Variability of Methylation Profiles of CpG Sites in microrNA Genes in Leukocytes and Vascular Tissues of Patients with Atherosclerosis
A. N. Kucher1,2*, M. S. Nazarenko1*, A. V. Markov1, I. A. Koroleva1, and O. L. Barbarash3
1Research Institute of Medical Genetics, Tomsk National Research Medical Center, Russian Academy of Sciences, 634050 Tomsk, Russia; E-mail: aksana.kucher@medgenetics.ru2National Research Tomsk State University, 634050 Tomsk, Russia
3Research Institute for Complex Issues of Cardiovascular Diseases, Siberian Branch of the Russian Academy of Medical Sciences, 650002 Kemerovo, Russia
* To whom correspondence should be addressed.
Received December 15, 2016; Revision received February 15, 2017
In this study, we for the first time described the variability of methylation levels of 71 CpG sites in microRNA genes in leukocytes and blood vessels (coronary artery atherosclerotic plaques, intact internal thoracic arteries, and great saphenous veins) in patients with atherosclerosis using the Infinium HumanMethylation27 BeadChip microarray. Most of the analyzed CpG sites were characterized by the low variability, and most of these low-variable sites were hypomethylated in all tissue samples. CpG sites in coronary artery atherosclerotic plaques and leukocytes were similar in their methylation status. The highest variability of CpG methylation levels between different tissues was found for the CpG sites of the MIR10B gene; the methylation levels of these sites in leukocytes and atherosclerotic arteries were lower than in intact blood vessels. We also found that several cardiovascular disease risk factors, as well as medications, might affect methylation levels of CpG sites in microRNAs.
KEY WORDS: atherosclerosis, microRNA, methylationDOI: 10.1134/S0006297917060062
DNA methylation and RNA-mediated regulatory mechanisms play an important role in the functioning of various human organs and systems in both healthy and diseased organisms. Epigenetic mechanisms, such as DNA methylation, histone modification, and gene regulation by small noncoding RNAs, in particular microRNAs, are essential for the regulation of expression of tissue-specific genes during ontogenesis. At the same time, expression of microRNA genes is controlled by methylation of DNA (CpG sites) and histones, whose methylation profiles are affected by the functional state of an organism or by various pathologies. The importance of microRNAs in tissue-specific gene expression [1], organism response to stimuli [2-7], and pathogenesis of various diseases [8-13] has been proven in several studies. This makes evaluation of the methylation profiles of regulatory elements of microRNA genes an important research task; however, such studies have been scarce so far [14, 15]. Thus, examination of epigenetic regulation of microRNA expression in human fibroblasts and mammary epithelial cells revealed that tissue-specific expression of microRNA genes depends on the epigenetic state of their promoters and can be repressed by DNA methylation (38%), H3 histone methylation (H3K27me3) (58%), or a combination of DNA and H3K27 methylation (21%) [14]. Both microRNA expression levels and epigenetic states of promoters showed strong interindividual concordance within a given tissue type.
Most in vivo studies on epigenetic regulation of microRNA genes in human tissues have been performed in cancer tumors [16], and only few such studies have accessed other types of pathologies [17]. Variability in DNA methylation is usually evaluated for the same tissue type in different individuals. For this reason, analysis of DNA methylation levels in different tissues from the same individual is of interest, because it might reveal the tissue-specificity of the methylation state in the same patient, as well as interindividual variability for various types of tissues. Data on the methylation state of microRNA genes are of both theoretical and practical significance, since they can provide a basis for the development of diagnostic approaches for various pathologies, search for new drugs, and optimization of the patient’s response to treatment.
The goal of this study was to evaluate the variability of the methylation levels of the CpG sites in microRNA genes in blood and vessel tissues of patients with atherosclerosis.
MATERIALS AND METHODS
The studied cohort included six male patients of the Vascular Surgery Department (average age, 55.5 ± 6.5 years) with coronary artery disease and arterial hypertension. Four of the patients had an acute myocardial infarction in the patient’s medical history; three patients were diagnosed with hypercholesterolemia; three patients had type 2 diabetes mellitus. All patients were accessed for angina functional class and heart failure. Two patients were smokers. The intake of prescription drugs (aspirin, ACE inhibitors/calcium channel blockers, loop diuretics, thiazides, spironolactone, nitrates, and insulin) was taken into consideration for all the individuals tested. All patients volunteered and gave signed consent to be in this study.
