared with normal human bladder epithelium cells (p < 0.05).
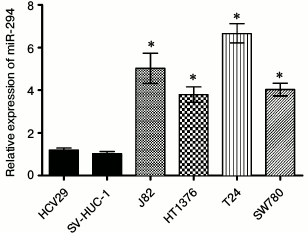
Correlation of miR-294 expression with T24 cell activity. As shown in Fig. 2a, the miR-294 expression of miR-294 mimic transfected T24 cells was significantly increased compared with that of the scramble control group (p < 0.05). However, the miR-294 expression level of miR-294 inhibitor transfected cells was significantly decreased (p < 0.05). Therefore, the miR-294 was effectively overexpressed by miR-294 mimic, and was inhibited by miR-294 inhibitor in T24 cells.
Fig. 2. Overexpression of miR-294 promoted T24 cell viability and colony formation and inhibited T24 cells apoptosis. a) Confirm the expression efficiency of miR-294 in T24 cells by qRT-PCR after being transfected with miR-294 mimetic, inhibitor, or scramble control, respectively. b) Cell viability of miR-trasfected T24 cells was detected using the Cell Counting Kit-8 assay. c) Survival fractions of miR-trasfected T24 cells was detected by clonogenic assay. d) Apoptosis of T24 cells was detected by flow cytometry. Error bars indicate means ± SD; * p < 0.05, ** p < 0.01, *** p < 0.001; ns, no significant.
In this study, we found that T24 cell viability and rate of T24 colony forming had the same trend as the expression of miR-294 (p < 0.05 or p < 0.01; Fig. 2, b and c). In addition, we found miR-294 inhibitor could increase the rate of T24 apoptosis compared with control, but there was no significant difference between them (p < 0.05) (Fig. 2d). This suggested that overexpression of miR-294 enhanced cell viability and survival, while inhibition of miR-294 expression promoted apoptosis in T24 cells.
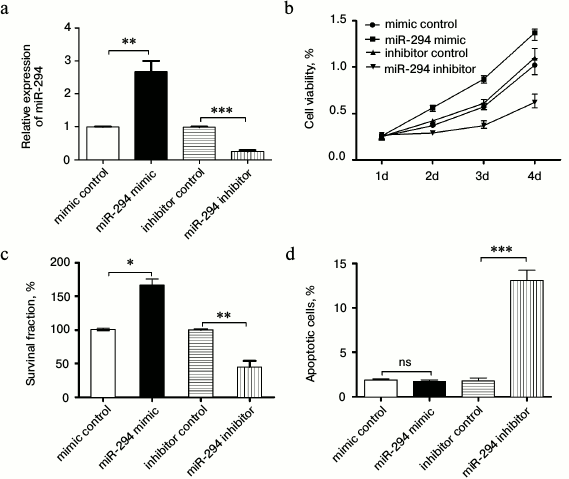
Correlation of miR-294 expression with cell migration and invasion. Wound healing assay was used to determine the migration ability of T24 cells after being treated (Fig. 3a). The results showed that the ability of migration was significantly increased in the miR-294 mimetic-treated group compared with that in the mimetic-control group (p < 0.05). However, miR-294 inhibitor could significantly inhibit the T24 cell migration compared with inhibitor control (p < 0.05). The rate of invasion showed the same trend as the cell migration of T24 cells (Fig. 3b). Therefore, the expression of miR-294 was positively related with cell migration and invasion.
Fig. 3. Overexpression of miR-294 promoted T24 cell migration and invasion. a) Wound healing assay and (b) invasion assay were carried out after T24 cells were transfected with miR-294 mimetic, inhibitor, or scramble control, respectively. Error bars indicate mean ± SD; * p < 0.05.
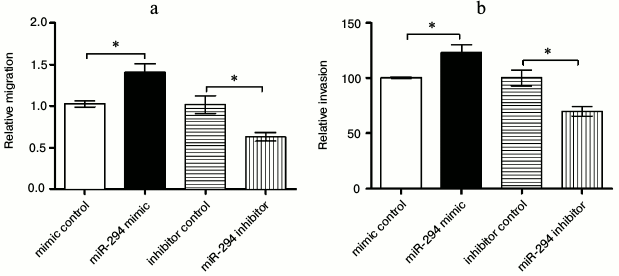
Correlation of miR-294 expression with inhibition effect of cisplatin on T24 cell viability. With increasing cisplatin concentration, the viability of T24 was decreased, while the drug resistance of T24 to cisplatin was increased in miR-294 mimetic-treated group compared with the control group (Fig. 4a). However, inhibition of miR-294 decreased the resistance of T24 to cisplatin compared with the inhibition-control group (Fig. 4b). We inferred that overexpression of miR-294 could efficiently increase the resistance of T24 cells.
