Effect of Anesthetics on Efficiency of Remote Ischemic Preconditioning
D. N. Silachev1#, E. A. Usatikova1#, I. B. Pevzner1, L. D. Zorova1,2, V. A. Babenko3, M. V. Gulyaev4, Yu. A. Pirogov5, E. Yu. Plotnikov1, and D. B. Zorov1*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; E-mail: zorov@genebee.msu.ru2International Laser Center, Lomonosov Moscow State University, 119992 Moscow, Russia
3Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119992 Moscow, Russia
4Lomonosov Moscow State University, Faculty of Fundamental Medicine, 119992 Moscow, Russia
5Lomonosov Moscow State University, Faculty of Physics, 119992 Moscow, Russia
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received October 28, 2016; Revision received May 4, 2017
Remote ischemic preconditioning of hind limbs (RIPC) is an effective method for preventing brain injury resulting from ischemia. However, in numerous studies RIPC has been used on the background of administered anesthetics, which also could exhibit neuroprotective properties. Therefore, investigation of the signaling pathways triggered by RIPC and the effect of anesthetics is important. In this study, we explored the effect of anesthetics (chloral hydrate and Zoletil) on the ability of RIPC to protect the brain from injury caused by ischemia and reperfusion. We found that RIPC without anesthesia resulted in statistically significant decrease in neurological deficit 24 h after ischemia, but did not affect the volume of brain injury. Administration of chloral hydrate or Zoletil one day prior to brain ischemia produced a preconditioning effect by their own, decreasing the degree of neurological deficit and lowering the volume of infarct with the use of Zoletil. The protective effects observed after RIPC with chloral hydrate or Zoletil were similar to those observed when only the respective anesthetic was used. RIPC was accompanied by significant increase in the level of brain proteins associated with the induction of ischemic tolerance such as pGSK-3β, BDNF, and HSP70. However, Zoletil did not affect the level of these proteins 24 h after injection, and chloral hydrate caused increase of only pGSK-3β. We conclude that RIPC, chloral hydrate, and Zoletil produce a significant neuroprotective effect, but the simultaneous use of anesthetics with RIPC does not enhance the degree of neuroprotection.
KEY WORDS: ischemia, brain, remote preconditioning, hind limb, chloral hydrate, ZoletilDOI: 10.1134/S0006297917090036
Abbreviations: BDNF, brain-derived neurotrophic factor; CH, chloral hydrate; EPO, erythropoietin; 5-HD, sodium 5-hydroxydecanoate; IPC, ischemic preconditioning; MCA, middle cerebral artery; MCAO, one-side occlusion of middle cerebral artery; MRI, magnetic resonance imaging; PMSF, phenylmethylsulfonyl fluoride; RIPC, remote ischemic preconditioning of hind limbs; ROS, reactive oxygen species; SO, sham-operated (animals); Zol, Zoletil.
Ischemic preconditioning (IPC) performed by application of short periods
of ischemia prior to an extended period of blood flow interruption is
one of the most efficient methods for protecting organs and tissues
from the effects of ischemia and reperfusion [1].
For the first time, this phenomenon was described for heart [2], but later similar protective mechanisms were
demonstrated for brain, kidneys [3], and skeletal
muscles [4]. Nevertheless, clinical application of
the brain IPC is limited due to the damaging effect of even short-term
brain ischemia and possible complications, e.g. vessels thrombosis. The
method of remote ischemic preconditioning (RIPC), which involves
short-term periods of ischemia/reperfusion of a distant organ (such as
limbs or kidney) resulting in the protection of brain from ischemic
injury, seems more reliable in this regard. It has been shown that
induction of IPC in limbs [5, 6] or kidney [7] afforded
protection of the brain from ischemic stroke. No doubt that the IPC
procedure conducted in limbs is safer and simpler than the procedure
conducted in other organs as it does not involve surgical intervention
and can be performed using, for example, the cuff of a tonometer.
However, data on the clinical efficiency of RIPC used under heart and
brain pathologies are contradictory. For example, the efficiency of
RIPC was demonstrated in several studies for heart failure and stroke
[8-10], while only
insignificant protective effect [11, 12] or total absence of effect [13] were observed in other studies.
Moreover, even in the case of proved efficiency of RIPC, such issues as the mechanism of transfer of the protective signals from a distant organ to the brain, involved signaling pathways, as well as the optimal scheme for ischemic tolerance induction (number of ischemia/reperfusion cycles and their duration) remain unclear [14]. Introduction of RIPC into clinical practice requires not only comprehensive study of neuroprotective mechanisms, but also assessment of interaction of RIPC with pharmacological vehicles such as anesthetics. Numerous experimental and clinical data indicate that some anesthetics protect such organs as heart, brain, and kidney from injuries caused by ischemia/reperfusion [15, 16]. It has been shown in recent studies that the mechanisms of the anesthetic protection effect share many components with the ischemic preconditioning signaling pathways [17].
It is worth mentioning that many studies on neuroprotective mechanisms of RIPC are conducted with anesthetized animals. Hence, the protective signals of anesthetics can interfere in a certain way with signaling induced by RIPC. Anesthesia is always used in invasive RIPC (surgical access to blood vessels involving tissue incision and clamping of the femoral artery), and it is used in the majority of noninvasive RIPC to immobilize the animal for the convenience of the experimentator. The use of either inhalational anesthetics such as isoflurane and sevoflurane or injectable ones such as chloral hydrate, tiletamine/zolazepam, and others complicate the situation in the analysis of the RIPC protective mechanism even further. While several signaling protective pathways associated with the induction of brain ischemic tolerance are known for the inhalational anesthetics [18-21], very little is known with regards to the infused ones. In particular, the preconditioning effect in the first (fast) therapeutic window has been shown recently for the injectable anesthetic used frequently in animal experiments [22]. A data on a possible anesthetic preconditioning with tiletamine/zolazepam are not yet available. Nevertheless, complex interference is possible between the mechanisms of acquiring ischemic tolerance mediated via RIPC or anesthetics, which requires special attention to adequately assess the efficiency of the neuroprotective effect of RIPC.
