Femtosecond and Picosecond Dynamics of Recombinant Bacteriorhodopsin Primary Reactions Compared to the Native Protein in Trimeric and Monomeric Forms
O. A. Smitienko1*, O. V. Nekrasova2,3, A. V. Kudriavtsev1,3, M. A. Yakovleva1, I. V. Shelaev4, F. E. Gostev4, D. A. Dolgikh2,3,5, I. B. Kolchugina3, V. A. Nadtochenko4, M. P. Kirpichnikov2,3, T. B. Feldman1,3,5, and M. A. Ostrovsky1,3
1Emanuel Institute of Biochemical Physics, Russian Academy of Sciences, 119334 Moscow, Russia; E-mail: djolia@gmail.com2Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia
3Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia
4Semenov Institute of Chemical Physics, Russian Academy of Sciences, 119991 Moscow, Russia
5Pirogov Russian National Research Medical University, 1117997 Moscow, Russia
* To whom correspondence should be addressed.
Received October 22, 2016; Revision received December 20, 2016
Photochemical reaction dynamics of the primary events in recombinant bacteriorhodopsin (bRrec) was studied by femtosecond laser absorption spectroscopy with 25-fs time resolution. bRrec was produced in an Escherichia coli expression system. Since bRrec was prepared in a DMPC–CHAPS micelle system in the monomeric form, its comparison with trimeric and monomeric forms of the native bacteriorhodopsin (bRtrim and bRmon, respectively) was carried out. We found that bRrec intermediate I (excited state of bR) was formed in the range of 100 fs, as in the case of bRtrim and bRmon. Further processes, namely the decay of the excited state I and the formation of intermediates J and K of bRrec, occurred more slowly compared to bRtrim, but similarly to bRmon. The lifetime of intermediate I, judging from the signal of ΔAESA(470-480 nm), was 0.68 ps (78%) and 4.4 ps (22%) for bRrec, 0.52 ps (73%) and 1.7 ps (27%) for bRmon, and 0.45 ps (90%) and 1.75 ps (10%) for bRtrim. The formation time of intermediate K, judging from the signal of ΔAGSA(625-635 nm), was 13.5 ps for bRrec, 9.8 ps for bRmon, and 4.3 ps for bRtrim. In addition, there was a decrease in the photoreaction efficiency of bRrec and bRmon as seen by a decrease in absorbance in the differential spectrum of the intermediate K by ~14%. Since photochemical properties of bRrec are similar to those of the monomeric form of the native protein, bRrec and its mutants can be considered as a basis for further studies of the mechanism of bacteriorhodopsin functioning.
KEY WORDS: bacteriorhodopsin, recombinant protein, primary reactions, femtosecond absorption laser spectroscopyDOI: 10.1134/S0006297917040113
Abbreviations: bR, bacteriorhodopsin; bRmon, native bacteriorhodopsin in the monomeric form; bRrec, recombinant bacteriorhodopsin; bRtrim, native bacteriorhodopsin in the trimeric form; CD, circular dichroism; DA, dark-adapted; ESA, excited state absorption for the S1 state of bR; FC, Franck–Condon; GSA, ground state absorption for the S0 state of bR or its photoreaction products; LA, light-adapted; SB, Schiff base; SE, stimulated emission from the S1 state of bR.
Bacteriorhodopsin (bR) is a membrane protein from Halobacterium
salinarum that carries out a photoenergetic function –
directed proton transport through the cellular membrane. It is a unique
object for studying molecular mechanisms of light energy conversion in
biological systems [1]. In membranes of
halobacteria, bR forms patches called purple membranes in which bR
trimers are arranged into a regular hexagonal structure [2]. The stability of this two-dimensional crystal is
supported by lipids tightly bound to the protein, such as phosphatidyl
glycerophosphate (50%) and sulfo-glycolipids (30%). Acting as a
light-dependent proton pump, bR generates electrochemical transmembrane
potential used for ATP synthesis [2-5]. bR is a chromoprotein with molecular weight of
26.5 kDa; it consists of apoprotein bacterioopsin and a
chromophore group, all-trans retinal, bound covalently to K-216
through a protonated Schiff base (SB). bR consists of 248 amino acids
tightly arranged in seven transmembrane α helices, some amino
acids of which form a chromophore center for retinal insertion.
Absorption of a light quantum causes chromophore isomerization into the 13-cis form and is accompanied by several reversible transformations of the bR molecule closed into a photocycle. The photocycle results in proton transport from the inner side of the cell membrane to the exterior medium. During the photocycle, intermediates I, J, K, L, MI, MII, N, and O are formed, which differ from the initial bR state in their physicochemical properties and may be differentiated spectrally [6-9]. Changes in the spectral properties of intermediates in the visible light region are determined by conformations of the chromophore and of its immediate protein environment. High levels of bR in native purple membranes, where it is the only protein component, allowed determination of its structure relatively quickly – first, with electron cryomicroscopy, and then with X-ray crystallographic analysis at high spatial resolution [10, 11]. Based on three-dimensional structures of bR and its numerous mutants, molecular mechanisms of light transformation into electrochemical potential of a cell are being studied [12-14]. Despite the large amount of accumulated data, bR studies on the molecular level remain relevant.
An important task in this regard is large-scale accumulation of bR and its mutant forms with altered physicochemical and spectral properties. In this regard, of interest is development of new approaches to obtaining recombinant bacteriorhodopsin, analysis of its properties, and of possibilities for its use and integration into various technical devices. In recent years, systems for expressing mutant bR forms in halobacterial cells have been developed. These systems are based on using deletion variants of halobacteria lacking the bR gene, which may be transformed by specific plasmid vectors containing a certain version of a modified gene [15]. Despite the possibility for obtaining genetically altered bR in the native three-dimensional form, these approaches involve complicated genetic engineering procedures as well as difficulties in cultivating halobacteria. The traditional method of bacteriorhodopsin isolation, developed by Oesterhelt and Stoeckenius in 1974 [16], is a labor- and time-consuming procedure involving ultracentrifugation in a sucrose density gradient with the yield of 20 to 50 mg of purple membranes from 1 liter of halobacterial culture. An alternative to this approach is obtaining bRrec in a bacterial expression system in E. coli. We developed one of variant of such a system characterized by a high level of bRrec production in cells, simplicity of cultivation, of isolation procedures for bRrec and renaturation with retinal in lipid–detergent micelles. The final yield of renatured bRrec was ~120 mg from 1 liter of culture [17]. A distinct feature of this expression system is monomeric organization of bR, unlike the native protein that is packed into trimers. The bRrec obtained by us was close to native bRmon in its spectral properties and was characterized by the appearance of a bathochromic shift during light adaptation and its capability for proton transport.
