An Alternative Pathway of Light-Induced Transmembrane Electron Transfer in Photosynthetic Reaction Centers of Rhodobacter sphaeroides
R. A. Khatypov*, A. M. Khristin, T. Yu. Fufina, and V. A. Shuvalov
Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: rgreen1@rambler.ru* To whom correspondence should be addressed.
Received October 25, 2016; Revision received March 1, 2017
In the absorption spectrum of Rhodobacter sphaeroides reaction centers, a minor absorption band was found with a maximum at 1053 nm. The amplitude of this band is ~10,000 times less and its half-width is comparable to that of the long-wavelength absorption band of the primary electron donor P870. When the primary electron donor is excited by femtosecond light pulses at 870 nm, the absorption band at 1053 nm is increased manifold during the earliest stages of charge separation. The growth of this absorption band in difference absorption spectra precedes the appearance of stimulated emission at 935 nm and the appearance of the absorption band of anion-radical BA– at 1020 nm, reported earlier by several researchers. When reaction centers are illuminated with 1064 nm light, the absorption spectrum undergoes changes indicating reduction of the primary electron acceptor QA, with the primary electron donor P870 remaining neutral. These photoinduced absorption changes reflect the formation of the long-lived radical state PBAHAQA–.
KEY WORDS: femtosecond spectroscopy, bacterial reaction centers, electron transferDOI: 10.1134/S0006297917060050
Abbreviations: BA and BB, accessory bacteriochlorophylls; BChl, bacteriochlorophyll; BPheo, bacteriopheophytin; ET, electron transfer; HA and HB, bacteriopheophytins serving as electron acceptors; P, BChl special pair (PA and PB) that is a primary electron donor; RC, reaction center.
Reaction centers (RC) of purple photosynthetic bacteria are
transmembrane pigment–protein complexes that carry out primary
conversion of light energy into electrochemical energy of separated
charges during photosynthesis. The spatial structure of RCs was
determined with high resolution [1, 2]. Rhodobacter sphaeroides RCs consists of
three protein subunits (L, M, and H) and 10 cofactors of electron
transfer: four bacteriochlorophyll (BChl) molecules, two of which form
a dimer of the electron donor special pair P, and two others are
accessory BChl (BA and BB); two
bacteriopheophytin (BPheo) molecules (HA and HB);
two ubiquinone molecules (QA and QB); an ion of
non-heme iron, and a carotenoid molecule. A key element of reaction
centers is the primary electron donor P, which, in its excited state
P*, can transfer an electron to the primary acceptor in a few
picoseconds, inducing all the following photosynthetic reactions. In
many laboratories in has been shown that within 3 ps under
illumination, a charge-separated state
P+HA– is formed in reaction
centers [3-7]. Electron
transfer from the excited donor P* to the HA molecule occurs
in two stages with an intermediate electron acceptor, the BA
molecule [8-13]. The kinetics
of rise and decay of the absorption band of the anion-radical
BA– at 1020 nm at room temperature is
consistent with a kinetic scheme in which an electron is transferred
from the excited donor P* to a BA molecule with a time
constant of 3.5 ps, and then from BA– to
HA with a time constant of 0.9 ps [4].
With a time delay of 200 ps, an electron crosses the membrane and is
localized at the primary quinone acceptor QA [14-16].
Much less is known about the processes taking place in reaction centers during the earliest, femtosecond delay times. This is because with time delays from 0 to ~200 fs, changes in light absorption are largely masked by a coherent action of ultrashort excitation and probing light pulses. The interaction of pulses causes perturbations that are expressed in difference absorption spectra as waves that distort or obscure minor changes in light absorption.
It was shown earlier that during excitation of primary electron donor P with ultrashort light pulses (~20 fs), irreversible electron transfer from the excited primary electron donor to the nearest acceptor, the BA molecule, is accompanied by a reversible, oscillating electron transfer [12, 13, 17, 18]. This oscillating process of electron transfer is caused by nuclear wave packet motion along the potential surfaces of the excited primary donor P* and the radical pair P+BA–. Due to this oscillating process, an electron localizes on a BA molecule already within 100 fs after the act of light absorption.