In this study, we evaluated the effect of anesthetics chloral hydrate and tiletamine/zolazepam (Zoletil) on the ability of RIPC to protect the brain from injury caused by ischemia/reperfusion.
MATERIALS AND METHODS
Modeling of brain ischemia. Experiments were performed on outbred white male rats (320-350 g). The animals had unlimited access to food and water and were kept in cages with a temperature-controlled environment (22 ± 2ºC) with light on from 9 AM to 9 PM. Experimental procedures were conducted in accordance with the European Community Council directives 2010/63/EU and the study was approved by the local institutional animal ethics committee.
The animals were anesthetized before surgery with chloral hydrate (300 mg/kg, i.p.). Ischemia was initiated by one-side occlusion of the middle cerebral artery (MCAO) with nylon filament coated with silicon [23]. An incision was made through a midline cervical area, and the left common carotid artery, external carotid artery, and internal carotid artery were isolated. A ligature was applied onto the external and internal carotid artery as well as a microvascular clip applied on the common carotid artery, and next the external carotid artery was cut distal to the thread application site. A heparinized 0.25-mm diameter silicon-coated nylon filament was introduced through the stump of the external carotid artery into the internal carotid artery to the depth of 19-20 mm (up to MCA) and fixed with a clip. Interruption of the blood flow was maintained for 60 min, followed by removal of the thread from the blood vessel, restoring blood flow in the MCA territory. The animal body temperature was maintained at 37.0 ± 0.5°C during and after surgery. Sham-operated (SO) animals were subjected to the same procedures excluding the blood vessel cutting and filament introduction.
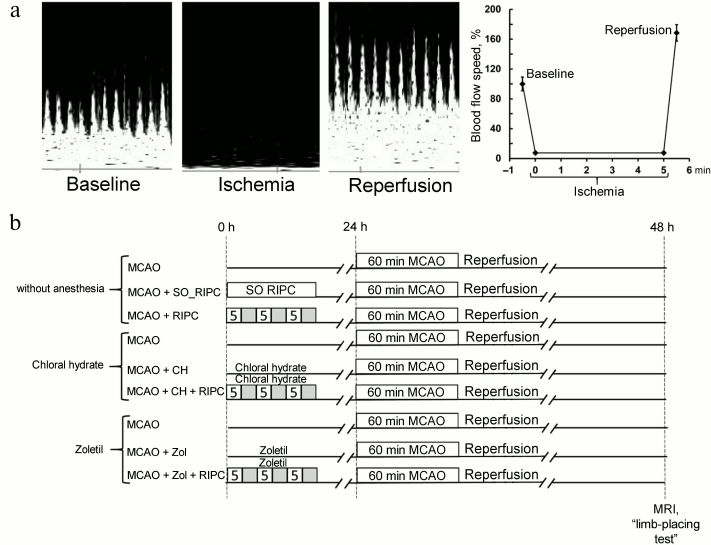
Remote ischemic preconditioning of hind limbs. IPC of hind limbs was conducted noninvasively via clamping of each limb with a nylon cable tie for 5 min followed by 5-min reperfusion interval repeated three times for each limb. The Doppler ultrasound 25 MHz (Doppler Minimax, Russia) was used to record the blood flow to the hind limbs. The blood flow was fully restored after 5-min occlusion (Fig. 1a). RIPC was performed 24 h prior to modeling of focal brain ischemia. Three experimental series were conducted (Fig. 1b). Animals from the first group were used for exploration of the effect of RIPC without anesthesia. Rats were placed into restrainer for 30 min per day during 3 days for habituation. RIPC was conducted on awake rat fixed in restrainer and sham RIPC was performed as regular RIPC but without hindlimbs clamping. The animals were randomly divided into three groups: MCAO (n = 11) – rats with cerebral ischemia control; MCAO + RIPC (n = 13) – ischemic, treated with RIPC; MCAO + SO_RIPC (n = 6) – ischemic, treated with sham RIPC rats.
Fig. 1. Effect of ischemic preconditioning on the blood flow in a hind limb. a) Representative recordings of a blood flow linear speed in the rat left hind limb following short-term ischemia/reperfusion (left panel) and calculation of linear speed of blood flow in the hind limb after one cycle of ischemia reperfusion (right panel). b) Scheme of three experimental series for investigation of neuroprotective effects of remote ischemic preconditioning of hind limbs (RIPC): without anesthesia (immobilization of awake animals in a restrainer), with anesthesia by chloral hydrate (CH, 300 mg/kg i.p.) or Zoletil (Zol, 40 mg/kg i.p.). Cerebral ischemia was induced by 60-min occlusion of the middle cerebral artery (MCAO) for all groups. RIPC was conducted by application of three cycles consisting of 5-min ischemia (light squares) and reperfusion (gray squares).
A specific inhibitor of KATP-channels – 5-hydroxydecanoate (5-HD) – was used to explore the role of this channel in remote ischemic preconditioning; the drug was injected intraperitoneally at a dose of 40 mg/kg 30 min before the preconditioning of hind limbs (group MCAO + RIPC + 5-HD, n = 6). The neurological status of animals was evaluated using the “limb-placing test” 24 h after the MCAO modeling.
In the second series of experiments, the effect of chloral hydrate anesthetic on RIPC was studied. Animals were also divided into three groups: MCAO (n = 7) – rats with cerebral ischemia; MCAO + CH (n = 6) – animals subjected to i.p. injection of chloral hydrate at a dose of 300 mg/kg 24 h prior to ischemia modeling; MCAO + CH + RIPC (n = 8) – rats that underwent limb preconditioning accompanied by anesthesia with chloral hydrate 24 h before the cerebral ischemia.
In the third series of experiments, the effect of tiletamine/zolazepam hydrochloride (trade name Zoletil) on the effects of RIPC was studied. Three experimental groups were as: MCAO (n = 11) – rats with cerebral ischemia; MCAO + Zol (n = 6) – animals intraperitoneally injected with Zoletil at dose 40 mg/kg 24 h before the ischemia modeling; MCAO + Zol + RIPC (n = 6) – animals subjected to preconditioning of limbs accompanied by the Zoletil-mediated anesthesia 24 h prior to cerebral ischemia.