The major spectral differences of the monomeric form of native bR are a shift of the absorption maximum to shorter wavelength from 568 to 552-553 nm for the light-adapted form (LA) and from 560 to 546 nm for the dark-adapted form (DA) [18-20], a decrease in extinction coefficient from 63,000 to 44,000 M–1·cm–1 [21, 22], and appearance of a monophasic curve on its circular dichroism (CD) spectrum in the visible range (in contrast to a biphasic curve for trimers) [23]. Additionally, bRmon is less thermally stable than bRtrim [24]. The differences in absorption spectra may be analyzed in two aspects: first, during bRtrim solubilization, protein–protein and protein–lipid interactions that stabilize bR folding in native membranes are disrupted and, as a result, changes occur in the molecular environment of the retinal binding site. Similar shifts in spectral maxima are observed in the cases of certain mutant bR forms, in which charge redistribution around a retinal molecule occurs [25]. Another reason for a spectral shift may be a change in the ratio of retinal isomers, all-trans/13-cis, from 90/10-95/5 to 62/38-84/16 for the LA form and from 45/55-50/50 to 40/60 for the DA form of trimer and monomer, respectively [21, 26-30]. On trimer → monomer transition, no qualitative differences in the bR photocycle are observed [31]. However, in bRmon the lifetimes of L and N intermediates are significantly shorter and, therefore, M formation occurs faster by an order of magnitude [8]. These kinetic features reflect the presence of a more flexible monomer conformation allowing the formation of a larger number of various bR conformational states.
The bRrec obtained earlier by expression in E. coli in lipid–detergent micelles [32, 33] (as well as bRmon) is characterized by a short-wavelength shift in the maximum of the absorption spectrum to 559 nm for the LA form and to 551 nm for the DA form, a decreased extinction coefficient of 52,000 M–1·cm–1, and by its capability for proton transport. The ratio of bRrec retinal isomers (all-trans/13-cis) – 96/4 (LA form) and 39/61 (DA form) – corresponds approximately to bRtrim.
In the last decade, ultrafast dynamics of the retinal excited state was studied by the method of pump–probe femtosecond laser absorption spectroscopy in both the native trimeric form and the monomeric form of bR [18, 34-39]. Currently, bRtrim primary reactions occurring in the femtosecond and picosecond time range are most often represented by the following scheme: bR568(hv) → FC(100-200 fs) → I460(500 fs) → J625(3-5 ps) → K590, where FC is the Franck–Condon state of the bR568 molecule. Under the action of a light quantum, the ground state of bRtrim transits into the excited FC state, which relaxes into intermediate I over the course of 100-200 fs with a prominent absorption signal (ΔAESA) in the range of 450-490 nm and a signal of stimulated emission (ΔASE) in the range of 800-1200 nm [34, 39]. Intermediate I transforms into the ground state over the course of 500 fs with formation of intermediate J (quantum yield of 0.64 [40]), accompanied by the transition of retinal from all-trans to the 13-cis form [37, 38]. After 3-5 ps, intermediate J transforms into the next intermediate K with a more relaxed 13-cis retinal [41, 42].
During bR monomerization, primary photocycle events change only slightly. The spectral maxima of the excited state I, as well as intermediates J and K, are practically identical for bRmon and bRtrim [18]. The main difference is in kinetic characteristics of the primary photocycle product formation for a trimer and a monomer, which are expressed as following. The transition of the excited state I into intermediate J is 0.45-0.55 ps for bRtrim (95-100%) [18, 43, 44]; in the case of a monomer, two components appear with lifetimes (relative amplitudes) of 0.63 ps (87%) and 3.51 ps (13%) [18]. Another characteristic of the primary photocycle events is stimulated emission of bR in the near-infrared range, which can be used to estimate the lifetime of the excited state. In the case of a trimer, the major fluorescent state relaxation time is 0.44 ps, corresponding to the major time of relaxation for intermediate I. In the case of a monomer, two relaxation components appear as well – 0.55 and 3.5 ps. The slow and fast components were attributed by the authors of [18] to differences in kinetics of relaxation of all-trans and 13-cis retinal isomers inside the protein. Thus, the ratio of amplitudes of fast and slow components, 83/13, corresponds approximately to the ratio of retinal isomers in the LA form of bR.
Therefore, isomerization of all-trans retinal in a trimer or a monomer occurs over the course of approximately the same time interval, within 0.5 ps. So, trimer destruction does not cause changes in photoisomerization dynamics of all-trans retinal and, therefore, the trimeric structure of bR is not a catalyst of the primary photocycle events.
To confirm the correct organization of the chromophore center and the whole bRrec protein, it was necessary to analyze the dynamics of primary reactions of bRrec photocycle in detail compared to the native protein. For the comparison, not only the trimeric, but also the monomeric form of native bR was used, since bRrec was obtained in a monomeric form.
The aim of this study was to analyze the spectral characteristics and formation dynamics of bRrec intermediates I, J, and K compared to the native protein in trimeric and monomeric forms by pump–probe femtosecond laser absorption spectroscopy with excitation by 560-nm pulses and time resolution of 25 fs.
MATERIALS AND METHODS
Preparation of bRrec samples. bRrec was obtained in a E. coli expression system according to the study of Nekrasova et al. [17]. After renaturation of bRrec with retinal in lipid–detergent micelles (1% DMPC, 1% CHAPS, 0.2% SDS), the protein was concentrated on Ni-Sepharose High Performance column (V = 2 ml) (GE Healthcare, Sigma-Aldrich) and then desalted on a PD-10 column (GE Healthcare) in buffer A (1% DMPC, 1% CHAPS, 50 mM Na-phosphate buffer, 150 mM KCl, pH 6.0). bRrec concentration was 2.1 mg/ml, with purity coefficient of A280/A559 = 1.49 and absorbance of A559 = 4.2 OD units.
Preparation of native bRtrim samples. Halobacteria (H. salinarum) were grown from the ET1001 strain in the Central Scientific-Research Institute of Technology “Tekhnomash”. Purple membranes were kindly provided by “Tekhnomash”. The sample was a suspension of purple membranes in buffer B (25 mM Na-phosphate buffer, pH 7.0). bRtrim concentration was 1.6 mg/ml, with purity coefficient of A280/A568 = 1.78 and absorbance of A568 = 3.7 OD units. Before the experiment, the sample was treated with ultrasound with power of 90 W for 2-3 min.
Preparation of native bRmon samples. The bR was monomerized by solubilizing native purple membranes in the nonionic detergent Triton X-100. With this type of preparation, the native lipid environment of the bR molecule remains partially preserved [45]. A bRmon sample was prepared under dimmed red light at room temperature. Stock solution of Triton X-100 prepared with Na-phosphate buffer (pH 6.05) was added to a suspension of purple membranes containing bRtrim to the final concentration of Triton X-100 – 2% and Na-phosphate – 25 mM. The sample was incubated at room temperature for 20 h with continuous stirring. Then the sample was centrifuged on a Beckman Coulter Optima TM XE centrifuge (80,000g, 18°C, 60 min). Extraction efficiency was ~85%, bRmon concentration was 2.5 mg/ml, and absorbance was A553 = 5.9 OD units. The purity coefficient could not be determined since the detergent strongly absorbs in the UV range.