Tissue samples from the right coronary arteries (CAAP, coronary artery atherosclerotic plaques), morphologically intact internal thoracic arteries (ITA), and great saphenous veins (GSV) were collected during coronary artery bypass surgery performed as a planned procedure for atherosclerosis treatment. Leukocytes (WBC) were isolated from venous blood drawn before the surgery. Samples of solid tissues were thoroughly washed with physiological saline, and all samples were frozen in liquid nitrogen and stored at –80°C.
Genomic DNA from leukocytes and vessel tissues was isolated by phenol/chloroform extraction after standard tissue treatment with proteinase K for 12 h at 37°C.
The methylation states of 71 CpG sites from 22 microRNA genes and one microRNA cluster (Table 1) were analyzed using the Infinium HumanMethylation27 BeadChip (Illumina, USA). Genomic DNA bisulfite modification was performed with EZ DNA Methylation Kit (Zymo Research, USA). Whole genome amplification, enzymatic DNA cleavage, DNA fragment purification, and hybridization were carried out as recommended by the manufacturer (Illumina) [18]. During hybridization, DNA fragments were annealed to the locus-specific DNA probes that corresponded to the methylated and unmethylated cytosine loci, respectively. Hybridization was followed by a single-base DNA extension with labeled ddNTPs. Signal intensity from the microarray was registered with an Illumina BeadArray Reader scanner (Illumina). Raw data were analyzed with the GenomeStudio Methylation Module program suite (Illumina). Methylation status of the interrogated CpG site was calculated as the ratio of signal from a methylated probe relative to the sum of signals from both methylated and unmethylated probes. This value, known as β, ranges continuously from 0 (unmethylated) to 1 (fully methylated). The data were deposited in the Gene Expression Omnibus database (accession number, GSE62867).
Table 1. Analyzed CpG sites of microRNA
genes and their location

1 CpG island coordinates are given according to the human
genome version GRCh37/hg19.
2 CpG site identifiers (IDs) are given according to the
annotation to the Illumina microchips; sites with the variability in
the β values below 0.1 for all studied samples (in all tissues
from all individuals) are shown in bold; low-variable CpG sites with
β < 0.2 are indicated with ↓; CpG sites with β >
0.8 are indicated with ↑.
Statistical analysis was performed using standard program packages from The R Project for Statistical Computing. Average DNA methylation levels in groups were compared using Student’s t-test for dependent samples with a correction for multiple testing by the Benjamini–Hochberg method. Correlations between DNA methylation levels and other quantitative parameters were analyzed using Spearman’s rank correlation coefficient. The significance (p) values for the Spearman’s rank correlation coefficient were computed using algorithm AS 89 [19]. The levels of similarity between DNA methylation profiles was estimated by multidimensional scaling and cluster analysis (using Euclidean coordinates as a distance measure and complete-linkage clustering for dendrogram generation) [20].
RESULTS AND DISCUSSION
Variability of methylation of CpG sites in microRNA genes in tissues and studied individuals. Out of 71 CpG sites analyzed, 51 sites (71.8%) displayed low variability in their methylation status: the difference in the methylation levels for these sites was below 0.1 in all tissues tested in all patients studied (Table 1), which could be related to existent DNA methylation conservatism of the corresponding genome regions. Almost all CpG sites with low variability were hypomethylated (β < 0.2; although in most cases, β did not exceed 0.1), except the only highly methylated cg17723549 in MIR423 gene (β > 0.90 in all samples).
Methylation levels of 20 CpG sites (28.2%) varied considerably (difference in the β values was over 0.1) (Tables 1 and 2 and Fig. 1). Eight of these sites were found in WBC; 11 – in GSV, 7 – in ITA, and 11 – in CAAP. CAAP samples displayed the lowest variability in the methylation levels – the difference between the β values in CAAP was 0.01 to 0.20, whereas in other tissues, this difference reached 0.30 and greater.