Investigation of signaling pathways involved in induction of ischemic tolerance. To explore the effect of RIPC and anesthetics on the signaling pathways involved in the induction of ischemic tolerance in brain, five groups of experimental animals were created: animals with RIPC and sham RIPC; animals subjected to i.p. injection of chloral hydrate at dose 300 mg/kg or Zoletil at dose 40 mg/kg, as well as a group of intact rats. Each group contained six animals. The rats were decapitated 24 h after treatment to obtain brain homogenate (see “Western blotting analysis” section).
Estimation of sensorimotor brain functions. The “limb-placing test” was used for evaluation of sensorimotor dysfunction. This procedure is based on techniques described by Jolkkonen and coauthors [24]. The rats were habituated for handling three days before surgery. Neurological status of animals was evaluated before the induction of ischemia as well as one postoperative day. Evaluation included seven tests assessing sensorimotor integration of front fore and hind limbs in response to tactile, proprioceptive, and visual stimulation. The following scores were used: rat performed the test normally – 2 points; rat performed the test with delay (>2 s) and/or not completely – 1 point; rat did not perform the test – 0 points.
Evaluation of brain injury volume. The brains of all experimental animals were investigated with magnetic resonance imaging (MRI) one day after surgery. All MRI experiments were performed as described previously [25] using a BioSpec 70/30 instrument (Bruker, Germany) with magnetic field induction of 7 T and a gradient system of 105 mT/m. Morphometric analysis of MR-images was performed using the ImageJ Program (National Institutes of Health, USA), and the volume of infarct was calculated and normalized for each group by the average value for the MCAO group. Brain swelling was also determined using the following formula:
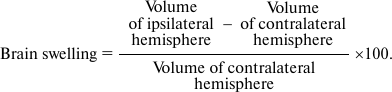
Determination of urinary erythropoietin content. The concentration of urinary erythropoietin (EPO) was determined using an immunoenzyme assay. Immediately after the RIPC procedure, animals were placed in metabolic cages, and urine was collected for the following 24 h. Next, the collected urine was concentrated by ultrafiltration in Centricon YM 30 tubes (Millipore, USA) using centrifugation according the manufacturer’s protocol. The final retentate was used for quantitative EPO level assay conducted according to the instructions of the test system manufacturer (R & D Systems, USA).
Western blotting analysis. Western blotting was used for determination of the levels of brain-derived neurotrophic factor (BDNF), phosphorylated glycogen synthase kinase-3 beta (pGSK-3β), and heat shock protein 70 (HSP70) in brain homogenates prepared 24 h after RIPC or intraperitoneal injection of chloral hydrate or Zoletil. The brain was removed immediately after decapitation and cooled in PBS. All brain structures were used for analysis except cerebellum and medulla; they were cut on small pieces and homogenized in 5 ml of PBS containing 1 mM of the PMSF protease inhibitor. Proteins in the brain homogenate were separated using electrophoresis on a 12.5% polyacrylamide gel under denaturing conditions. After electrophoresis, gels were blotted onto PVDF membranes (Amersham Pharmacia Biotech, UK), which were blocked for 12 h at 4°C in Tris buffer by 5% non-fat milk and subsequently incubated with primary rabbit anti-BDNF, -pGSK-3β, or -HSP70 antibodies at dilution 1 : 1000. Membranes were stained with secondary antibodies anti-rabbit IgG conjugated with horseradish peroxidase at a dilution 1 : 10,000 (Jackson ImmunoResearch, USA). Signal was recorded using chemiluminescent substrate of ECL (enhanced chemiluminescence system) (Amersham Pharmacia Biotech, UK) on a V3 Western Blot Imager (BioRad, USA). The images were analyzed using the ImageLab program.
Statistical analysis. Statistical analyses were performed using Statistica 7.0 for Windows (StatSoft, Inc., USA). Normality of the parameter distribution was estimated using the Shapiro–Wilk criterion W. All data were presented as means ± standard error of means while neurological deficit scores were expressed as median ± interquartile ranges (the 25th to 75th percentile are shown in the parentheses). Statistical differences between groups in the data of infarct volume and brain swelling were analyzed using one-way ANOVA with Tukey’s post hoc test. Statistical differences in limb-placing tests between groups were analyzed using Kruskal–Wallis test with the Mann–Whitney U-test with Bonferroni correction post hoc. Statistical differences of the Western blotting data were analyzed using Student’s t-test. The values p < 0.05 were considered as statistically significant.
RESULTS
Neuroprotective effects of RIPC. By using MRI, after 24-h exposure of the rat brain to ischemia/reperfusion we observed extensive injury of the sensorimotor cortex and striatum as well as partial damages to the hypothalamus and amygdala located outside the vascular basin of the middle cerebral artery (Fig. 2a). The average volume of damage in the MCAO group was 261 ± 20 mm3, and the swelling of the damaged hemisphere was on average 16.9 ± 2.6% (Fig. 2c). Preconditioning of the hind limbs 24 h prior to MCAO did not affect the volume of injury or the degree of brain swelling (Fig. 2, a-c), which were 261 ± 20 mm3 and 16.4 ± 1.4%, respectively. In addition, MCAO caused significant sensorimotor deficit observed in the contralateral limbs with respect to the damaged hemisphere. While before the induction of ischemia the intact rats scored 14 (14-14) in the limb-placing test and the sham-operated (SO) rats scored 13.75 (13.2-14.0), the rats after ischemia demonstrated neurological status of only 2 (2.0-2.5) points. RIPC was able to induce a significant improvement of the total score in the limb-placing test to 4 (4-6) points (p < 0.05) (Fig. 2d). Sham-preconditioned animals did not have any differences from the MCAO group.