Stationary absorption spectra for samples were registered with a UV 1700 spectrophotometer (Shimadzu, Japan). Light adaptation was carried out by illumination with a halogen lamp (KGM24-250, 24 V, 250 W) with filters YGL-19 and GL-8 passing the spectral range ~500-600 nm. Dark adaptation was conducted for three days at 4°C or for one day at room temperature.
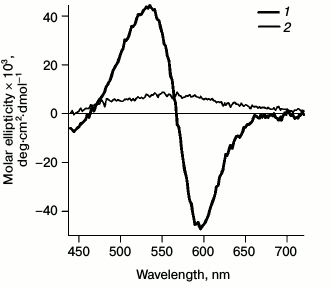
Circular dichroism. Since formation of the monomeric bR form from the trimeric one is accompanied by a rapid change in the CD spectrum [23], to monitor bR monomerization the following spectra were registered (Fig. 1). Registration was carried out with a SKD-2M device (Institute of Spectroscopy, Russian Academy of Sciences) with a thermostatted cell at room temperature. To increase the signal-to-noise ratio, every spectrum was recorded four times in increments of 2 nm. A distinct shift was observed from the biphasic CD spectrum of bRtrim with maxima of the positive band at 535 nm and of the negative band at 595 nm to the monophasic spectrum of bRmon with a non-distinct maximum at 560 nm. This indicated destruction of the bRtrim hexagonal lattice and trimeric structure and formation of detergent micelles containing single bRmon molecules.
Fig. 1. CD spectra registered for bRtrim (1) and bRmon (2) in the spectral range of 440-720 nm.
HPLC analysis of retinal isomers. Retinal isomers were extracted following the method described in [46]. A 2-ml sample was treated with 1 ml of formaldehyde, mixed, and incubated for 2 min. Then 1.5 ml of methylene chloride was added, and the mixture was intensively shaken and incubated for 10 min. Then 1.5 ml of hexane was added, and the mixture was shaken and centrifuged with a MLW K 26 D centrifuge (680g, 10 min). The upper layer containing methylene chloride, hexane, and free retinal isomers was collected and evaporated using the vacuum of a water-jet pump. All procedures were carried out under dim red light.
Chromatographic analysis was carried out on a HPLC system Smartline 1000 (Knauer, Germany) using a K-2500 variable-wavelength spectrophotometric detector. Measurements were taken at the wavelength of 365 nm. The retinal isomers were separated on a Silica column (7 µm, 250 × 4.6 mm; IBM Instruments, USA). Hexane–ethyl acetate solvent mixture (7% v/v) with the addition of absolute methanol (100 µl for 1 liter of the mixture) was used as the eluant at flow rate 1 ml/min.
Femtosecond spectroscopy. Photoinduced absorption spectra were measured with a femtosecond setup using the pump–probe method [44-47]. The sample was excited by Gaussian pulses with recurrence frequency of 60 Hz, duration of 25 fs, wavelength of 560 nm, and energy of 120 nJ. The diameter of the excitation beam in the sample was 300 µm. For the probe pulse, a supercontinuum was generated in a quartz cuvette containing water. The probing pulse energy did not exceed 10 nJ. The diameter of the probing beam was 100 µm. Polarization of the probing pulse in relation to the excitation pulse was rotated by 54.7° (“magic angle”). After the sample, the supercontinuum pulse was directed to an Acton SP-300 polychromator (Princeton Instruments, USA) and detected by a CCB-camera Roper Scientific SPEC-10 (Roper Scientific, Germany). Differential absorption spectra ΔA(λ) were recorded in the spectral range of 400-900 nm.
In spectral ranges of 535-600 and 790-835 nm the ΔA signal could not be registered due to a high level of noise caused by specific features of preparation of the excitation 560 nm pulse. The probing supercontinuum pulse was applied with delay time of 0-3 ps in increments of 3.3 fs, and 3-10 ps in increments of 10 fs with accumulation of signals from 50 spectra. Control differential spectra were also registered with the delay time of 100 ps with accumulation of signals from 5000 spectra to track sample degradation during the experiment. All experiments were carried out at 21°C in a 0.5-mm quartz flow cuvette with window thickness of 0.1 mm. The flow rate allowed the sample to be completely refreshed after each laser pulse. For bR molecules to be in the LA form during excitation by a fs-pulse, the sample circulating in the flow system was constantly illuminated by a continuous laser with wavelength λ = 532 nm, power P = 0.02 mW/cm2, and diameter d = 0.8 cm in the range other than that of the excitation pulse. Photoadaptation of a sample was verified spectrally.
Experimental data were processed with Span software written in the language environment of Matlab.
RESULTS
Spectral properties of bRtrim, bRmon, and bRrec. Stationary absorption spectra were registered for samples of bacteriorhodopsin in trimeric and monomeric forms and of the recombinant protein (Fig. 2).
Fig. 2. Stationary absorption spectra for LA samples of bRtrim (1), bRmon (2), and bRrec (3). The bRmon spectrum was registered only in the 305-700 nm range since Triton X-100 detergent has intense absorption at wavelength <300 nm. The spectra were normalized by the α-band intensity.
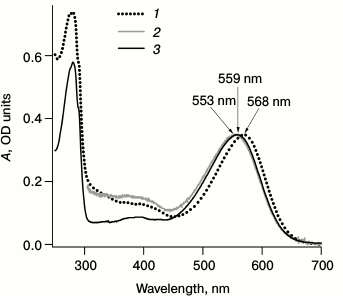
Figure 2 shows the maxima of sample absorption spectra – 568 nm for bRtrim, 553 nm for bRmon, and 559 nm for bRrec, which corresponds to data from the literature [18, 20, 27, 32, 33, 48]. Differences in absorption spectra may be explained by the fact that during bRtrim solubilization, protein–protein and protein–lipid interactions are disrupted, which stabilize bR folding in native membranes. This results in changes in the molecular environment of the retinal binding site and in the conformation of retinal itself, increasing the energy of the S0→S1 phototransition. It should be noted that the bRrec absorption spectrum is more like bRmon in the red range (Fig. 2), which may indicate a similar structure of the chromophore center and chromophore conformation.
HPLC analysis of retinal isomers. To estimate the ratio of all-trans and 13-cis retinal in the analyzed samples by HPLC, chromatograms were registered for retinals extracted from LA and DA samples of bRtrim, bRmon, and bRrec (Fig. 3). The percentage ratio of retinal isomers is provided in Table 1.