Table 2. CpG sites with varying methylation
levels (β) in the studied samples and interindividual variability
of β values for these sites in the studied tissues
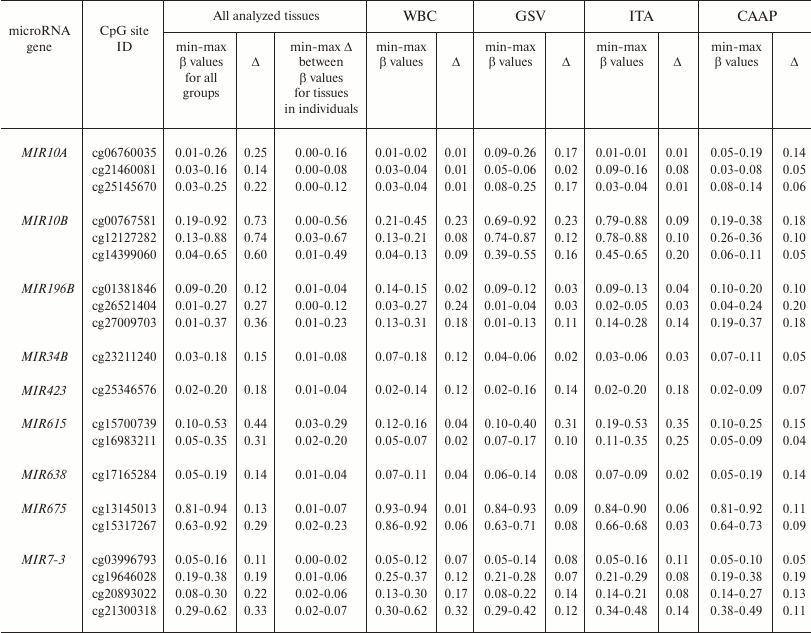
Note: min-max, minimal and maximal registered β values,
respectively; Δ, difference between the maximal and minimal
β values of the index.
Fig. 1. DNA methylation heatmap for CpG sites with varying methylation levels in the studied tissues.
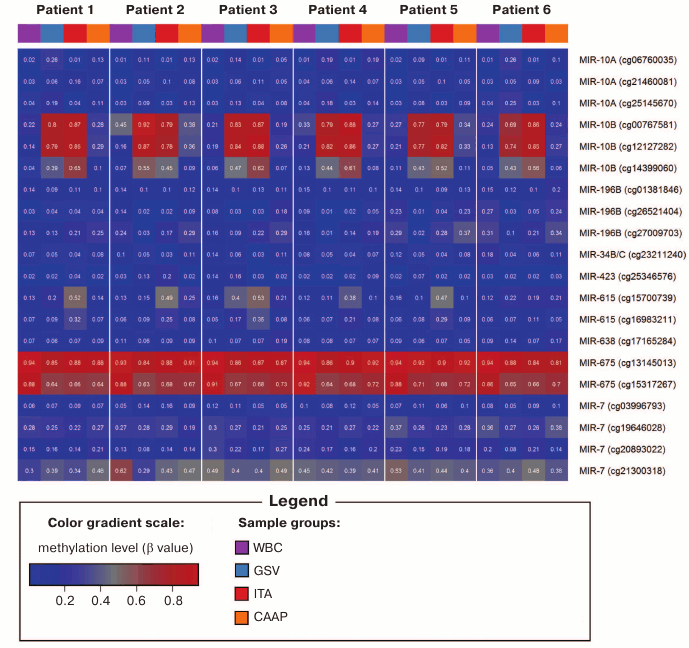
Analyzed CpG sites were unevenly represented on the Infinium Human Methylation27 BeadChip for different microRNA genes. Thus, eight genes (including miR17/92 cluster) were represented by only one CpG site; 18 genes were represented by two to eight CpG sites (Table 1). All analyzed CpG sites were hypomethylated in eight microRNA genes (MIR219A1, MIR320A, MIR564 – two sites; MIR330, MIR92B – three sites; MIR632 – four sites; MIR611 – six sites; MIR639 – eight sites) in all tissues. For two microRNA genes (MIR10B – three sites; MIR615 – two sites), the levels of CpG methylation varied in different samples. The highest variability for the methylation levels in different tissues was observed for the CpG sites of the MIR10B gene. Only a single site (cg13145013 in the MIR675 gene) was hypermethylated in all samples. Both low-variable (hypo- or hypermethylated) and variable CpG sites were found in six microRNA genes (Tables 1 and 2). In several cases (MIR10A, MIR196B, MIR34B, MIR423, MIR638, and MIR7-3), the same CpG islands contained CpG sites that differed in their methylation status, although the methylation patterns were similar in tested patients (Fig. 1).
Aavik et al. [21] observed decreased DNA methylation levels in tissues of atherosclerotic femoral arteries as compared to intact ITA. In addition, DNA hypomethylation in the imprinted 14q32 locus induced transcription of a microRNA gene cluster in this locus [21]. In our study, we found that several CpG sites in the MIR10B gene were less methylated in CAAP than in intact vessels (similar to the results of [21]); the methylation levels of CpG sites in atherosclerotic arteries were similar to the DNA methylation levels in WBC. This was typical for cg12127282 located close to the 5′-UTR (difference between mean β values in the compared groups, Δβ = 0.55; p < 0.001), as well as for cg00767581 (Δβ = 0.56; p = 0.001) and cg14399060 (Δβ = 0.48; p < 0.001), located in the coding region and close to the 3′-UTR, respectively.