Fig. 2. Neuroprotective effects of RIPC without anesthesia. a) Representative T2-weighted MR-images obtained 24 h after MCAO (sampling thickness 0.8 mm). Hyperintense regions (light area) refer to ischemic areas. Infarct volume (b) and brain swelling (c) evaluated by using MRI with analysis of T2-weighted MR-images. d) Neurological status evaluated by a limb-placing test. Data are presented as median (thick black line), interquartile ranges, and minimum and maximum of the statistically significant data set; * denotes significant difference from the MCAO group p < 0.05; one-way ANOVA, followed by Tukey’s post hoc analysis for (b) and (c); Kruskal–Wallis test with the Mann–Whitney U-test for (d).
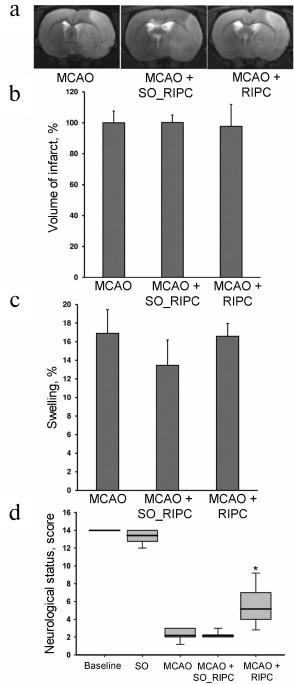
Neuroprotective effects of RIPC with use of chloral hydrate during preconditioning. Pretreatment with chloral hydrate by itself had no effect on an infarct volume and brain edema, and RIPC did not affect these factors as well (Fig. 3, a-c). However, both chloral hydrate and RIPC accompanied with chloral hydrate administration caused restoration of neurological functions to 8 (5-8) and 7 (5.25-7.25) points, respectively (p < 0.05) (Fig. 3d).
Fig. 3. Neuroprotective effects of RIPC using chloral hydrate during preconditioning. a) Representative T2-weighted MR-images obtained 24 h after MCAO (sampling thickness 0.8 mm). Hyperintense regions (light area) refer to ischemic areas. Infarct volume (b) and brain swelling (c) evaluated by using MRI with analysis of T2-weighted MR-images. d) Neurological status evaluated by a limb-placing test. Data are presented as median (thick black line), interquartile ranges, and minimum and maximum of the statistically significant data set; * denotes significant difference from the MCAO group p < 0.05; one-way ANOVA, followed by Tukey’s post hoc analysis for (b) and (c); Kruskal–Wallis test with the Mann–Whitney U-test for (d).
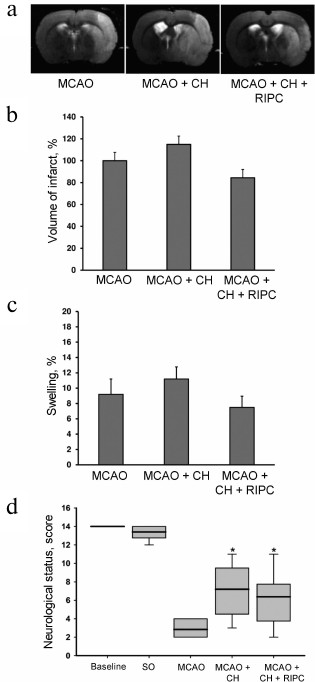
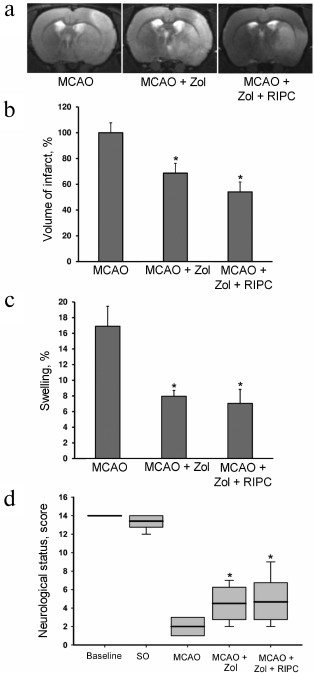
Neuroprotective effects of RIPC using Zoletil during preconditioning. Both Zoletil and RIPC with Zoletil caused lesser brain damage after MCAO reaching 68.7 ± 7.6% (p < 0.05) and 54 ± 7.6% (p < 0.05) in comparison with the MCAO group (Fig. 4, a and b). Moreover, the pretreatment with Zoletil produced statistically significant decrease in brain swelling (p < 0.05), and similar 2-fold decrease in swelling was observed when RIPC was used (Fig. 4c). The neurological score after 24 h of reperfusion demonstrates that Zoletil significantly decreases neurological deficit of the ischemic animals to 4.5 (3.25-5.75) score as well as RIPC using Zoletil during preconditioning to 4 (3.0-5.75) score when compared with MCAO group (p < 0.05) (Fig. 4d).
Fig. 4. Neuroprotective effects of RIPC using Zoletil during preconditioning. a) Representative T2-weighted MR-images obtained 24 h after MCAO (sampling thickness 0.8 mm). Hyperintense regions (light area) refer to ischemic areas. Infarct volume (b) and brain swelling (c) evaluated by using MRI with analysis of T2-weighted MR-images. d) Neurological status was evaluated by a limb-placing test. Data are presented as median (thick black line), interquartile ranges, and minimum and maximum of the statistically significant data set; * denotes significant difference from the MCAO group p < 0.05, using one-way ANOVA, followed by Tukey’s post hoc analysis for (b) and (c); Kruskal–Wallis test with the Mann–Whitney U-test for (d).