Fig. 3. Normalized chromatograms of retinals extracted from LA and DA samples of bRtrim, bRmon, and bRrec. Absorbance was detected at the 365 nm wavelength.
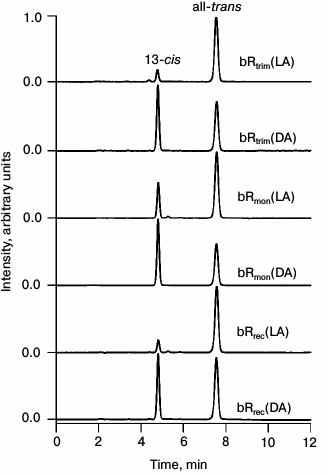
Table 1. All-trans and 13-cis
retinal content in LA and DA samples of bRtrim,
bRmon, and bRrec and the maxima for stationary
absorption spectra of the samples
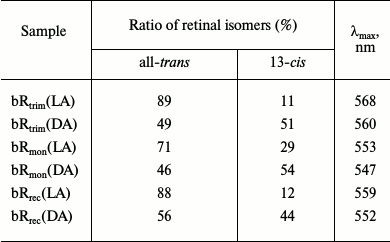
As seen in Table 1, the ratio of all-trans/13-cis retinals (in percent) in the LA sample of bRtrim was 89/11, which is slightly less than in other studies [29, 30]. A similar ratio was found for LA samples of bRrec – 88/12, while for bRmon it was 71/29, which is in accordance with published studies [21, 28]. In DA samples of bR the ratio of all-trans/13-cis retinals decreased to 49/51 for bRtrim, 46/54 for bRmon, and 56/44 for bRrec.
Comparison of photoinduced absorption spectra of bRtrim, bRmon, and bRrec. Figure 4 shows normalized spectra of photoinduced absorption for three LA samples of bR with delay times of 0.1, 0.6, 1.3, and 9 ps.
Fig. 4. Normalized spectra of photoinduced absorption for LA samples of bRtrim (1), bRmon (2), and bRrec (3) for delay times of 0.1 (a), 0.6 (b), 1.3 (c), and 9 ps (d) in the spectral ranges of 400-890 nm (a-c) and 460-730 nm (d). The curves were normalized to the maximum intensity of the intermediate I absorption band in the differential spectra.
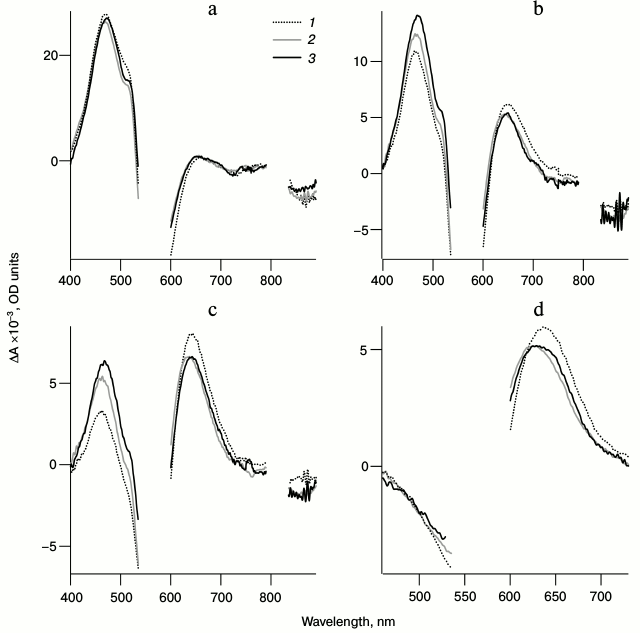
Figure 4a shows that within 100 fs after excitation the formation of a pronounced excited state absorption band is observed (ΔAESA) in the 450-490 nm range for all three samples, which characterizes intermediate I formation. During the next 1.3 ps this band decreases significantly, and in the 650-nm range the absorption band of the next intermediate J (ΔAGSA) is formed. In the 530-640 nm range at the time of 100 fs, a bR bleaching band is observed (ΔABL) (Fig. 4a), which decreases significantly in the picosecond time range due to the formation of intermediate J, which contributes positively to absorption in this range, and to the transition of a part of bR molecules from the excited to the initial state (Fig. 4, b-d). Within early times in the long-wavelength ranges of 700-790 nm and 835-890 nm, a negative band of stimulated emission of state I (ΔASE) is observed (Fig. 4, a and b), which disappears in the picosecond time range. By 9 ps after excitation, the differential spectra consist only of the absorption band of intermediate K ΔAGSA(t = 9 ps) and the bleaching band of bR ΔABL(t = 9 ps) (Fig. 4d). The J → K transition is accompanied by a shift of ΔAGSA signal to shorter wavelength by approximately 20 nm and a slight decrease in absorption intensity.
Comparison of differential spectra of the studied samples shows that formation time, absorption maximum, and the form of intermediate I absorption band are practically identical for all three samples (λmax = 470 nm), except the bRrec; the maximum of this band is shifted to longer wavelength by 7 nm (Fig. 4a and Table 2). The maximum intensity for intermediate I absorption band in a differential spectrum ΔA(λmax(I)) was used to normalize the spectra and kinetic curves represented in Figs. 4 and 5. Within times of 0.6 and 1.3 ps (Fig. 4, b and c), the maximum decrease in absorption is observed for intermediate I and, therefore, the maximum increase in absorption for intermediate J of bRtrim compared to bRmon and bRrec. This indicates increased time of the I → J transition in these samples. As seen in Fig. 4 (c and d), absorption bands of intermediates J and K for the bRmon and bRrec samples are shifted to shorter wavelength compared to bRtrim. The absorption spectra maxima of intermediate K, registered with delay time of 9 ps, are 625 nm (bRmon), 630 nm (bRrec), and 635 nm (bRtrim) (Fig. 4d and Table 2). A shift to shorter wavelength of intermediates J and K absorption spectra for bRmon and bRrec samples approximately correlates with the shift in stationary absorption spectra by 15 and 9 nm, respectively (Fig. 2). A similar correlation of absorption spectra maxima for the initial state of bR and the K form was observed earlier for bRmon [49] and for some bR mutants [42, 50]. It can also be noted that signal ΔAGSA intensity for intermediates J and K for bRmon and bRrec samples is decreased by 13-14% compared to that of bRtrim (Fig. 4b and Table 2), which may indicate a less effective formation of the J form and, subsequently, K form in the photoreaction process.
Table 2. Maxima of absorption bands of
intermediates I and K in differential
spectra of bRtrim, bRmon, and bRrec
samples registered with delay times of 0.1 and 9 ps, respectively.