On the other hand, within the same early time delays of ~100-200 fs, absorption changes were observed in Rba. sphaeroides reaction centers that reflect charge separation within the dimer of the special pair and the formation of a short-lived state with change transfer, P* (PAδ+PBδ–) [19, 20].
The same conclusion was reached by other authors [21] who found a spectral component that may reflect charge separation within the excited primary electron donor in the infrared range (1000-1600 cm–1) of the absorption spectrum of Rba. sphaeroides reaction centers within 200 fs after light absorption.
Recently, Australian researchers discovered a minor band with a maximum at 705 nm within the long-wavelength absorption spectrum region of photosystem 2 complexes [22]. Excitation of complexes in this absorption band caused photochemical charge separation.
Zhu et al. [23] detected reduction of active bacteriopheophytin HA at unexpectedly short delay times of ≤500 fs, which does not correspond to the generally accepted simple scheme of two-stage electron transfer with rate constants of 3 and 1 ps.
Thus, the processes taking place in reaction centers of Rba. sphaeroides during the earliest femtosecond delay times remain unclear.
MATERIALS AND METHODS
Wild-type reaction centers were isolated from the corresponding strain of purple bacteria Rba. sphaeroides as described earlier [24], diluted in 20 mM Tris-HCl buffer (pH 8.0) with 0.1% Triton X-100, and concentrated to optical density 0.5 at 760 nm (with 1-mm optical pathlength). Absorption spectra for the reaction centers were measured at room temperature with a UV1600PC spectrophotometer (Shimadzu, Japan). The long-wavelength range of the absorption spectrum, 950-1100 nm, was measured on samples with optical density of 5 at 870 nm (with 10-mm optical pathlength) and accumulated to improve signal-to-noise ratio. The spectra were averaged over ten measurements and normalized by the optical pathlength.
To carry out femtosecond measurements, 5 mM sodium dithionite was added to the reaction centers and they were illuminated with low intensity white light for 5 min to reduce the primary electron acceptor QA. Femtosecond difference absorption spectra were obtained using a pump–probe spectrometer. A MaiTai SP titanium-sapphire laser (Spectra-Physics, USA) was used to obtain pulses of ~25 fs duration with wavelength 800 nm and repetition rate 84 MHz. Generator pulses were routed into a Spitfire Ace optical regenerative amplifier (Spectra-Physics), and ~35 fs pulses were obtained at the output with energy ~4 mJ and repetition rate 50 Hz. The light beam coming out of the amplifier was split into two beams. The first beam was attenuated to the energy of ∼0.3 µJ/pulse and focused in a water-filled cuvette with 5-mm thickness to generate a continuum, which was used for probing pulses. The second beam with the energy of ~17 µJ/pulse was used to generate a continuum on a sapphire with ~2-mm thickness. From the resulting continuum, light with wavelength >850 nm was cut-off by an RG850 light filter (Newport, USA) and used for sample excitation. Excitation and probing pulses were focused in the plane of a rotating quartz cuvette with 1-mm thickness containing the sample and overlapped at the same point within the sample volume. Relative polarization of excitation pulse was parallel to measuring pulse. A time delay between pumping and probing pulses was varied with a PI-M531.DD computer-controlled optical delay (Physik Instrumente, Germany). After the sample cuvette, the probing pulse was directed to the entrance slit of a Spectra Pro 2300i spectrograph (Acton Research Corporation, USA). The spectra were registered with a Pixis 400BR CCD camera (Princeton Instruments, USA). To obtain a difference absorption spectrum, the excitation pulse was blocked by a 3501 chopper (NewFocus, USA) during each time delay. Approximately 10% of primary electron donors in reaction centers were excited by the pulse. Difference absorption spectra for each time delay were obtained by averaging 1000-5000 measurements.