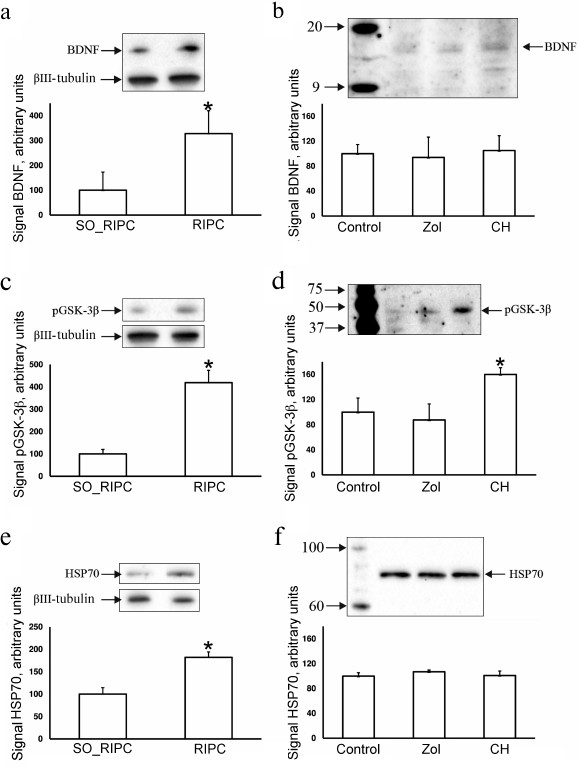
Investigation of ischemic tolerance signaling pathways. Taking into account data on the improvement of neurological outcomes after ischemia following pretreatment with RIPC, chloral hydrate, and Zoletil we analyzed some signaling pathways involved in the induction of brain ischemic tolerance. The levels of pGSK-3β, HSP70, and BDNF in the brain were measured 24 h after RIPC or intraperitoneal injection of anesthetics. The levels of BDNF and HSP70 increased up to 320 and 180%, respectively, in the animals exposed to RIPC (Fig. 5, a and e), and the brain level of pGSK-3β increased 4-fold in comparison with the SO_RIPC group (Fig. 5c). The level of EPO estimated using ELISA was 78 ± 18 pg/liter in the urine from animals after RIPC, which was not significantly different in comparison with the animals after sham preconditioning (104 ± 22 pg/liter). Chloral hydrate caused increase in only pGSK-3β by 60% (Fig. 5d) in comparison with the control animals, while Zoletil did not affect the levels of pGSK-3β, HSP70, and BDNF in the brain (Fig. 5, b, d, and f).
Fig. 5. Exploration of ischemic tolerance signaling pathways. Evaluation of concentration of BDNF (a, b), pGSK-3β (c, d), and HSP70 (e, f) in the brain 24 h after RIPC without anesthesia or intraperitoneal injection of anesthetics using Western blotting. Representative Western blots with averaged corresponding densitometry are presented. Band densities are normalized to the density of βIII-tubulin band.
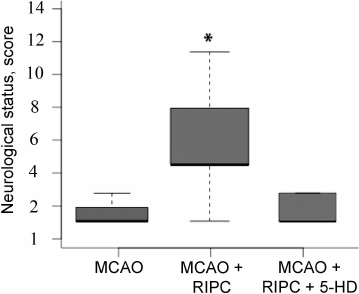
Administration of the KATP-channel inhibitor 5-HD completely abolishes the neuroprotective effect of RIPC, resulting in decrease in the score in neurological testing to the levels observed in animals from the MCAO group (Fig. 6). The volume of damage did not change significantly, remaining at a level similar to the MCAO group.
Fig. 6. 5-Hydroxydecanoate (5-HD) abolishes the neuroprotective effect of RIPC. Data are presented as median (black line), interquartile range, and maximum and minimum for the statistically significant data set; * p < 0.05 in comparison with the MCAO group (Mann–Whitney U-test).
DISCUSSION
In this study, we confirmed the possibility of brain protection from adverse effects of ischemia/reperfusion by the technique of remote ischemic preconditioning of hind limbs. Although we were able to separate the effects of RIPC from the effects of anesthesia used during preconditioning, our results indicated the possibility of complex interactions between the protective effects of anesthetics and preconditioning itself.
According to our results, RIPC performed 24 h prior to the ischemia modeling in the absence of anesthetics did not affect the infarct volume and the level of brain swelling; however, it improved significantly the sensorimotor function one day after the stroke. It is important to mention that the improvement in sensorimotor functions occurred in all groups subjected to RIPC either with anesthesia or without it, but there were no statistically significant differences observed in the improvement of the neurological status between these groups of animals. Hence, it can be stated that there is no synergism or antagonism in the interaction of the investigated anesthetics and RIPC.
Chloral hydrate administered to rats 24 h before induction of ischemia did not affect the degree of brain injury similarly to RIPC, but it improved sensorimotor functions. Based on these data, we suggest that chloral hydrate itself produces a preconditioning effect with efficiency similar to RIPC. The data on neuroprotective effects of chloral hydrate are in agreement and corroborate the results of earlier study of Liu and coauthors, where it was demonstrated that the i.p. injection of chloral hydrate 1 h prior to MCAO significantly improved neurological status of the rats, as well as decreased the degree of brain injury and swelling after ischemia via increase in the level of annexin A1 [22]. In addition, there are indications of the systemic antiinflammatory action of chloral hydrate, which can be involved in the neuroprotective mechanism of chloral hydrate [26]. A study using cardiomyocytes also demonstrated that chloral hydrate potentiated opening of KATP-channels [27], which could be the part of its neuroprotective mechanism, as the KATP-channels play an important role in the initial phase of ischemic preconditioning [1].
The other explored anesthetic – Zoletil – containing equal parts of an NMDA-receptor antagonist (tiletamine) and GABA-receptor agonist (zolazepam) is often used in experimental surgery due to several advantages, such as rapid anesthesia induction, highly efficient muscle relaxation, and smooth recovery from anesthesia [28]. In our experiments, Zoletil provided pronounced neuroprotective effect, decreasing the volume of infarct and brain swelling. Similar results were obtained for the RIPC group with the use of Zoletil. Equal and statistically significant decrease in neurological deficit was also demonstrated for both groups subjected to administration of only Zoletil or Zoletil with preconditioning. Considering that Zoletil is a two-component drug, the mechanism of its preconditioning action can be complex. On one hand, NMDA-receptor antagonists can induce signals similar to preconditioning [29, 30]. On the other hand, GABA-receptor antagonists can exhibit action similar to preconditioning and afford protective effect to heart; however, agonists such as zolazepam do not demonstrate this property [31]. It is worth mentioning that in this study Zoletil was used at a relatively high dose (40 mg/kg), which can inhibit various sites in the respiratory chain in addition to the effect on NMDA- and GABA-receptors. Such inhibition can enhance reactive oxygen species (ROS) generation, which in turn can induce ischemic tolerance via signaling mediated by protein kinase C and KATP-channels [32, 33].