Relative intensities of the K intermediate absorption
band in differential spectra
ΔAGSA(λmax(K))
of bRtrim, bRmon, and bRrec samples,
given in % from
ΔAGSA(λmax(K))
of bRtrim with the delay time of 9 ps
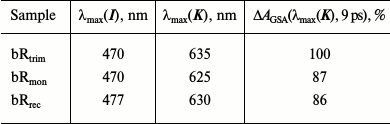
Fig. 5. Normalized kinetic curves of photoinduced absorption for LA samples of bRtrim (1), bRmon (2), and bRrec (3), registered at the following probing wavelengths: a) 470 nm (1, 2) and 477 nm (3); b) 625 nm (2), 630 nm (3) and 635 nm (1); c) 850 nm. Normalization was to the maximum intensity of intermediate I absorption band in the differential spectrum.
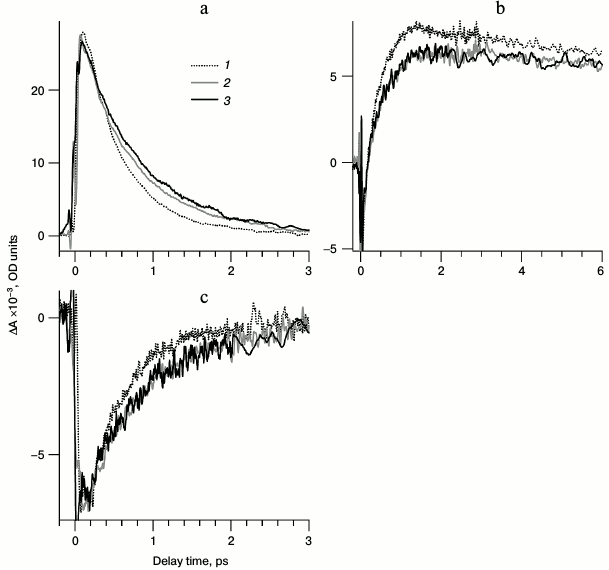
Comparison of kinetic curves of photoinduced absorption for bRtrim, bRmon, and bRrec. Based on the differential spectra (Fig. 4), the time-resolved signals of photoinduced absorption were compared for different LA samples of bR at characteristic wavelengths (Fig. 5, curves 1-3). For comparison of formation and decay dynamics of intermediate I, kinetic curves were analyzed at wavelengths corresponding to absorption maxima for this intermediate in the differential spectrum (ΔAESA) (Fig. 5a and Table 2), as well as in a stimulated emission band of bR (ΔASE) (Fig. 5c). For comparison of intermediates J and K formation dynamics for the different bR samples, kinetic curves were analyzed at wavelengths corresponding to absorption maxima of K in the differential spectrum with a delay time of 9 ps (Fig. 5b and Table 2). Characteristic times of the analyzed processes were estimated by plotting model biexponential curves (Table 3).
Table 3. Parameters (characteristic times
and their contributions) of kinetic curves of photoinduced absorption
for bRtrim, bRmon, and bRrec obtained
by plotting model biexponential curves in time ranges of 0.1-9 ps
(470-477 nm) and 0.15-9 ps (625-850 nm) at characteristic
probing wavelengths
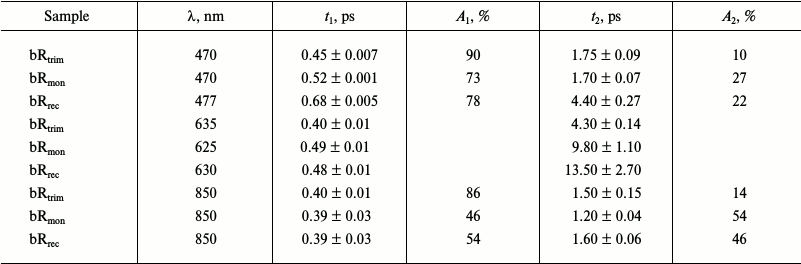
As seen from the dynamics of the ΔAESA signal (Fig. 5a), the appearance of intermediate I absorption band occurs within 100 fs after excitation for bRtrim, 70 fs for bRmon, and 90 fs for bRrec. The decrease in ΔAESA signal occurs faster for bRtrim than for bRmon and bRrec, as was already observed in spectral dynamics of photoinduced absorption for bR samples (Fig. 4, a-c). This process in bRtrim is characterized by a fast time component of 0.45 ps (90%) and a slow component of 1.75 ps (10%). In other samples, the fast component increases slightly to 0.52 ps (bRmon) and 0.68 ps (bRrec), and its contribution decreases to ~75%. The slow component for bRmon (1.7 ps) is practically unchanged compared to bRtrim (1.75 ps), while for bRrec it is significantly increased (4.4 ps) (Table 3).
In the stimulated emission range of bR at 850 nm, the negative signal ΔASE for all samples reaches approximately the same intensity by 100-150 fs, after which it begins to decrease in the picosecond time range (Fig. 5c). As well as the ΔAESA signal, the ΔASE signal decreases more slowly in bRmon and bRrec compared to bRtrim. The fast component of this process is practically the same for all samples being 0.39-0.4 ps, but the contribution of this component decreases sharply for bRmon (46%) and bRrec (54%) compared to bRtrim (86%). The slow component is 1.5 ps for bRtrim, 1.2 ps for bRmon, and 1.6 ps for bRrec (Table 3).
In the absorption range of intermediates J and K, the kinetic curves demonstrate the appearance of a negative signal at early times <50 fs, most likely related to both ΔABL and ΔASE (Fig. 5b), which shifts to a positive ΔAGSA signal with two characteristic time constants. The first, fast component of signal increase could not be determined due to a significant contribution from an artifact signal at times <90 fs, but it may be approximated as 50-100 fs for all three samples. The following slower rise of the ΔAGSA signal in this spectral range reflects, at times up to 1-2 ps, the dynamics of intermediate J formation. For bRtrim, the characteristic time of this process was 0.4 ps, and the ΔAGSA signal reached its maximum by 1.3 ps (Fig. 5b and Table 3). For the bRmon and bRrec samples, the characteristic time of this process was slightly more, 0.48-0.49 ps, and the ΔAGSA signal reached its maximum by 1.8 ps. Further, in the picosecond time range, a slight decrease in absorption is observed, reflecting the process of K form formation with the absorption maximum shifted to shorter wavelength. The characteristic time of this process for bRtrim was 4.3 ps, while for bRmon and bRrec samples, this time increased to 9.8 and 13.5 ps, respectively. Unfortunately, the times of 9.8 and 13.5 ps were determined with low precision, since the kinetic curves were obtained only for times up to 9 ps. The kinetic curves presented in Fig. 5b, as well as differential spectra (Fig. 4d), demonstrated, with times up to 9 ps, reduced ΔAGSA signal by 13-14% for bRmon and bRrec compared to bRtrim. As stated earlier, this is most likely caused by a slightly decreased quantum yield of the photoreaction for these samples.