Fig. 4. Correlation of miR-294 expression with inhibition effect of cisplatin on T24 cell viability. T24 cells was treated with different concentrations of cisplatin (0-50 µg/ml) and then transfected with miR-294 mimetic, inhibitor, or scramble as control. a) Drug resistance of miR-294 mimic-transfected T24 cells to cisplatin was assessed by cell viability assay. b) Inhibition of miR-294 expression effect on T24 cell viability was analyzed to test drug resistance of T24 cells to cisplatin. Error bars indicate mean ± SD; * p < 0.05.
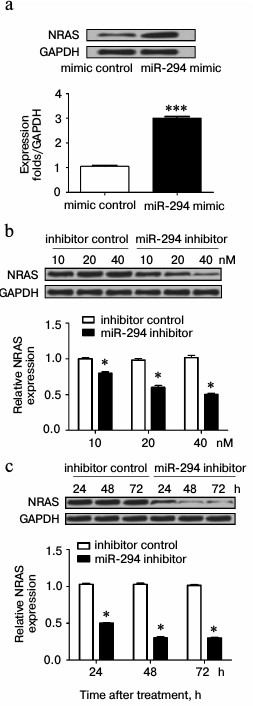
miR-294 regulated expression of NRAS in T24 cells. As shown in Fig. 5a, the expression of NRAS was increased in miR-294 mimic-transfected group compared with scramble-control group. However, the expression of NRAS was decreased in miR-294 inhibitor-treated group, especially at 40 nM (Fig. 5b). Besides, Fig. 5b illustrated that the expression of NRAS progressively decreased with time after suppression of miR-294.
Fig. 5. miR-294 regulates the expression of NRAS. The expression of NRAS in miR-294 transfected T24 cells was detected by Western blot. a) Protein immunoblots and quantification of NRAS expression in miR-294-transfected T24 cells. b) Protein immunoblots and quantification of NRAS expression in T24 cells after being transfected by miR-294 inhibitor or scramble control with concentration gradients 10, 20, and 40 nM. c) Protein immunoblots and quantification of NRAS expression in T24 cells for 24, 48, and 72 h after being transfected with 40 nM miR-294 inhibitor or scramble control. Error bars indicate mean ± SD; * p < 0.05, *** p < 0.001.
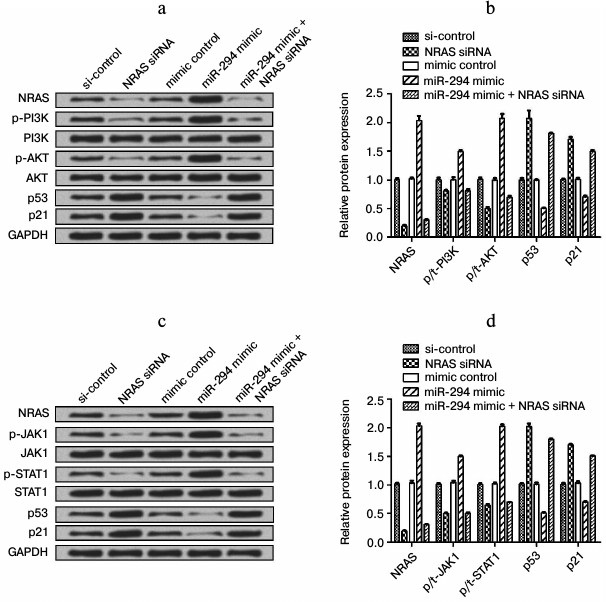
miR-294 regulated the expressions of factors in PI3K/AKT and JAK/STAT signal pathway. We found that the expression level of NRAS was decreased in NRAS siRNA-treated group compared to negative-control group, as well as p-PI3K and p-AKT expression (Fig. 6, a and b). The expression of p53 and p21 was increased in the NRAS siRNA-treated group compared to the negative-control group (Fig. 6, a and b). In addition, the expression of those factors in only miR-294 mimic-transfected group showed the reverse trend with NRAS siRNA-treated group, while the expression of those factors was decreased after cotransfection of miR-294 mimic and NRAS siRNA as same trend with only NRAS siRNA treated group. The expression level of p-JAK1, p-STAT1, and NRAS was decreased in NRAS siRNA-treated group compared with negative-control group (Fig. 6, c and d). In addition, the expression of those factors in miR-294 mimic-transfected group showed the reverse trend with the NRAS siRNA-treated group, while the expression of those factors was decreased as same as NRAS siRNA-treated group after cotransfection with NRAS siRNA and miR-294 mimic. These results indicated that miR-294 could activate the PI3K/AKT and JAK/STAT pathways via upregulation of NRAS.