Stationary photoinduced difference absorption spectra for P+QA–/PQA were recorded on a UV1800 spectrophotometer (Shimadzu, Japan) during illumination of the sample with excitation light in the direction perpendicular to that of the measuring beam. The sample was illuminated during spectrum registration, the duration of which was ~80 s. A series of pulses from a Nd:YAG LS-2130 laser was used as the excitation light source (pulse duration 10 ns; repetition rate 20 Hz; wavelength 532 or 1064 nm).
RESULTS AND DISCUSSION
Figure 1 presents the room temperature absorption spectrum of reaction centers of the purple bacteria Rba. sphaeroides. The absorption band with maximum at 870 nm reflects a low-energy transition within the dimer of the special pair P; the absorption maxima at 805 and 760 nm correspond to absorption of accessory bacteriochlorophyll (BA and BB) and bacteriopheophytin (HA and HB), respectively. In the long-wavelength spectral range, positive absorption is observed; it is caused by light scattering of reaction center (RC) pigment–protein complexes. Against the background of RC complex light scattering, a minor absorption band can be observed with maximum at 1053 nm (Fig. 1, inset). This absorption band remains in the absorption spectra of reaction centers that were adapted to the dark during a long time and corresponds specifically to the reaction center complex, since its amplitude changes proportionally to concentration of complexes, just as the amplitudes of other absorption bands of reaction centers do (Fig. 1, dashed line). The amplitude of the absorption band at 1053 nm is 9000 times less than that of the primary electron donor absorption band at 870 nm. At the same time, half-widths of these bands are practically the same, which may indicate an association of the band at 1053 nm to the low-energy Qy transition of P. Therefore, our data suggest the presence of an electron transition not described earlier in Rba. sphaeroides RC that is lower by 0.2 eV compared to the Qy transition of P.
Fig. 1. Absorption spectrum for reaction centers (RC) of wild-type Rhodobacter sphaeroides measured at room temperature with 1-mm optical pathlength. The dashed lines show absorption spectra measured with increased RC concentration. Inset: absorption spectra in 980-1100 nm range obtained by accumulating 10 measurements.
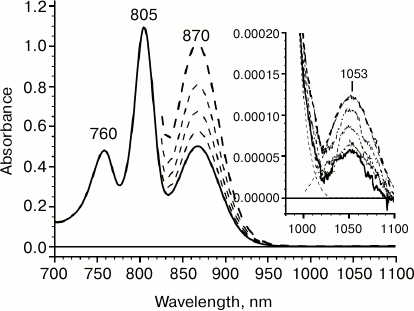
The extremely small amplitude of the absorption band at 1053 nm indicates that it corresponds to a low-intensity optical transition. In particular, it can be proposed that the absorption band at 1053 nm reflects a “hot” transition from one vibrational level of the ground state S0 of the primary electron donor P to its lowest singlet excited level S1. The proposed vibrational level is located 2050 cm–1 higher than S0. According to Boltzmann temperature distribution, the population of such a vibrational level at room temperature would be ~10–4, which corresponds to the experimentally measured ratio of amplitudes of absorption bands at 1053 and 870 nm. With this assumption, the fact that half-widths of these absorption bands match would also be explained.
Another and perhaps more probable proposition is attributing the absorption band at 1053 nm to a spin-forbidden singlet–triplet (S0 → T1) P transition. It was shown earlier that the maximum of P phosphorescence spectrum in Rba. sphaeroides RCs is located at 1318 nm [25, 26]. Given these data, the model considered here assumes the existence of a Stokes shift of ~260 nm. Even though this value appears to be unusually high, such a possibility should not be excluded, since a S0 → T1 transition was not detected experimentally due to it being spin-forbidden. It should be noted that in the triplet state with the lifetime of ~1 µs the probability for relaxation of vibronic levels is significantly higher than that for a singlet state.
Excitation light induces transmembrane electron transfer from the excited primary electron donor P* to the primary quinone acceptor QA in reaction centers. Various stages of this process can be directly observed by changes in the absorption of the reaction centers occurring at different time delays after a femtosecond excitation of the primary electron donor. Figure 2 presents difference absorption spectra of Rba. sphaeroides RCs reflecting evolution of light energy conversion into the energy of charge separated states. For instance, in reaction centers with pre-reduced quinone, the P+HA– state is formed within 12-15 ps (Fig. 2d).