DISCUSSION
In this study, we obtained differential spectra and kinetic curves of photoinduced absorption for bRtrim, bRmon, and bRrec samples over a wide spectral probe range, 400-900 nm, within times up to 9 ps. The data obtained for bRtrim and bRmon correlate with data from the literature [18, 35, 36, 39]. bRrec demonstrates primary reaction dynamics with characteristics close to those of bRmon, but slightly differing from bRtrim. They are: (i) an increase in the excited state lifetime evident in the ΔAESA band: for the fast component (from 0.45 ps for bRtrim to 0.68 ps) and the slow component (from 1.75 ps for bRtrim to 4.4 ps) with a decrease in contribution from the fast component (from 90% for bRtrim to 78%) (Fig. 5a and Table 3); (ii) an increase in formation time for intermediates J (from 0.4 ps for bRtrim to 0.48 ps) and K (from 4.3 ps for bRtrim to 13.5 ps) (Fig. 5b and Table 3); (iii) a minor decrease in efficiency of trans-cis retinal isomerization reaction in bRrec, judging by a decrease in intensity of intermediate K absorption band in the differential spectrum by 14% (Figs. 4d and 5b and Table 2). The observed changes in primary reaction dynamics for bRrec and bRmon compared to the native form of bRtrim contained in purple membranes and forming a dimeric hexagonal structure are probably connected to the monomeric protein form.
The protein environment is known to strongly control the reaction of retinal isomerization in bR, ensuring its speed, efficiency, and strict selectivity compared to the same reaction in solution [51] and in the gas phase [52], and minor changes in amino acid location and composition in the chromophore center may alter the potential energy surfaces, the magnitude of S0→S1 transition, and, eventually, its whole dynamics. bR monomerization apparently leads to certain changes in the bRrec and bRmon chromophore center, as seen already at the level of a shift in a maximum of their stationary absorption spectra by 9 and 15 nm, respectively. This hypsochromic shift in the stationary absorption spectra is attributed in the literature to a change in the ratio of all-trans/13-cis retinals observed during bR solubilization [21, 28]. bR containing 13-cis retinal, for example, 50% in DA samples of bRtrim, has a shorter wavelength absorption band (560 nm) and a lower extinction coefficient (55,400 M–1·cm–1) [27]. However, according to data from [33] and to our data from HPLC analysis, bRrec contains the same amount of all-trans retinal isomer (88%) as bRtrim (89%), while bRmon contains only 71% of it (Fig. 3 and Table 1). However, the shift of the maximum during bRmon solubilization is quite significant, from 568 to 553 nm, and 13-cis retinal level increases to 29% of total retinal content (Table 1), while during dark adaptation of bRtrim the maximum shifts from 568 to 560 nm, and 13-cis retinal level increases to approximately 60%. Therefore, the shift of the maximum of the stationary absorption spectrum and the change in photoreaction dynamics for bRrec and bRmon compared to bRtrim are apparently connected not to changes in isomeric composition of the retinal, but to some other processes the nature of which is to be clarified.
As stated above, the inner environment structure of retinal in opsin affects spectral localizations of its energy levels. The greatest effect on absorption maximum is exhibited by interaction of the protonated Schiff base of retinal with charged amino acid residues, specifically with a complex counter-ion consisting of D-212, D-85, and three water molecules [53]. This counter-ion in the monomeric form of bR is probably located closer to the protonated SB of retinal, which leads to an increase in the energy of the S0→S1 transition [54]. It can be proposed that inner environment structure of retinal in monomer and trimer differs. This causes a perturbation in electron levels of the retinal molecule during the transition from trimer to monomer. A perturbation in electron levels is expressed by changes in reaction dynamics for a transition from electron-excited state to primary product. In particular, the rate of this reaction slows slightly, and the quantum yield decreases slightly (the reaction becomes less effective). Unfortunately, X-ray structures of bR obtained to date in large number with satisfactory spatial resolution [11, 55] do not allow comparison of the chromophore centers of bR trimer and monomer even though many crystals pass a monomerization stage during preparation [55]. This is caused by tight molecule packing in the crystal regardless of the symmetry type, allowing the use of known X-ray structures only for bR in purple membranes.
Even though bRmon and bRrec molecules are located in different environments, in detergent micelles of Triton X-100 and lipid–detergent micelles of DMPC-CHAPS respectively, this most likely affects the dynamics of their primary reactions only slightly. It can be assumed that the nature of photoreactions in a monomeric form of bR is affected above all by the absence of specific natural protein–protein and protein–lipid interactions, and to a lesser degree by the composition of the artificial environment of detergents and lipid–detergent complexes.
Without having information on the structure of the chromophore center of bR in the monomeric form, what can be said about the reasons for an increase in the lifetime of bRmon and bRrec excited state and about the general slowing of the isomerization reaction compared to bRtrim? The observed rate of the photoisomerization reaction may also be affected by isomeric composition of retinals inside the bR chromophore center. The authors of a study [18] explained the two-component nature of bRmon kinetic curves, describing the decay of the excited state, and an increase in the time of the fast component, compared to bRtrim, by the presence of higher 13-cis retinal content in bRmon (~15%). However, it was shown [39] that the reaction of 13-cis retinal isomerization in the DA form of bR is barrierless in the excited state and significantly faster (~200 fs) than that of the all-trans retinal in the LA form (500 fs), and it strongly resembles in its characteristics the photoreaction of 11-cis retinal in visual pigment rhodopsin. This indicates that an increase in 13-cis retinal content in bRmon, which was also observed in the current study, should have decreased decay time of intermediate I and not increased, as shown in the experiment. At the same time, as discussed earlier, the level of 13-cis retinal increases only in bRmon, and kinetic parameters for the formation of primary photocycle intermediates of bRrec and bRmon are practically the same. This fact is most likely explained by a strong effect of protein environment on retinal, which changes during solubilization. Further studies using mutant forms of bR with substitutions of amino acids in the immediate vicinity of retinal could bring us closer to understanding this effect.
Therefore, we conclude that recombinant bR obtained in the E. coli expression system is similar in dynamics of primary reactions to native bacteriorhodopsin in the monomeric form. All differences of bR in the monomeric form (recombinant and native) are caused by effects of the protein environment, which directs and regulates the reaction of all-trans→13-cis isomerization of retinal in bR.
Acknowledgements
The authors would like to thank the Central Scientific-Research Institute of Technology “Tekhnomash” for the kindly provided purple membranes, and V. A. Kuzmin and N. A. Durandin for assistance with registration of CD spectra of bRtrim and bRmon samples.
This study was supported by the Russian Foundation for Basic Research (project No. 15-04-05816), Program of Fundamental Research Support of Presidium of the Russian Academy of Sciences No. 1 “Nanostructures: physics, chemistry, biology, basis of technologies”, as well as National Task of Pirogov Russian National Research Medical University of the Ministry of Health of Russian Federation for 2015-2017.