The observed differences between the DNA methylation levels in the atherosclerotic and intact arteries in the same individual, on one hand, and in the same type of tissues in different subjects, on the other hand, might reflect different functional states of these tissues and organisms. In particular, low DNA methylation levels might result in transcription activation of the corresponding microRNAs, which would affect the posttranslational regulation (protein synthesis suppression) of the target genes.
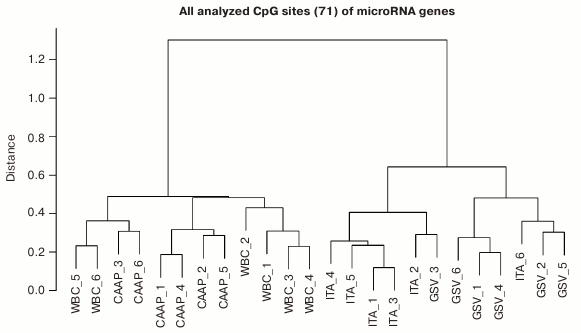
Based on the methylation levels of the analyzed CpG sites, all studied microRNAs could be distributed into two major clusters (Fig. 2), one of them including WBC and CAAP samples, and the other – GSV and ITA samples. Interestingly, samples most similar in the CpG methylation levels were from CAAP and WBC and could be subdivided into three subclusters: only CAAP, only WBC, and a mixed group. The cluster that included samples from GSV and ITA contained two distinct subclusters corresponding to the types of blood vessels (Fig. 2).
Fig. 2. Dendrogram reflecting tissue sample clusterization based on methylation levels of 71 CpG sites of microRNA genes (numbers correspond to individual patients).
Pronounced difference in the methylation levels of CpG sites in intact vessels and WBC (the difference in the mean β values for each tissue type, >0.2) was observed for three CpG sites in the MIR10B gene and cg15317267 in the MIR675 gene; in CAAP and GSV – for the same sites in the MIR10B gene and cg15700739 in the MIR615 gene (for these sites, the CpG methylation levels in CAAP were close to those in WBC) (Table 2).
The differences in the methylation levels of CpG sites in intact vessels (GSV and ITA) correlate with existing concepts on the tissue-specificity of epigenetic genome modifications [22]. The observed differences between the CpG methylation states in atherosclerotic and intact vessels were not completely unexpected, because some of the analyzed microRNA genes with variable CpG sites have been found to participate in various stages of atherogenesis (see reviews [23, 24]). Similar methylation levels of CpG sites in CAAP and WBC could be explained by the inflammation of atherosclerotic tissues.
Factors associated with methylation levels of CpG sites in microRNA genes. Clusterization of the analyzed CpG sites according to their methylation levels in the studied individuals was different in different tissues, which might reflect the effects of individual factors and/or functional states (e.g. pathologies) of the patients tested. For example, the WBC + CAAP cluster could be subdivided into three groups: CAAP only, WBC only, and a mixed group (Fig. 2). In this last mixed group, CAAP samples that clustered with WBC samples, were obtained from the smokers. Samples from the same patients clustered “incorrectly” in the GSV + ITA cluster: the ITA sample from one of the patients fell into the GSV group, while the GSV sample from the other patient fell into the ITA group. The highest correlation between the methylation status and smoking was observed for cg17165284 in the MIR638 gene – the methylation level of this site was 10% higher in the CAAP samples from smokers than from nonsmokers (Table 3). This microRNA plays an important role in the development of emphysematous lung destruction in smokers with chronic obstructive bronchitis [25]. It should be noted that not one but multiple factors can affect methylation status of CpG sites in microRNA genes, and combined action of such factors in individuals will complicate elucidation of the contribution of factors to the regulation of DNA methylation.
Table 3. Factors associated with the
methylation level of studied variable CpG sites in microRNA genes
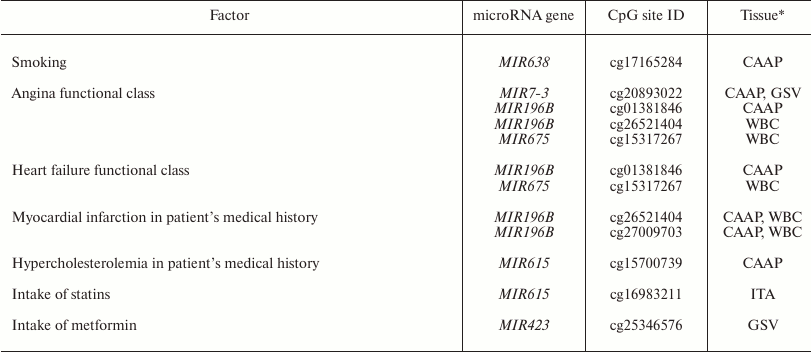
* Only tissues, in which the studied factor affected methylation levels,
are shown.