Fig. 2. Difference light-minus-dark absorption spectra for reaction centers of Rhodobacter sphaeroides measured at room temperature: a) from –80 to 80 fs; b) from 80 to 200 fs; c) from 200 fs to 3 ps; d) from 3 to 12 ps. The spectra were obtained by accumulating 5000 measurements.
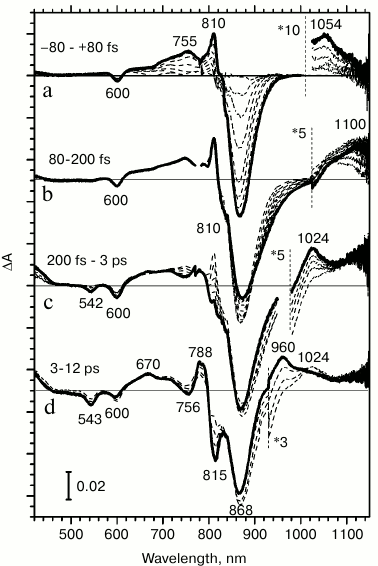
With a time delay of 200 fs, an excited electron leaves the special pair dimer and is transferred to the nearest acceptor, the BA molecule. This is indicated by an increase in the anion-radical BA– absorption band in difference spectra at 1024 nm, which occurs in the ion-radical pair P+BA– within 2.5 ps (Fig. 2c). Absorption changes with time delays of less than 200 fs apparently occur in the excited primary electron donor P870.
At time delays from 80 to 200 fs after light absorption, stimulated emission increases at 935 nm, and absorption increases at 1100 nm (Fig. 2b). Earlier, we associated these changes in absorption to the formation of a charge transfer complex P* (PAδ+PBδ–), since the absorption band at 1100 nm is absent in the dark absorption spectrum, and it appears simultaneously with the increase in stimulated emission at 935 nm in femtosecond difference absorption spectra [20, 21].
During the earliest time delays, from –80 to 80 fs, changes in difference absorption spectra are due to the transition of P870 to the excited state (Fig. 2a). A bleaching of P absorption bands at 870 and 600 nm occurs due to the primary donor transitioning to the lowest exciton excited level S1 and to an increase in stimulated emission of the excited state at 870 nm. Positive absorption bands appear at 755, 810, and 1053 nm, which reflect the properties of absorption of the excited primary electron donor. The absorption band at 1053 nm appears in difference spectra during delay times that are as small as if it were one of the absorption bands of the excited primary electron donor P*.
From the fact that the minor absorption band at 1053 nm is observed in a steady-state absorption spectrum (Fig. 1), it follows that this band reflects the S0 → Sx* transition. If this absorption band corresponds to the primary electron donor, then an absorption decrease might be expected in this range on difference absorption spectra (Fig. 2a). However, during primary electron donor excitation by 870 nm light, the absorption band at 1053 nm is increased multifold within the earliest time delays. Such a time delay is so small that it is comparable to the magnitude of light dispersion in the probing pulse.
We can evaluate the degree of increase in absorption band at 1053 nm. Optical density of the absorption band at 1053 nm is ~0.00006 OD in the dark absorption spectrum. It increases up to ~0.004 OD in femtosecond difference spectra that is by ~100-fold. Because a primary electron donor is excited only in ~10% of RCs during femtosecond measurements, this number should be increased by an order of magnitude. Therefore, during the excitation of a primary electron donor, absorption at 1053 nm increases by three orders of magnitude.