We explored the levels of BDNF, HSP70, and pGSK-3β proteins associated with the induction of brain ischemic tolerance caused by RIPC and anesthetics to establish the architecture of the protective signaling pathways. It is well known that BDNF belongs to the neurotrophin family, members of which facilitate growth, differentiation, and survival of neurons. Ischemic or hypoxic brain preconditioning increases the expression of BDNF mRNA in rat brain [34, 35], and the introduction of BDNF into the brain ventricles causes neuroprotective effect demonstrated in different models of brain ischemia including MCAO [36]. We also showed that the BDNF levels in brain increased several-fold one day after RIPC, which could initiate neuroprotective mechanisms induced by BDNF. In addition, RIPC resulted in an increase in HSP70 expression in the brain. Earlier, similar results were obtained in the model of global ischemia, where RIPC caused an increase in HSP70 content in the CA1 region of the hippocampus, while HSP70 inhibitors (quercetin or SB 203580) abolished the protective effect of preconditioning [37]. In numerous studies it has been shown that increase in HSP70 levels in brain was associated with neuroprotection; for example, hyperexpression of HSP70 in transgenic mice increased the survival of neurons following focal ischemia [38]. Participation of HSP70 in the induction of ischemic tolerance following ischemic preconditioning has been clearly demonstrated [39-41].
Most authors consider the mitochondrial KATP-channel as one of the key members of ischemic tolerance pathway in brain and other organs. Opening of these channels causes slight swelling of mitochondria that facilitates generation of signaling ROS, stimulates protein kinase C, and leads to phosphorylation of the GSK-3β, which in turn inhibits opening of the mitochondrial pore [42]. The role of GSK-3β in RIPC remains poorly understood, but the increase in the amount of the phosphorylated form of GSK-3β in kidneys one day after short-term limb ischemia has been demonstrated in a recent study [43]. Previously, we showed that increase in phosphorylation of this kinase is the basis for the action of some nephro- and neuroprotectors, as well as it is observed in the kidney preconditioning [7, 44]. In this work, we also studied the involvement of this mechanism in the induction of neuroprotection, demonstrating that the levels of pGSK-3β increased one day after RIPC induction or chloral hydrate injection. We suggest that phosphorylation of GSK-3β causes the increase in threshold of mitochondrial nonspecific pore opening and protection of cells from death induced by ischemia/reperfusion.
We have previously shown that the increase in the levels of EPO in kidneys is one of the possible mechanisms of generation of protective stimuli in response to remote kidney preconditioning [7]. Similar results were obtained in a study deciphering the mechanisms of RIPC [45], where the authors explained the increase in EPO by the decrease in renal blood flow as a response to RIPC, which resulted in activation of the HIF-1α-dependent pathway caused by EPO. In our study, we did not observe changes in EPO levels in the 24-h urine sample of animals, which was in agreement with data obtained by Malhotra and colleagues [46]. Such contradictions of experimental results could likely be related to the use of different schemes of the preconditioning protocol and administration of anesthetics, which again emphasizes the need for investigation of the effect of pharmacological intervention on the performance of the RIPC.
We conclude that remote ischemic preconditioning of hind limbs without prior use of anesthetics results in restoration of brain sensorimotor functions in ischemia/reperfusion modeling. On one hand, preconditioning of hind limbs accompanied by administered anesthetics does not enhance neuroprotection. On the other hand, the protective effects of RIPC cannot be detected because the anesthetics exhibit the similar effect. These data indicate that for clean investigation of the protective effects and signaling pathways associated with RIPC in experimental studies, prior usage of anesthetics must be avoided.
The fact that anesthetics themselves demonstrate protective effect without added RIPC can be explained by the implementation of their neuroprotective action via similar mechanisms. This similarity can lie in the fact that neuroprotection correlates with the level of phosphorylation of GSK-3β in brain tissue, which according to the numerous data is the convergence point of different protective pathways. Eventually, all the signaling protective pathways of ischemic and pharmacological preconditioning (including the ones observed in this study during RIPC and administration of chloral hydrate) keep this enzyme in a phosphorylated (inhibited) form as the terminal element of the pathway [42]. Previous data pointing that the main role of GSK-3β is confined by the prevention of induction of nonspecific mitochondrial permeability, which is a point of no return in the cell death cascade, brings both this kinase and the mitochondrion as a whole to the center of the researcher’s attention as a target for using known and developing novel protective pharmacological agents.
We did not observe any changes in the level of GSK-3β phosphorylation in the animals subjected to Zoletil injections, but this drug exhibited a pronounced neuroprotective effect, causing decrease in brain injury volume and swelling, unlike the RIPC and chloral hydrate. These facts could imply that Zoletil exhibits another mechanism of neuroprotective action. Hence, separate extended studies of the effects of anesthetics on the signaling pathways associated with the induction of ischemic tolerance are required. Clinical and experimental studies of the phenomenon of ischemic preconditioning must be conducted with consideration of the possible effect of anesthetics on the final clinical effect.
Acknowledgments
This work was financially supported by the Russian Foundation for Basic Research (project No. 15-34-20074, stroke modeling and neurological studies) and by the Russian Science Foundation (project No. 14-24-00107, investigation of brain with magnetic resonance imaging).
REFERENCES
1.Silachev, D. N., Plotnikov, E. Y., Pevzner, I. B.,
Zorova, L. D., Babenko, V. A., Zorov, S. D., Popkov, V. A., Jankauskas,
S. S., Zinchenko, V. P., Sukhikh, G. T., and Zorov, D. B. (2014) The
mitochondrion as a key regulator of ischaemic tolerance and injury,
Heart Lung Circ., 23, 897-904.