It is known that expression of some microRNAs is determined not only by the tissue type, but also by the functional state of cells and the organism as a whole. It might also be affected by drug treatment of the patients [2-7]. We found that in WBC, methylation level of the MIR10A gene cg06760035 site correlated with the low-density lipoprotein cholesterol content, and methylation of the MIR10B gene cg00767581 site correlated with the triglyceride content and body mass index (Table 4). MIR10A and MIR10B are located within the HOX gene cluster (HOXB3/4 and HOXD3/4, respectively) and display pleiotropic effects that are not limited to the cross-regulation of their expression [26]. The content of circulating miR-10a is associated with hyperlipidemia [27]. mir-10b is involved in lipid metabolism regulation and can presumably regulate in vitro levels of triglycerides via interacting with PPAR-α [28, 29].
Table 4. Clinical factors that correlate
with the methylation levels of analyzed variable CpG sites in microRNA
genes
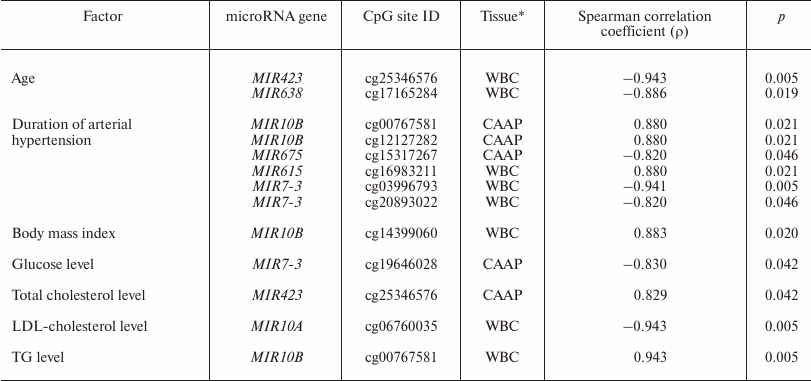
Note: LDL-cholesterol, low-density lipoprotein cholesterol; TG,
triglycerides.
* Only tissues, in which studied factor affected DNA methylation levels,
are shown.
In CAAP samples, methylation levels of cg25346576 in the MIR423 gene positively correlated with the total blood cholesterol content, and methylation levels of cg19646028 in the MIR7-3 gene negatively correlated with the glucose content. Prabu et al. [30] revealed weak negative correlation between the levels of circulating miR-423-5p and high-density lipoprotein cholesterol concentration in the blood of patients with impaired glucose tolerance and type 2 diabetes mellitus. However, we failed to identify any association between the methylation levels of MIR423 CpG sites and these pathologies.
All the studied patients had arterial hypertension. The methylation levels of six CpG sites in CAAP and WBC correlated either positively (three CpG sites in MIR10B and MIR615) or negatively (three CpG sites in MIR675 and MIR7-3) with the disease duration (Table 4). Among the products of these genes, only miR-615-5p was found in increased concentrations in the blood of patients with essential hypertension [31]. This microRNA also promotes the phagocytic capacity of splenic macrophages in cirrhosis-related portal hypertension [32].
Recent studies in human epigenetics have focused on the age dynamics of DNA methylation and its association with aging [33]. We found that methylation levels of CpG sites in MIR423 (cg25346576) and MIR638 (cg17165284) correlate with the patient’s age (48 to 67 years in our study) (Table 4). It is interesting to note that there were interactions between MIR423 rs6505162 and age group among the patients with esophageal cancer [34]. The cg25346576 site is located in the CpG island in the MIR423 gene promoter and, therefore, can be involved in the gene expression regulation. At the same time, it has been found before [35] that miR-423-5p expression decreases in aging human epithelial cells, which contradicts the results of our study. However, it should be remembered that the methylation status of a gene can differ in different tissues and cells. The MIR638 gene, which contains a CpG site whose methylation level negatively correlates with the patient’s age, is a part of the CpG island, and according to [36], expression of miR-638 is activated during replicative senescence of human fibroblasts.