REFERENCES
1.Voet, D., and Voet, J. G. (2004) in
Biochemistry (Hoboken, N. J., ed.) 3rd Edn., Wiley & Sons,
New York.
2.Henderson, R., and Shotton, D. (1980)
Crystallization of purple membrane in three dimensions, J. Mol.
Biol., 139, 99-109.
3.Bogomolni, R. A., Baker, R. A., Lozier, R. H., and
Stoeckenius, W. (1976) Light-driven proton translocations in
Halobacterium halobium, Biochim. Biophys. Acta,
440, 68-88.
4.Hartmann, R., Sickinger, H. D., and Oesterhelt, D.
(1977) Quantitative aspects of energy conversion in halobacteria,
FEBS Lett., 82, 1-6.
5.Drachev, L. A., Kaulen, A. D., and Skulachev, V. P.
(1984) Correlation of photochemical cycle, H+ release and
uptake, and electric events in bacteriorhodopsin, FEBS Lett.,
178, 331-335.
6.Lozier, R. H., Bogomolni, R. A., and Stoeckenius,
W. (1975) Bacteriorhodopsin: a light-driven proton pump in
Halobacterium halobium, Biophys. J., 15,
955-962.
7.Danshina, S. V., Drachev, L. A., and Kaulen, A. D.
(1992) The inward H+ pathway in bacteriorhodopsin: the role
of M412 and P(N)560 intermediates, Photochem. Photobiol.,
55, 735-740.
8.Varo, G., and Lanyi, J. K. (1991) Thermodynamics
and energy coupling in the bacteriorhodopsin photocycle,
Biochemistry, 30, 5016-5022.
9.Edman, K., Nollert, P., Royant, A., Belrhali, H.,
Pebay-Peyroula, E., Hajdu, J., Neutze, R., and Landau, E. M. (1999)
High-resolution X-ray structure of an early intermediate in the
bacteriorhodopsin photocycle, Nature, 401, 822-826.
10.Henderson, R., Baldwin, J. M., Ceska, T. A.,
Zemlin, F., Beckmann, F. E., and Downing, K. H. (1990) Model for the
structure of bacteriorhodopsin based on high-resolution electron
cryo-microscopy, J. Mol. Biol., 213, 899-929.
11.Luecke, H., Schobert, B., Richter, H. T.,
Cartailler, J. P., and Lanyi, J. K. (1999) Structure of
bacteriorhodopsin at 1.55 Å resolution, J. Mol. Biol.,
291, 899-911.
12.Lanyi, J. K. (1999) Progress toward an explicit
mechanistic model for the light-driven pump, bacteriorhodopsin, FEBS
Lett., 464, 103-107.
13.Kandori, A., Kanzaki, H., Miyatake, K.,
Hashimoto, S., Itoh, S., Tanaka, N., Miyashita, T., and Tsukada, K.
(2001) A method for detecting myocardial abnormality by using a total
current-vector calculated from ST-segment deviation of a
magnetocardiogram signal, Med. Biol. Eng. Comp., 39,
21-28.
14.Wang, J., and El-Sayed, M. A. (2001)
Time-resolved Fourier transform infrared spectroscopy of the
polarizable proton continua and the proton pump mechanism of
bacteriorhodopsin, Biophys. J., 80, 961-971.
15.Sanz, C., Lazarova, T., Sepulcre, F.,
Gonzalez-Moreno, R., Bourdelande, J. L., Querol, E., and Padros, E.
(1999) Opening the Schiff base moiety of bacteriorhodopsin by mutation
of the four extracellular Glu side chains, FEBS Lett.,
456, 191-195.
16.Oesterhelt, D., and Stoeckenius, W. (1974)
Isolation of the cell membrane of Halobacterium halobium and its
fractionation into red and purple membrane, Methods Enzymol.,
31, 667-678.
17.Nekrasova, O. V., Wulfson, A. N., Tikhonov, R.
V., Yakimov, S. A., Simonova, T. N., Tagvey, A. I., Dolgikh, D. A.,
Ostrovsky, M. A., and Kirpichnikov, M. P. (2010) A new hybrid protein
for production of recombinant bacteriorhodopsin in Escherichia
coli, J. Biotech., 147, 145-150.
18.Wang, J., Link, S., Heyes, C. D., and El-Sayed,
M. A. (2002) Comparison of the dynamics of the primary events of
bacteriorhodopsin in its trimeric and monomeric states, Biophys.
J., 83, 1557-1566.
19.Dencher, N. A., and Heyn, M. P. (1982)
Preparation and properties of monomeric bacteriorhodopsin, Meth.
Enzymol., 88, 5-10.
20.Kovacs, I., Hollos-Nagy, K., and Varo, G. (1995)
Dark adaptation and spectral changes in Triton-X-100-treated
bacteriorhodopsin, J. Photochem. Photobiol. B, 27,
21-25.
21.Gonzalez-Manas, J. M., Montoya, G.,
Rodriguez-Fernandez, C., Gurtubay, J. I. G., and Goni, F. M. (1990) The
interaction of Triton X-100 with purple membrane: effect of light-dark
adaptation, Biochim. Biophys. Acta, 1019, 167-169.
22.Gergely, C., Zimanyi, L., and Varo, G. (1997)
Bacteriorhodopsin intermediate spectra determined over a wide pH range,
J. Phys. Chem. B, 101, 9390-9395.
23.Pescitelli, G., and Woody, R. W. (2012) The
exciton origin of the visible circular dichroism spectrum of
bacteriorhodopsin, J. Phys. Chem. B, 116, 6751-6763.
24.Brouillette, C. G., McMichens, R. B., Stern, L.
J., and Khorana, H. G. (1989) Structure and thermal stability of
monomeric bacteriorhodopsin in mixed phospholipid/detergent micelles,
Proteins, 5, 38-46.
25.Krebs, M. P., and Khorana, H. G. (1993) Mechanism
of light-dependent proton translocation by bacteriorhodopsin, J.
Bacteriol., 175, 1555-1560.
26.Tsuda, M., and Ebrey, T. G. (1980) Effect of high
pressure on the absorption spectrum and isomeric composition of
bacteriorhodopsin, Biophys. J., 30, 149-157.
27.Casadio, R., Gutowitz, H., Mowery, P., Taylor,
M., and Stoeckenius, W. (1980) Light-dark adaptation of
bacteriorhodopsin in Triton-treated purple membrane, Biochim.
Biophys. Acta, 590, 13-23.
28.Dencher, N. A., Kohl, K. D., and Heyn, M. P.
(1983) Photochemical cycle and light-dark adaptation of monomeric and
aggregated bacteriorhodopsin in various lipid environments,
Biochemistry, 22, 1323-1334.