It is known that by illuminating RC with continuous light, absorption changes can be observed that are caused by the formation of the charge-separated state, P+QA–, whose lifetime is approximately 100 ms at room temperature. For instance, during RC illumination by 532 nm light, the difference absorption spectrum is observed that is shown in Fig. 3a. The spectrum includes: (i) bleaching of primary electron donor absorption bands at 870 and 600 nm and the appearance of a positive band in the absorption range of quinone QA– anion-radical at 448 nm; (ii) positive absorption changes in the long-wavelength spectrum range, corresponding to P+; (iii) electrochromic shifts of accessory bacteriochlorophyll absorption bands at 800 nm and of bacteriopheophytin at 760 nm. All these changes indicate formation of a charge-separated state P+QA–. The same difference absorption spectrum is observed during excitation of reaction centers with light of any other wavelength.
Fig. 3. Difference light-minus-dark absorption spectra for reaction centers of Rhodobacter sphaeroides measured at room temperature under constant illumination by nanosecond light pulses with frequency 20 Hz: a) excitation light with wavelength 532 nm; b) double-difference spectrum, excitation light with wavelength 1064 nm. The spectra were obtained by accumulating 10 measurements.
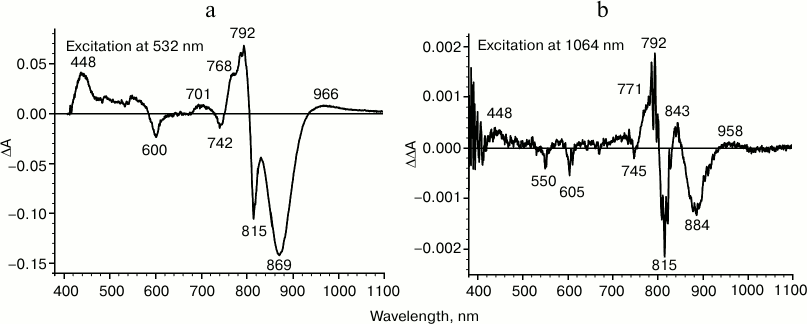
If the absorption band at 1053 nm corresponds to the primary electron donor or to the complex with charge transfer, PAδ+PBδ–, then during excitation of reaction centers in this absorption band, oxidation of the primary electron donor might be expected if the quantum energy were not so low.
During excitation by 1064 nm light, at least two processes take place in reaction centers: (i) long-lived absorption changes, including short-wavelength shifts of primary electron absorption bands at 870 nm and of accessory bacteriochlorophyll at 800 nm, similar to electrochromic shifts. It is however significant that no positive absorption bands of known ion-radical RC pigment forms are observed in the spectrum (data not shown). These changes in absorption are apparently not connected to electron transfer, but may rather be due to a local variation in sample temperature. Alongside these absorption changes in RCs, a second process occurs – accumulation of a state in which a positive absorption band is appeared at 450 nm on the minute time scale. To minimize the contribution of the first process, we measured a double-difference absorption spectrum after illuminating RCs by 1064 nm light, as shown in Fig. 3b. In this spectrum, positive changes in absorption at 450 nm and a long-wavelength H shift at 760 nm seem to indicate quinone reduction. In the long-wavelength absorption range, there is no significant bleaching of primary electron donor absorption band associated to a transition of P into the cation-radical form, but rather a short-wavelength shift is observed, which is apparently caused by the contribution from the first process. The lifetimes of the absorption changes shown in the figure increase up to tens of minutes. These results suggest that photoinduced quinone reduction takes place; however, there are no facts indicating whether the reducing electron belongs to the dimer of the special pair or some other component of the RC complex.
Thus, during the excitation of reaction centers with 1064 nm light, changes occur in the absorption spectrum indicating reduction of the primary electron acceptor QA, while the primary electron donor P870 apparently remains neutral. Absence of charge recombination suggests that these photoinduced changes in absorption reflect the formation of the long-lived state PQA–.
Summarizing these data, we note the following. Earlier we discovered an absorption band at 1100 nm, which we interpreted as a mixed state of the excited primary electron donor and the complex with charge transfer. However, according to the results represented in the current study, the nature of evolution of the primary electron donor excited state is apparently a more complex process. We found difference spectra of excited state P* to contain also a shorter wavelength band at 1053 nm registered at ultrashort time delays (~100 fs). It is significant that a low-intensity optical transition in this spectrum range is also observed on the long-wavelength QY slope of the absorption band of RC primary electron donor. Selective RC excitation within this absorption band at 1064 nm causes photoinduced formation of a primary quinone acceptor anion-radical. Although the nature of the electron donor and the mechanism of QA– formation remain unknown in this case, further investigations in this field may facilitate a more detailed understanding of characteristics of the primary charge separation mechanism in bacterial RCs.