Other factors that could affect the methylation status of some CpG sites are pathologies and drug intake (Table 3). In our study, these were the CpG sites of the MIR196B gene: differences in the methylation levels of these sites were observed in WBC and CAAP in patients with different functional classes of angina and patients that had suffered myocardium infarction in the past. In case of CAAP, these differences were also found in patients with functional classes of heart failure. Similarly, the difference in the methylation levels of cg15317267 site of the MIR675 gene was found in WBC of patients with angina and heart failure. It is interesting to note that in mice, the development of hyperhomocysteinemia (an independent factor in pathogenesis of cardiovascular disorders) was accompanied by a decrease in the methylation levels of the differentially methylated domain of the H19 gene (host gene for MIR675) in the liver, but caused an increase in the methylation levels of the same gene in the brain and aorta [37]. This allowed the authors to suggest that methylation of the differentially methylated domain of the H19 gene in hyperhomocysteinemia is a tissue-specific process. The effects of hyperhomocysteinemia and folic acid on DNA methylation were demonstrated in humans as well [38].
Based on these results, we suggest that intake of certain drugs might affect the methylation levels of CpG sites in microRNA genes. Thus, we found that intake of statins affected CpG methylation level in MIR615 in intact arteries, and intake of metformin affected CpG methylation in MIR423 in GSV. The fact that expression of microRNA genes might be modified by various supplements and drugs has been confirmed in other studies. However, CpG methylation levels changed differently in different tissues in response to the drug treatment, which corresponds well to the studies of other research groups [37]. Although our results were obtained using a relatively small cohort of patients, the observed effect of drugs and pathologies on the methylation of CpG sites in microRNA genes has been corroborated in other studies, which makes the problem of the effect of clinical factors on DNA methylation worth further study.
In conclusion, we demonstrated that out of 71 studied CpG sites in microRNA genes, 51 sites (71.8%) exhibited low variability of their methylation state in all analyzed samples: 50 of these sites were hypomethylated, and one site was hypermethylated. Twenty CpG sites (28.2%) varied in their methylation levels. The number of variable CpG sites in CAAP and GSV was higher than in WBC and ITA. Only two CpG sites were highly variable in all sample tissues, but they displayed different methylation levels. The highest variability between tissue samples was observed for the CpG sites in the MIR10B gene (the methylation levels of these sites in WBC and CAAP were lower than in intact ITA). We also identified several clinical factors that might affect methylation of CpG sites in microRNA genes.
Acknowledgments
This work was supported by the Russian Science Foundation (project No. 16-15-10150).
REFERENCES
1.Song, G., Xu, G., Ji, C., Shi, C., Shen, Y., Chen,
L., Zhu, L., Yang, L., Zhao, Y., and Guo, X. (2014) The role of
microRNA-26b in human adipocyte differentiation and proliferation,
Gene, 533, 481-487.
2.Virtue, A., Mai, J., Yin, Y., Meng, S., Tran, T.,
Jiang, X., Wang, H., and Yang, X.-F. (2013) Structural evidence of
anti-atherogenic microRNAs, Front. Biosci., 16,
3133-3145.
3.Jiang, Y., Wang, H. Y., Li, Y., Guo, S. H., Zhang,
L., and Cai, J. H. (2014) Peripheral blood miRNAs as a biomarker for
chronic cardiovascular diseases, Sci. Rep., 4, 5026.
4.Guo, W., Liu, H., Li, L., Yang, M., and Du, A.
(2014) Regulation of lovastatin on a key inflammation-related microRNA
in myocardial cells, Chin. Med. J., 127, 2977-2981.
5.Landgraf, P., Rusu, M., Sheridan, R., Sewer, A.,
Iovino, N., Aravin, A., Pfeffer, S., Rice, A., Kamphorst, A. O.,
Landthaler, M., Lin, C., Socci, N. D., Hermida, L., Fulci, V.,
Chiaretti, S., Foà, R., Schliwka, J., Fuchs, U., Novosel, A.,
Müller, R. U., Schermer, B., Bissels, U., Inman, J., Phan, Q.,
Chien, M., Weir, D. B., Choksi, R., De Vita, G., Frezzetti, D.,
Trompeter, H. I., Hornung, V., Teng, G., Hartmann, G., Palkovits, M.,
Di Lauro, R., Wernet, P., Macino, G., Rogler, C. E., Nagle, J. W., Ju,
J., Papavasiliou, F. N., Benzing, T., Lichter, P., Tam, W., Brownstein,
M. J., Bosio, A., Borkhardt, A., Russo, J. J., Sander, C., Zavolan, M.,
and Tuschl, T. (2007) A mammalian microRNA expression atlas based on
small RNA library sequencing, Cell, 129, 1401-1414.
6.He, D. X., Gu, F., Gao, F., Hao, J. J., Gong, D.,
Gu, X. T., Mao, A. Q., Jin, J., Fu, L., and Ma, X. (2016) Genome-wide
profiles of methylation, microRNAs, and gene expression in
chemoresistant breast cancer, Sci. Rep., 6, 24706.