Fig. 6. Expression level of related factors in PI3K/AKT and JAK/STAT signal pathways. Protein expression was determined by Western blot. a) Protein immunoblots of main factors in the PI3K/AKT signal pathway. b) Quantification of expression of the main factors in the PI3K/AKT signal pathway. c) Protein immunoblots of main factors in the JAK/STAT signal pathway. d) Quantification of expression of main factors in the JAK/STAT signal pathway.
DISCUSSION
Bladder cancer is the most common tumor of the urinary system with a great threat to the survival of patients in China [19]. There are many factors that could lead to bladder cancer, including associated mutations, oncogene expression or overexpression, the surrounding matrix, systemic factors, outside carcinogens, etc. [1]. Carcinogens stimulate normal bladder cells by DNA damage, in which up- or downregulated activation of oncogenes or anti-oncogenes play an important role [20]. Tumor suppressor is an important regulating factor in the process of differentiation and proliferation. Once the tumor suppressor is mutated or inactivated, cell cycle regulation equilibrium will be broken and tend to abnormal proliferation of cells leading to tumorigenesis [21-23]. Therefore, exogenous expression of wild-type tumor suppressor gene to repair the defect and restore the function of tumor suppressor genes is one method of bladder cancer treatment [24].
In recent years, the abnormal expression of miRNAs in bladder cancer has being gradually clarified, and this indicated that miRNAs have an important effect on oncogene or tumor suppressor genes in the development of bladder cancer. Many scholars believe that changes in expression of miRNAs play a key role in the development of human tumors, and someone even proposed that the abnormal expression of miRNAs is the most important reason for tumor occurrence and development. It was reported that the abnormal expression of miRNAs was related with many tumors, including lung cancer, breast cancer, urinary tract cancer, leukemia, and lymphoma [25]. In the present study, we found miR-294 was highly expressed in several bladder cancer cell lines. It has also been reported that miR-200b and miR-429 were upregulated in bladder cancer tissue [26]. However, some miRNAs were lowly expressed in bladder cancer cell lines, such as miR-143 [27], miR-145 [28], miR-582-3p/5p, etc. [29].
The miRNAs change the development of bladder cancer primarily through regulating the proliferation, migration, and apoptosis of cells. miR-409-3p has been reported to be downregulated in bladder cancer, and overexpression of miR-409-3p inhibited cell migration and invasion [30]. miR-23b was reported to act as a tumor-suppressive factor that inhibited cancer cell proliferation and colony formation [31]. Moreover, miR-135a was found to be upregulated in bladder cancer and promoted cell proliferation [32]. Our study revealed that miR-294 could promote the proliferation and migration of cells, and also decrease the apoptosis of T24 cells. These results indicated that miR-294 was positively correlated with the development of bladder cancer and acted as an oncogenic miRNA in T24 cells.
To further analyze the mechanism by which miR-294 functioned as an oncogenic miRNA in bladder cancer, we analyzed the relationship of miR-294 and NRAS in bladder cancer and found that the regulatory function of miR-294 in bladder cancer cells was dependent on regulating NRAS expression. NRAS is a member of the RAS family, which codes for a small GTP-binding protein [33]. NRAS mutation leads to excess activation of carcinoma cells and make cells abnormally proliferate and migrate. NRAS gene mutation was found in several human tumors, including adrenocortical carcinomas [34], cutaneous melanomas [33], colorectal cancer [35], multiple myeloma [36], etc. After binding with GTP, RAS transmitted signals through a variety of effectors in downstream pathways, such as the PI3K/AKT and JAK/STAT cascades [37]. In the present study, we found that miR-294 could promote PI3K/AKT and JAK/STAT phosphorylation by upregulating NRAS.