Acknowledgments
This study was supported by grants of the Program of Fundamental Research Support of the Russian Academy of Sciences Presidium “Molecular and cell biology” and by the Russian Science Foundation (project No. 14-14-00789).
REFERENCES
1.Allen, J. P., Feher, G., Yeates, T. O., Komiya, H.,
and Rees, D. C. (1987) Structure of the reaction center from
Rhodobacter sphaeroides R-26: the cofactors, Proc. Natl.
Acad. Sci. USA, 84, 5730-5734.
2.Koepke, J., Krammer, E. M., Klingen, A. R., Sebban,
P., Ullmann, G. M., and Fritzsch, G. (2007) pH modulates the quinone
position in the photosynthetic reaction center from Rhodobacter
sphaeroides in the neutral and charge separated states, J. Mol.
Biol., 371, 396-409.
3.Shuvalov, V. A., Klevanik, A. V., Sharkov, A. V.,
Matveetz, Yu. A., and Krukov, P. G. (1978) Picosecond detection of
BChl-800 as an intermediate electron carrier between selectively
excited P870 and bacteriopheophytin in Rhodospirillum
rubrum reaction centers, FEBS Lett., 91,
135-139.
4.Arlt, T., Schmidt, S., Kaiser, W., Lauterwasser,
C., Meyer, M., Scheer, H., and Zinth, W. (1993) The accessory
bacteriochlorophyll: a real electron carrier in primary photosynthesis,
Proc. Natl. Acad. Sci. USA, 90, 11757-11761.
5.Holzwarth, A. R., and Muller, M. G. (1996)
Energetics and kinetics of radical pairs in reaction centers from
Rhodobacter sphaeroides. A femtosecond transient absorption
study, Biochemistry, 35, 11820-11831.
6.Van Stokkum, I. H. M., Beekman, L. M., Jones, M.
R., Van Brederode, M. E., and Van Grondelle, R. (1997) Primary electron
transfer kinetics in membrane-bound Rhodobacter sphaeroides
reaction centers: a global and target analysis, Biochemistry,
36, 11360-11368.
7.Sporlein, S., Zinth, W., and Wachtveitl, J. (1998)
Vibrational coherence in photosynthetic reaction centers observed in
the bacteriochlorophyll anion band, J. Phys. Chem. B,
102, 7492-7496.
8.Kennis, J. T., Shkuropatov, A. Y., van Stokkum, I.
H. M., Gast, P., Hoff, A. J., Shuvalov, V. A., and Aartsma, T. J.
(1997) Formation of a long-lived
P+BA– state in plant
pheophytin-exchanged reaction centers of Rhodobacter sphaeroides
R26 at low temperature, Biochemistry, 36,
16231-16238.
9.Schmidt, S., Arlt, T., Hamm, P., Huber, H., Nagele,
T., Wachtveitl, J., Zinth, W., Meyer, M., and Scheer, H. (1995) Primary
electron-transfer dynamics in modified bacterial reaction centers
containing pheophytin a instead of bacteriopheophytin a,
Spectrochim. Acta, 51, 1565-1578.
10.Shkuropatov, A. Ya., and Shuvalov, V. A. (1993)
Electron transfer in pheophytin a-modified reaction centers from
Rhodobacter sphaeroides (R-26), FEBS Lett., 322,
168-172.
11.Shuvalov, V. A., and Duysens, L. N. M. (1986)
Primary electron transfer reactions in modified reaction centers from
Rhodopseudomonas sphaeroides, Proc. Natl. Acad. Sci. USA,
83, 1690-1694.