7.Shi, F., Chen, X., Fu, A., Hansen, J., Stevens, R.,
Tjonneland, A., Vogel, U. B., Zheng, T., and Zhu, Y. (2013) Aberrant
DNA methylation of miR-219 promoter in long-term night shiftworkers,
Environ. Mol. Mutagen., 54, 406-413.
8.Cao, D., Hu, L., Lei, D., Fang, X., Zhang, Z.,
Wang, T., Lin, M., Huang, J., Yang, H., Zhou, X., and Zhong, L. (2015)
MicroRNA-196b promotes cell proliferation and suppress cell
differentiation in vitro, Biochem. Biophys. Res. Commun.,
457, 1-6.
9.Camkurt, M. A., Acar, S., Coskun, S., Gunes, M.,
Gunes, S., Yılmaz, M. F., Gorur, A., and Tamer, L. (2015)
Comparison of plasma microRNA levels in drug naive, first episode
depressed patients and healthy controls, J. Psychiatr. Res.,
69, 67-71.
10.Liu, R., Jacobs, D. I., Hansen, J., Fu, A.,
Stevens, R. G., and Zhu, Y. (2014) Aberrant methylation of miR-34b is
associated with long-term shiftwork: a potential mechanism for
increased breast cancer susceptibility, Cancer Causes Control,
26, 171-178.
11.Kucher, A. N., and Babushkina, N. P. (2011) Role
of microRNAs and genes involved in microRNA biogenesis and functioning
in various human pathologies, Med. Genet., 1, 3-13.
12.Burmistrova, O. A., Goltsov, A. Y., Abramova, L.
I., Kaleda, V. G., Orlova, V. A., and Rogaev, E. I. (2007) MicroRNA in
schizophrenia: genetic and expression analysis of miR-130b (22q11),
Biochemistry (Moscow), 72, 860-865.
13.Filatova, E. V., Alieva, A. Kh., Shadrina, M. I.,
and Slonimsky, P. A. (2012) MicroRNAs: possible role in pathogenesis of
Parkinson’s disease, Biochemistry (Moscow), 77,
981-988.
14.Vrba, L., Garbe, J. C., Stampfer, M. R., and
Futscher, B. W. (2011) Epigenetic regulation of normal human mammary
cell type-specific miRNAs, Genome Res., 21,
2026-2037.
15.Renteria, M. E., Coolen, M. W., Statham, A. L.,
Choi, R. S., Qu, W., Campbell, M. J., Smith, S., Henders, A. K.,
Montgomery, G. W., Clark, S. J., Martin, N. G., and Medland, S. E.
(2013) GWAS of DNA methylation variation within imprinting control
regions suggests parent-of-origin association, Twin Res. Hum.
Genet., 16, 767-781.
16.Loginov, V. I., Rykov, S. V., Fridman, M. V., and
Braga, E. A. (2015) Methylation of miRNA genes and oncogenesis,
Biochemistry (Moscow), 80, 145-162.
17.Piletic, K., and Kunej, T. (2016) MicroRNA
epigenetic signatures in human disease, Arch. Toxicol.,
90, 2405-2419.
18.Bibikova, M., Le, J., Barnes, B.,
Saedinia-Melnyk, S., Zhou, L., Shen, R., and Gunderson, K. L. (2009)
Genome-wide DNA methylation profiling using Infinium assay,
Epigenomics, 1, 177-200.
19.Best, D. J., and Roberts, D. E. (1975) Algorithm
AS 89: the upper tail probabilities of Spearman’s rho, Appl.
Stat., 24, 377-379.
20.Everitt, B. S. (2011) Cluster Analysis
(Everitt, B. S., Landau, S., Leese, M., and Stahl, D., eds.) 5th Edn.,
John Wiley, p. 330.
21.Aavik, E., Lumivuori, H., Leppanen, O., Wirth,
T., Hakkinen, S. K., Brasen, J. H., Beschorner, U., Zeller, T.,
Braspenning, M., Van Criekinge, W., Makinen, K., and Yla-Herttuala, S.
(2015) Global DNA methylation analysis of human atherosclerotic plaques
reveals extensive genomic hypomethylation and reactivation at imprinted
locus 14q32 involving induction of a miRNA cluster, Eur. Heart
J., 36, 993-1000.