PI3K is recruited and activated by cell surface receptors of peptide growth factors and cytokines and then makes exogenous peptide growth factors directly activate intracellular survival-related pathways. It was reported that abnormal PI3K/AKT activity was linked to the formation and metastasis of tumors [38-41]. JAK (Janus kinase)/STAT (signal transducer and activator of transcription) is a critical signal transduction pathway of cytokines. It was initially found in cell proliferation, and then this pathway was found to play important roles in cell proliferation, immune function, and apoptosis. Once activated, the JAK/STAT signal transduction pathway can pass signals from cell membrane to cytoplasm and cell nucleus, then induce some biological effects involved in physiological and pathological reactions in humans. Abnormally activated STAT can promote the proliferation and differentiation of cells and suppress the apoptosis of cells, which leads to occurrence of some diseases [42-44]. Our study showed that abnormally activated PI3K/AKT and JAK/STAT signaling pathways could promote T24 cell proliferation, migration, and invasion.
In conclusion, we explored the functions of miR-294 as an oncogenic miRNA in bladder cancer by regulating NRAS expression and activating the PI3K/AKT and JAK/STAT signaling pathways, and thus improving the survival ability of T24 cells. These results might provide novel insights for approaches to bladder cancer diagnosis and therapy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81572835; 81500587) and by the Nature Science Foundation of Shandong Province (Grant No. ZR2015HM021).
REFERENCES
1.Jemal, A., Siegel, R., Ward, E., Hao, Y., Xu, J.,
and Thun, M. J. (2009) Cancer statistics, 2009 CA, Cancer J.
Clin., 59, 225-249.
2.Cheung, G., Sahai, A., Billia, M., Dasgupta, P.,
and Khan, M. S. (2013) Recent advances in the diagnosis and treatment
of bladder cancer, BMC Med., 11, 1-8.
3.Martyn-Hemphill, C., Mak, D., Khan, M. S.,
Challacombe, B. J., and Bishop, C. V. (2013) Recent advances in
diagnosis and treatment of transitional cell carcinoma of the bladder,
Int. J. Surg., 11, 749-752.
4.Moyer, V. A. (2011) Screening for bladder cancer:
U.S. Preventive Services Task Force (USPSTF) recommendation statement,
Ann. Int. Med., 155, 246-251.
5.Yafi, F. A., Aprikian, A. G., and Chin, J. L.
(2011) Contemporary outcomes of 2287 patients with bladder cancer who
were treated with radical cystectomy: a Canadian multicentre
experience, BJU Int., 108, 539-545.
6.Dalbagni, G., Vora, K., Kaag, M., Cronin, A.,
Bochner, B., Donat, S. M., and Herr, H. W. (2009) Clinical outcome in a
contemporary series of restaged patients with clinical T1 bladder
cancer, Eur. Urol., 56, 903-910.
7.Richards, K. A., Smith, N. D., and Steinberg, G. D.
(2014) The importance of transurethral resection of bladder tumor in
the management of nonmuscle invasive bladder cancer: a systematic
review of novel technologies, J. Urol., 191,
1655-1664.
8.Herr, H. W., and Donat, S. M. (2008) Quality
control in transurethral resection of bladder tumours, BJU Int.,
102, 1242-1246.
9.Filipowicz, W., Bhattacharyya, S. N., and
Sonenberg, N. (2008) Mechanisms of post-transcriptional regulation by
microRNAs: are the answers in sight? Nat. Rev. Genet., 9,
102-114.
10.Lagos-Quintana, M., Rauhut, R., Lendeckel, W.,
and Tuschl, T. (2001) Identification of novel genes coding for small
expressed RNAs, Science, 294, 853-858.
11.Lawrie, C. H. (2013) MicroRNAs as Oncogenes
and Tumor Suppressors. MicroRNAs in Medicine, Wiley,
Hoboken, NJ, pp. 223-243.
12.Feng, Y., Liu, J., Kang, Y., He, Y., Liang, B.,
Yang, P., and Yu, Z. (2014) miR-19a acts as an oncogenic microRNA and
is up-regulated in bladder cancer, J. Exp. Clin. Cancer Res.,
33, 1-10.
13.Gan, Y., Yao, W., Wei, X., Li, H., Hua, X., and
Lang, B. (2014) MicroRNA-34a functions as an anti-metastatic microRNA
and suppresses angiogenesis in bladder cancer by directly targeting
CD44, J. Exp. Clin. Cancer Res., 33, 1-13.
14.Dyrskjot, L., Ostenfeld, M. S., Bramsen, J. B.,
Silahtaroglu, A. N., Lamy, P., Ramanathan, R., Fristrup, N., Jensen, J.