2.Schott, R. J., Rohmann, S., Braun, E. R., and
Schaper, W. (1990) Ischemic preconditioning reduces infarct size in
swine myocardium, Circ. Res., 66, 1133-1142.
3.Cochrane, J., Williams, B. T., Banerjee, A.,
Harken, A. H., Burke, T. J., Cairns, C. B., and Shapiro, J. I. (1999)
Ischemic preconditioning attenuates functional, metabolic, and
morphologic injury from ischemic acute renal failure in the rat,
Ren. Fail, 21, 135-145.
4.Mounsey, R. A., Pang, C. Y., Boyd, J. B., and
Forrest, C. (1992) Augmentation of skeletal muscle survival in the
latissimus dorsi porcine model using acute ischemic preconditioning,
J. Otolaryngol., 21, 315-320.
5.Malhotra, S., Naggar, I., Stewart, M., and
Rosenbaum, D. M. (2011) Neurogenic pathway mediated remote
preconditioning protects the brain from transient focal ischemic
injury, Brain Res., 1386, 184-190.
6.Ren, C., Gao, X., Steinberg, G. K., and Zhao, H.
(2008) Limb remote-preconditioning protects against focal ischemia in
rats and contradicts the dogma of therapeutic time windows for
preconditioning, Neuroscience, 151, 1099-1103.
7.Silachev, D. N., Isaev, N. K., Pevzner, I. B.,
Zorova, L. D., Stelmashook, E. V., Novikova, S. V., Plotnikov, E. Y.,
Skulachev, V. P., and Zorov, D. B. (2012) The mitochondria-targeted
antioxidants and remote kidney preconditioning ameliorate brain damage
through kidney-to-brain cross-talk, PLoS One, 7,
e51553.
8.Koch, S., Katsnelson, M., Dong, C., and
Perez-Pinzon, M. (2011) Remote ischemic limb preconditioning after
subarachnoid hemorrhage: a phase Ib study of safety and feasibility,
Stroke, 42, 1387-1391.
9.Hougaard, K. D., Hjort, N., Zeidler, D., Sorensen,
L., Norgaard, A., Hansen, T. M., Von Weitzel-Mudersbach, P., Simonsen,
C. Z., Damgaard, D., Gottrup, H., Svendsen, K., Rasmussen, P. V., Ribe,
L. R., Mikkelsen, I. K., Nagenthiraja, K., Cho, T. H., Redington, A.
N., Botker, H. E., Ostergaard, L., Mouridsen, K., and Andersen, G.
(2014) Remote ischemic preconditioning as an adjunct therapy to
thrombolysis in patients with acute ischemic stroke: a randomized
trial, Stroke, 45, 159-167.
10.Candilio, L., Malik, A., Ariti, C., Barnard, M.,
Di Salvo, C., Lawrence, D., Hayward, M., Yap, J., Roberts, N., Sheikh,
A., Kolvekar, S., Hausenloy, D. J., and Yellon, D. M. (2015) Effect of
remote ischaemic preconditioning on clinical outcomes in patients
undergoing cardiac bypass surgery: a randomized controlled clinical
trial, Heart, 101, 185-192.
11.Meybohm, P., Bein, B., Brosteanu, O., Cremer, J.,
Gruenewald, M., Stoppe, C., Coburn, M., Schaelte, G., Boning, A.,
Niemann, B., Roesner, J., Kletzin, F., Strouhal, U., Reyher, C.,
Laufenberg-Feldmann, R., Ferner, M., Brandes, I. F., Bauer, M., Stehr,
S. N., Kortgen, A., Wittmann, M., Baumgarten, G., Meyer-Treschan, T.,
Kienbaum, P., Heringlake, M., Schon, J., Sander, M., Treskatsch, S.,
Smul, T., Wolwender, E., Schilling, T., Fuernau, G., Hasenclever, D.,
and Zacharowski, K. (2015) A multicenter trial of remote ischemic
preconditioning for heart surgery, N. Engl. J. Med., 373,
1397-1407.
12.Hausenloy, D. J., Candilio, L., Evans, R., Ariti,
C., Jenkins, D. P., Kolvekar, S., Knight, R., Kunst, G., Laing, C.,
Nicholas, J., Pepper, J., Robertson, S., Xenou, M., Clayton, T., and
Yellon, D. M. (2015) Remote ischemic preconditioning and outcomes of
cardiac surgery, N. Engl. J. Med., 373, 1408-1417.
13.Jones, B. O., Pepe, S., Sheeran, F. L., Donath,
S., Hardy, P., Shekerdemian, L., Penny, D. J., McKenzie, I., Horton,
S., Brizard, C. P., d’Udekem, Y., Konstantinov, I. E., and
Cheung, M. M. (2013) Remote ischemic preconditioning in cyanosed
neonates undergoing cardiopulmonary bypass: a randomized controlled
trial, J. Thorac. Cardiovasc. Surg., 146, 1334-1340.
14.Hess, D. C., Blauenfeldt, R. A., Andersen, G.,
Hougaard, K. D., Hoda, M. N., Ding, Y., and Ji, X. (2015) Remote
ischaemic conditioning – a new paradigm of self-protection in the
brain, Nat. Rev. Neurol., 11, 698-710.
15.Zangrillo, A., Musu, M., Greco, T., Di Prima, A.
L., Matteazzi, A., Testa, V., Nardelli, P., Febres, D., Monaco, F.,
Calabro, M. G., Ma, J., Finco, G., and Landoni, G. (2015) Additive
effect on survival of anesthetic cardiac protection and remote ischemic
preconditioning in cardiac surgery: a Bayesian network meta-analysis of
randomized trials, PLoS One, 10, e0134264.
16.Zwerus, R., and Absalom, A. (2015) Update on
anesthetic neuroprotection, Curr. Opin. Anaesthesiol.,
28, 424-430.
17.Swyers, T., Redford, D., and Larson, D. F. (2014)
Volatile anesthetic-induced preconditioning, Perfusion,
29, 10-15.