22.Varley, K. E., Gertz, J., Bowling, K. M., Parker,
S. L., Reddy, T. E., Pauli-Behn, F., Cross, M. K., Williams, B. A.,
Stamatoyannopoulos, J. A., Crawford, G. E., Absher, D. M., Wold, B. J.,
and Myers, R. M. (2013) Dynamic DNA methylation across diverse human
cell lines and tissues, Genome Res., 23, 555-567.
23.Kucher, A. N., and Nazarenko, M. S. (2017) Role
of microRNAs in atherogenesis, Kardiologiya, 57, No. 8,
in print.
24.Kucher, A. N., and Babushkina, N. P. (2012) Role
of microRNAs in the development of cardiovascular disorders, Mol.
Med., 1, 10-17.
25.Christenson, S. A., Brandsma, J. D., Campbell, J.
D., Knight, D. A., Pechkovsky, D. V., Hogg, J. C., Timens, W., Postma,
D. S., Lenburg, M., and Spira, A. (2013) miR-638 regulates gene
expression networks associated with emphysematous lung destruction,
Genome Med., 5, 114.
26.Tehler, D., Hoyland-Kroghsbo, N. M., and Lund, A.
H. (2011) The miR-10 microRNA precursor family, RNA Biol.,
8, 728-734.
27.Simionescu, N., Niculescu, L. S., Sanda, G. M.,
Margina, D., and Sima, A. V. (2014) Analysis of circulating microRNAs
that are specifically increased in hyperlipidemic and/or hyperglycemic
sera, Mol. Biol. Rep., 41, 5765-5773.
28.Zheng, L., Lv, G. C., Sheng, J., and Yang, Y. D.
(2010) Effect of miRNA-10b in regulating cellular steatosis level by
targeting PPAR-alpha expression, a novel mechanism for the pathogenesis
of NAFLD, J. Gastroenterol. Hepatol., 25, 156-163.
29.Wang, D., Xia, M., Yan, X., Li, D., Wang, L., Xu,
Y., Jin, T., and Ling, W. (2012) Gut microbiota metabolism of
anthocyanin promotes reverse cholesterol transport in mice via
repressing miRNA-10b, Circ. Res., 111, 967-981.
30.Prabu, P., Rome, S., Sathishkumar, C., Aravind,
S., Mahalingam, B., Shanthirani, C. S., Gastebois, C., Villard, A.,
Mohan, V., and Balasubramanyam, M. (2015) Circulating miRNAs of
“Asian Indian phenotype” identified in subjects with
impaired glucose tolerance and patients with type 2 diabetes, PLoS
One, 10, e0128372.
31.Li, S., Zhu, J., Zhang, W., Chen, Y., Zhang, K.,
Popescu, L. M., Ma, X., Lau, W. B., Rong, R., Yu, X., Wang, B., Li, Y.,
Xiao, C., Zhang, M., Wang, S., Yu, L., Chen, A. F., Yang, X., and Cai,
J. (2011) Signature microRNA expression profile of essential
hypertension and its novel link to human cytomegalovirus infection,
Circulation, 124, 175-184.
32.Jiang, A., Zhang, S., Li, Z., Liang, R., Ren, S.,
Li, J., Pu, Y., and Yang, J. (2011) miR-615-3p promotes the phagocytic
capacity of splenic macrophages by targeting ligand-dependent nuclear
receptor corepressor in cirrhosis-related portal hypertension, Exp.
Biol. Med. (Maywood), 236, 672-680.
33.Pal, S., and Tyler, J. K. (2016) Epigenetics and
aging, Sci. Adv., 2, e1600584.
34.Ye, Y., Wang, K. K., Gu, J., Yang, H., Lin, J.,
Ajani, J. A., and Wu, X. (2008) Genetic variations in microRNA-related
genes are novel susceptibility loci for esophageal cancer risk,
Cancer Prev. Res. (Phila), 1, 460-469.
35.Taguchi, Y. H. (2012) Inference of target gene
regulation via miRNAs during cell senescence by using the MiRaGE
server, Aging Dis., 3, 301-306.
36.Maes, O. C., Sarojini, H., and Wang, E. (2009)
Stepwise up-regulation of microRNA expression levels from replicating
to reversible and irreversible growth arrest states in WI-38 human
fibroblasts, J. Cell Physiol., 221, 109-119.
37.Devlin, A. M., Bottiglieri, T., Domann, F. E.,
and Lentz, S. R. (2005) Tissue-specific changes in H19 methylation and
expression in mice with hyperhomocysteinemia, J. Biol. Chem.,
280, 25506-25511.
38.Ingrosso, D., and Perna, A. F. (2009) Epigenetics
in hyperhomocysteinemic states. A special focus on uremia, Biochim.
Biophys. Acta, 1790, 892-899.