L., Andersen, C. L., Zieger, K., Kauppinen, S., Ulhoi, B. P., Kjems,
J., Borre, M., and Orntoft, T. F. (2009) Genomic profiling of microRNAs
in bladder cancer: miR-129 is associated with poor outcome and promotes
cell death in vitro, Cancer Res., 69,
4851-4860.
15.Ichimi, T., Enokida, H., Okuno, Y., Kunimoto, R.,
Chiyomaru, T., Kawamoto, K., Kawahara, K., Toki, K., Kawakami, K.,
Nishiyama, K., Tsujimoto, G., Nakagawa, M., and Seki, N. (2009)
Identification of novel microRNA targets based on microRNA signatures
in bladder cancer, Int. J. Cancer, 125, 345-352.
16.Chen, C., Ridzon, D., Lee, C. T., Blake, J., Sun,
Y., and Strauss, W. M. (2007) Defining embryonic stem cell identity
using differentiation-related microRNAs and their potential targets,
Mammal. Genome (Official J. Int. Mammal. Genome Soc.),
18, 316-327.
17.Houbaviy, H. B., Dennis, L., Jaenisch, R., and
Sharp, P. A. (2005) Characterization of a highly variable eutherian
microRNA gene, RNA (A Publ. RNA Soc.), 11,
1245-1257.
18.Wang, Y., Baskerville, S., Shenoy, A., Babiarz,
J. E., Baehner, L., and Blelloch, R. (2008) Embryonic stem
cell-specific microRNAs regulate the G1–S transition and promote
rapid proliferation, Nat. Genet., 40, 1478-1483.
19.Parkin, D. M., Bray, F., Ferlay, J., and Pisani,
P. (2005) Global cancer statistics, 2002, Cancer J. Clin.,
55, 74-108.
20.Nobori, T. (1994) Deletions of the
cyclin-dependent kinase-4 inhibitor gene in multiple human cancers,
Nature, 368, 753-756.
21.Tricoli, J. V., and Jacobson, J. W. (2007)
MicroRNA: potential for cancer detection, diagnosis, and prognosis,
Cancer Res., 67, 4553-4555.
22.Hammond, S. M. (2007) MicroRNAs as tumor
suppressors, Nat. Genet., 39, 582-583.
23.Lee, B. R., Feinbaum, R., and Ambros, V. (2012)
The C. elegans heterochronic gene lin-4 encodes small RNAs with
antisense complementarity to lin-14, Cell, 75,
843-854.
24.Kamb, A. (1994) A cell regulator potentially
involved in genesis of many tumor types, Science, 264,
436-440.
25.Androulakakis, P. A., Davaris, P., Karayannis,
A., Michael, V., and Aghioutantis, C. (1992) Urothelial tumors of the
bladder, Child Nephrol. Urol., 12, 32-34.
26.Han, Y., Chen, J., Zhao, X., Liang, C., Wang, Y.,
Sun, L., Jiang, Z., Zhang, Z., Yang, R., and Chen, J. (2011) MicroRNA
expression signatures of bladder cancer revealed by deep sequencing,
PLoS One, 6, e18286.
27.Lin, T., Dong, W., Huang, J., Pan, Q., Fan, X.,
Zhang, C., and Huang, L. (2009) MicroRNA-143 as a tumor suppressor for
bladder cancer, J. Urol., 181, 1372-1380.
28.Noguchi, S., Yasui, Y., Iwasaki, J., Kumazaki,
M., Yamada, N., Naito, S., and Akao, Y. (2012) Replacement treatment
with microRNA-143 and -145 induces synergistic inhibition of the growth
of human bladder cancer cells by regulating PI3K/Akt and MAPK signaling
pathways, Cancer Lett., 328, 353-361.
29.Uchino, K., Takeshita, F., Takahashi, R. U.,
Kosaka, N., Fujiwara, K., Naruoka, H., Sonoke, S., Yano, J., Sasaki,
H., Nozawa, S., et al. (2013) Therapeutic effects of microRNA-582-5p
and -3p on the inhibition of bladder cancer progression, Mol.
Ther. (J. Am. Soc. Gene Ther.), 21, 610-619.
30.Xu, X., Chen, H., Lin, Y., Hu, Z., Mao, Y., Wu,
J., Xu, X., Zhu, Y., Li, S., and Zheng, X. (2013) MicroRNA-409-3p
inhibits migration and invasion of bladder cancer cells via targeting
c-Met, Mol. Cells, 36, 62-68.