12.Yakovlev, A. G., Shkuropatov, A. Y., and
Shuvalov, V. A. (2000) Nuclear wavepacket motion producing a reversible
charge separation in bacterial reaction centers, FEBS Lett.,
466, 209-212.
13.Yakovlev, A., and Shuvalov, V. (2000) Formation
of bacteriochlorophyll anion band at 1020 nm produced by nuclear
wavepacket motion in bacterial reaction centers, J. Chin. Chem.
Soc., 47, 709-714.
14.Kaufmann, K. J., Petty, K. M., Dutton, P. L., and
Rentzepis, P. M. (1975) Picosecond kinetics of events leading to
reaction center bacteriochlorophyll oxidation, Science,
188, 1301-1304.
15.Rockley, M. G., Windsor, M. W., Cogdell, R. J.,
and Parson, W. W. (1975) Picosecond detection of an intermediate in the
photochemical reaction of bacterial photosynthesis, Proc. Natl.
Acad. Sci. USA, 72, 2251-2255.
16.Holten, D., Windsor, M. W., Parson, W. W., and
Thornber, J. P. (1978) Primary photochemical processes in isolated
reaction centers of Rhodopseudomonas viridis, Biochim.
Biophys. Acta, 501, 112-126.
17.Yakovlev, A. G., Shkuropatov, A. Y., and
Shuvalov, V. A. (2002) Nuclear wavepacket motion between P* and
P+BA– potential surfaces with
subsequent electron transfer to HA in bacterial reaction
centers. 1. Room temperature, Biochemistry, 41,
2667-2674.
18.Shuvalov, V. A., and Yakovlev, A. G. (2003)
Coupling of nuclear wavepacket motion and charge separation in
bacterial reaction centers, FEBS Lett., 540, 26-34.
19.Khatypov, R. A., Khmelnitskiy, A. Y., Khristin,
A. M., and Shuvalov, V. A. (2010) Femtosecond absorption band formation
at 1080 and 1020 nm as an indication of charge-separated states
PAδ+PBδ–
and P+BA– in photosynthetic
reaction centers of the purple bacterium Rhodobacter
sphaeroides, Dokl. Biochem. Biophys., 430, 24-28.
20.Khatypov, R. A., Khmelnitskiy, A. Y., Khristin,
A. M., Fufina, T. Y., Vasilieva, L. G., and Shuvalov, V. A. (2012)
Primary charge separation within P870* in wild type and heterodimer
mutants in femtosecond time domain, Biochim. Biophys. Acta,
1817, 1392-1398.
21.Hamm, P., and Zinth, W. (1995) Ultrafast initial
reaction in bacterial photosynthesis revealed by femtosecond infrared
spectroscopy, J. Phys. Chem., 99, 13537-13544.
22.Hughes, J. L., Smith, P., Pace, R., and Krausz,
E. (2006) Charge separation in photosystem II core complexes induced by
690-730 nm excitation at 1.7 K, Biochim. Biophys. Acta,
1757, 841-851.
23.Zhu, J., van Stokkum, I. H. M., Paparelli, L.,
Jones, M. R., and Groot, M. L. (2013) Early bacteriopheophytin
reduction in charge separation in reaction centers of Rhodobacter
sphaeroides, Biophys. J., 104, 2493-2502.
24.Shuvalov, V. A., Shkuropatov, A. Ya., Kulakova,
S. M., Ismailov, M. A., and Shkuropatova, V. A. (1986) Photoreactions
of bacteriopheophytins and bacteriochlorophylls in reaction centers of
Rhodopseudomonas sphaeroides and Chloroflexus
aurantiacus, Biochim. Biophys. Acta, 849,
337-346.
25.Takiff, L., and Boxer, S. G. (1988)
Phosphorescence spectra of bacteriochlorophylls, J. Am. Soc.,
110, 4425-4426.
26.Takiff, L., and Boxer, S. G. (1988)
Phosphorescence from the primary electron donor in Rhodobacter
sphaeroides and Rhodopseudomonas viridis reaction centers,
Biochim. Biophys. Acta, 932, 325-334.