29.Scherrer, P., Mathew, M. K., Sperling, W., and
Stoeckenius, W. (1989) Retinal isomer ratio in dark-adapted purple
membrane and bacteriorhodopsin monomers, Biochemistry,
28, 829-834.
30.Song, L., Yang, D., El-Sayed, M. A., and Lanyi,
J. K. (1995) Retinal isomer composition in some bacteriorhodopsin
mutants under light and dark adaptation conditions, J. Phys.
Chem., 99, 10052-10055.
31.Milder, S. J., Thorgeirsson, T. E., Miercke, L.
J. W., Stroud, R. M., and Kliger, D. S. (1991) Effects of detergent
environments on the photocycle of purified monomeric bacteriorhodopsin,
Biochemistry, 30, 1751-1761.
32.Braiman, M. S., Stern, L. J., Chao, B. H., and
Khorana, H. G. (1987) Structure-function studies on bacteriorhodopsin.
IV. Purification and renaturation of bacterio-opsin polypeptide
expressed in Escherichia coli, J. Biol. Chem.,
262, 9271-9276.
33.Greenhalgh, D. A., Farrens, D. L., Subramaniam,
S., and Khorana, H. G. (1993) Hydrophobic amino acids in the
retinal-binding pocket of bacteriorhodopsin, J. Biol. Chem.,
268, 20305-20311.
34.Sharkov, A. V., Pakulev, A. V., Chekalin, S. V.,
and Matveetz, Y. A. (1985) Primary events in bacteriorhodopsin probed
by subpicosecond spectroscopy, Biochim. Biophys. Acta,
88, 94-102.
35.Dobler, J., Zinth, W., Kaiser, W., and
Oesterhelt, D. (1988) Excited-state reaction dynamics of
bacteriorhodopsin studied by femtosecond spectroscopy, Chem. Phys.
Lett., 144, 215-220.
36.Gai, F., Hasson, K. C., McDonald, J. C., and
Anfinrud, P. A. (1998) Chemical dynamics in proteins: the
photoisomerization of retinal in bacteriorhodopsin, Science,
279, 1886-1891.
37.Kobayashi, T., Saito, T., and Ohtani, H. (2001)
Real-time spectroscopy of transition states in bacteriorhodopsin during
retinal isomerization, Nature, 414, 531-534.
38.Yabushita, A., and Kobayashi, T. (2009) Primary
conformation change in bacteriorhodopsin on photoexcitation,
Biophys. J., 96, 1447-1461.
39.Wand, A., Friedman, N., Sheves, M., and Ruhman,
S. (2012) Ultrafast photochemistry of light-adapted and dark-adapted
bacteriorhodopsin: effects of the initial retinal configuration, J.
Phys. Chem. B, 116, 10444-10452.
40.Govindjee, R., Balashov, S. P., and Ebrey, T. G.
(1990) Quantum efficiency of the photochemical cycle of
bacteriorhodopsin, Biophys. J., 58, 597-608.
41.Ye, T., Friedman, N., Gat, Y., Atkinson, G. H.,
Sheves, J. M., Ottolenghi, M., and Ruhman, S. (1999) On the nature of
the primary light-induced events in bacteriorhodopsin: ultrafast
spectroscopy of native and C13=C14 locked pigments, J. Phys. Chem.
B, 103, 5122-5130.
42.Briand, J., Leonard, J., and Haacke, S. (2010)
Ultrafast photo-induced reaction dynamics in bacteriorhodopsin and its
Trp mutants, J. Opt., 12, 1-14.
43.Mathies, R. A., Brito-Cruz, C. H., Pollard, W.
T., and Shank, C. V. (1988) Direct observation of the femtosecond
excited-state cis-trans isomerization in
bacteriorhodopsin, Science, 240, 777-779.
44.Song, L., El-Sayed, M. A., and Lanyi, J. K.
(1993) Protein catalysis of the retinal subpicosecond
photoisomerization in the primary process of bacteriorhodopsin
photosynthesis, Science, 261, 891-894.
45.Reyenolds, J. A., and Stoeckenius, W. (1977)
Molecular weight of bacteriorhodopsin solubilized in Triton X-100,
Proc. Natl. Acad. Sci. USA, 74, 2803-2804.
46.Pepe, I. M., and Schwemer, J. (1987) An improved
HPLC method for the separation of retinaldehyde isomers from visual
pigments, Photochem. Photobiol., 45, 679-687.
47.Shelaev, I. V., Gostev, F. E., Mamedov, M. D.,
Sarkisov, O. M., Nadtochenko, V. A., Shuvalov, V. A., and Semenov, A.
Yu. (2010) Femtosecond primary charge separation in
Synechocystis sp. PCC 6803 photosystem I, Biochim. Biophys.
Acta, 8, 1410-1420.
48.Dencher, N. A., and Heyn, M. P. (1978) Formation
and properties of bacteriorhodopsin monomers in the non-ionic
detergents octyl-β-D-glucoside and Triton X-100, FEBS
Lett., 96, 322-326.
49.Varo, G., and Lanyi, J. K. (1991) Kinetic and
spectroscopic evidence for an irreversible step between deprotonation
and reprotonation of the Schiff base in the bacteriorhodopsin
photocycle, Biochemistry, 30, 5008-5015.
50.Ahl, P. L., Stern, L. J., Düring, D., Mogi,
T., Khorana, H. G., and Rothschild, K. J. (1988) Effects of amino acid
substitutions in the F helix of bacteriorhodopsin. Low temperature
ultraviolet/visible difference spectroscopy, J. Biol. Chem.,
263, 13594-13601.
51.Hamm, P., Zurek, M., Roschinger, T., Patzelt, H.,
Oesterhelt, D., and Zinth, W. (1996) Femtosecond spectroscopy of the
photoisomerization of the protonated Schiff base of all-trans
retinal, Chem. Phys. Lett., 263, 613-621.
52.Coughlan, N. J. A., Catani, K. J., Adamson, B.
D., Wille, U., and Bieske, E. J. (2014) Photoisomerization action
spectrum of retinal protonated Schiff base in the gas phase, J.
Chem. Phys., 140, 164-307.
53.Ernst, O. P., Lodowski, D. T., Elstner, M.,
Hegemann, P., Brown, L. S., and Kandori, H. (2014) Microbial and animal
rhodopsins: structures, functions, and molecular mechanisms, Chem.
Rev., 114, 126-163.
54.Hayashi, T., Matsuura, A., Sato, H., and Sakurai,
M. (2012) Full-quantum chemical calculation of the absorption maximum
of bacteriorhodopsin: a comprehensive analysis of the amino acid
residues contributing to the opsin shift, Biophysics, 8,
115-125.
55.Faham, S., and Bowie, J. U. (2002) Bicelle
crystallization: a new method for crystallizing membrane proteins
yields a monomeric bacteriorhodopsin structure, J. Mol. Biol.,
316, 1-6.