31.Majid, S., Dar, A. A., Saini, S., Deng, G.,
Chang, I., Greene, K., Tanaka, Y., Dahiya, R., and Yamamura, S. (2013)
MicroRNA-23b functions as a tumor suppressor by regulating Zeb1 in
bladder cancer, PLoS One, 8, e67686.
32.Mao, X. P., Zhang, L. S., Huang, B., Zhou, S. Y.,
Liao, J., Chen, L. W., Qiu, S. P., and Chen, J. X. (2015) Mir-135a
enhances cellular proliferation through post-transcriptionally
regulating PHLPP2 and FOXO1 in human bladder cancer, J. Transl.
Med., 13, 1-13.
33.Jakob, J. A., Bassett, R. L., Jr., Ng, C. S.,
Curry, J. L., Joseph, R. W., Alvarado, G. C., Rohlfs, M. L., Richard,
J., Gershenwald, J. E., Kim, K. B., Lazar, A. J., Hwu, P., and Davies,
M. A. (2012) NRAS mutation status is an independent prognostic factor
in metastatic melanoma, Cancer, 118, 4014-4023.
34.Kotoula, V., Sozopoulos, E., Litsiou, H.,
Fanourakis, G., Koletsa, T., Voutsinas, G., Tseleni-Balafouta, S.,
Mitsiades, C. S., Wellmann, A., and Mitsiades, N. (2009) Mutational
analysis of the BRAF, RAS and EGFR genes in human adrenocortical
carcinomas, Endocrine Rel. Cancer, 16, 565-572.
35.Vaughn, C. P., Zobell, S. D., Furtado, L. V.,
Baker, C. L., and Samowitz, W. S. (2011) Frequency of KRAS, BRAF, and
NRAS mutations in colorectal cancer, Genes Chromosomes Cancer,
50, 307-312.
36.Mulligan, G., Lichter, D. I., Bacco, A. D.,
Blakemore, S. J., Berger, A., Koenig, E., Bernard, H., Trepicchio, W.,
Li, B., and Neuwirth, R. (2014) Mutation of NRAS but not KRAS
significantly reduces myeloma sensitivity to single-agent bortezomib
therapy, Blood, 123, 632-639.
37.Irahara, N., Baba, Y. K., Shima, K., Yan, L.,
Dias, S. D., Iafrate, A. J., Fuchs, C. S., Haigis, K. M., and Ogino, S.
(2010) NRAS mutations are rare in colorectal cancer, Diagn. Mol.
Pathol. (Am. J. Surg. Pathol.), Part B, 19, 157-163.
38.Fry, M. J. (2000) Phosphoinositide 3-kinase
signalling in breast cancer: how big a role might it play? Breast
Cancer Res., 3, 304-312.
39.Martinez, L., Anel, A., Sierra, J., Pineiro, A.,
Naval, J., and Alava, M. (2000) Tyrosine phosphorylation of the p85
subunit of phosphatidylinositol 3-kinase correlates with high
proliferation rates in sublines derived from the Jurkat leukemia,
Int. J. Biochem. Cell Biol., 32, 435-445.
40.Krasilnikov, M., Adler, V., Fuchs, S. Y., Zheng,
D., Haimovitz-Friedman, A., Herlyn, M., and Ronai, Z. E. (1999)
Contribution of phosphatidylinositol 3-kinase to radiation resistance
in human melanoma cells, Mol. Carcinogen., 24, 64-69.
41.Lin, X., Bohle, A., Dohrmann, P., Leuschner, I.,
Schulz, A., Kremer, B., and Fandrich, F. (2001) Overexpression of
phosphatidylinositol 3-kinase in human lung cancer,
Langenbeck’s Arch. Surg., 386, 293-301.
42.Kaminska, B., and Swiatek-Machado, K. (2014)
Targeting signaling pathways with small molecules to treat autoimmune
disorders, Exp. Rev. Clin. Immunol., 4, 93-112.
43.Jackson, M., Howle, S. E. M., Weller, R. J.,
Sabin, E., Hunter, J. A. A., and Mckenzie, R. C. (1999) Psoriatic
keratinocytes show reduced IRF-1 and STAT-1α activation in
response to γ-IFN, FASEB, 13, 495-502.
44.Yamazaki, F., Aragane, Y., Maeda, A., Matsushita,
K., Ueno, K., Yudate, T., Kawada, A., and Tezuka, T. (2002)
Overactivation of IL-4-induced activator protein-1 in atopic
dermatitis, J. Dermatol. Sci., 28, 227-233.