18.Kapinya, K. J., Lowl, D., Futterer, C., Maurer,
M., Waschke, K. F., Isaev, N. K., and Dirnagl, U. (2002) Tolerance
against ischemic neuronal injury can be induced by volatile anesthetics
and is inducible NO synthase dependent, Stroke, 33,
1889-1898.
19.Zhang, H., Xiong, X., Liu, J., Gu, L., Li, F.,
Wan, Y., and Xu, S. (2016) Emulsified isoflurane protects against
transient focal cerebral ischemia injury in rats via the PI3K/Akt
signaling pathway, Anesth. Analg., 122, 1377-1384.
20.Bickler, P. E., and Fahlman, C. S. (2006) The
inhaled anesthetic, isoflurane, enhances Ca2+-dependent
survival signaling in cortical neurons and modulates MAP kinases,
apoptosis proteins and transcription factors during hypoxia, Anesth.
Analg., 103, 419-429.
21.Ye, Z., Xia, P., Cheng, Z. G., and Guo, Q. (2015)
Neuroprotection induced by sevoflurane-delayed post-conditioning is
attributable to increased phosphorylation of mitochondrial GSK-3β
through the PI3K/Akt survival pathway, J. Neurol. Sci.,
348, 216-225.
22.Liu, J. H., Feng, D., Zhang, Y. F., Shang, Y.,
Wu, Y., Li, X. F., and Pei, L. (2015) Chloral hydrate preconditioning
protects against ischemic stroke via upregulating annexin A1, CNS
Neurosci. Ther., 21, 718-726.
23.Longa, E. Z., Weinstein, P. R., Carlson, S., and
Cummins, R. (1989) Reversible middle cerebral artery occlusion without
craniectomy in rats, Stroke, 20, 84-91.
24.Jolkkonen, J., Puurunen, K., Rantakomi, S.,
Harkonen, A., Haapalinna, A., and Sivenius, J. (2000) Behavioral
effects of the alpha(2)-adrenoceptor antagonist, atipamezole, after
focal cerebral ischemia in rats, Eur. J. Pharmacol., 400,
211-219.
25.Silachev, D. N., Uchevatkin, A. A., Pirogov, Y.
A., Zorov, D. B., and Isaev, N. K. (2009) Comparative evaluation of two
methods for studies of experimental focal ischemia: magnetic resonance
tomography and triphenyltetrazolium detection of brain injuries,
Bull. Exp. Biol. Med., 147, 269-272.
26.Pan, Q., Liu, Y., Zheng, J., Lu, X., Wu, S., Zhu,
P., and Fu, N. (2010) Protective effect of chloral hydrate against
lipopolysaccharide/D-galactosamine-induced acute lethal liver injury
and zymosan-induced peritonitis in mice, Int. Immunopharmacol.,
10, 967-977.
27.Zaugg, M., Lucchinetti, E., Spahn, D. R., Pasch,
T., Garcia, C., and Schaub, M. C. (2002) Differential effects of
anesthetics on mitochondrial KATP channel activity and
cardiomyocyte protection, Anesthesiology, 97, 15-23.
28.Ferrari, L., Turrini, G., Rostello, C., Guidi,
A., Casartelli, A., Piaia, A., and Sartori, M. (2005) Evaluation of two
combinations of Domitor, Zoletil 100, and Euthatal to obtain long-term
nonrecovery anesthesia in Sprague–Dawley rats, Comp. Med.,
55, 256-264.
29.Slomka, M., Kuszczyk, M., Lazarewicz, J. W., and
Makarewicz, D. (2014) NMDA receptor antagonists MK-801 and memantine
induce tolerance to oxygen and glucose deprivation in primary cultures
of rat cerebellar granule cells, Acta Neurobiol. Exp. (Wars.),
74, 396-404.
30.Makarewicz, D., Sulejczak, D., Duszczyk, M.,
Malek, M., Slomka, M., and Lazarewicz, J. W. (2014) Delayed
preconditioning with NMDA receptor antagonists in a rat model of
perinatal asphyxia, Folia Neuropathol., 52, 270-284.
31.Leducq, N., Bono, F., Sulpice, T., Vin, V.,
Janiak, P., Fur, G. L., O’Connor, S. E., and Herbert, J. M.
(2003) Role of peripheral benzodiazepine receptors in mitochondrial,
cellular, and cardiac damage induced by oxidative stress and
ischemia-reperfusion, J. Pharmacol. Exp. Ther., 306,
828-837.
32.Rivo, J., Raphael, J., Drenger, B., Berenshtein,
E., Chevion, M., and Gozal, Y. (2006) Flumazenil mimics whereas
midazolam abolishes ischemic preconditioning in a rabbit heart model of
ischemia-reperfusion, Anesthesiology, 105, 65-71.
33.Zhang, H. Y., McPherson, B. C., Liu, H., Baman,
T. S., Rock, P., and Yao, Z. (2002) H2O2 opens
mitochondrial KATP channels and inhibits GABA receptors via
protein kinase C-epsilon in cardiomyocytes, Am. J. Physiol. Heart
Circ. Physiol., 282, H1395-1403.
34.Neumann, J. T., Thompson, J. W., Raval, A. P.,
Cohan, C. H., Koronowski, K. B., and Perez-Pinzon, M. A. (2015)
Increased BDNF protein expression after ischemic or PKC epsilon
preconditioning promotes electrophysiologic changes that lead to
neuroprotection, J. Cereb. Blood Flow Metab., 35,
121-130.
35.Samoilov, M., Churilova, A., Gluschenko, T., and
Rybnikova, E. (2014) Neocortical pCREB and BDNF expression under
different modes of hypobaric hypoxia: role in brain hypoxic tolerance
in rats, Acta Histochem., 116, 949-957.
36.Schabitz, W. R., Schwab, S., Spranger, M., and
Hacke, W. (1997) Intraventricular brain-derived neurotrophic factor
reduces infarct size after focal cerebral ischemia in rats, J.
Cereb. Blood Flow Metab., 17, 500-506.
37.Sun, X. C., Xian, X. H., Li, W. B., Li, L., Yan,
C. Z., Li, Q. J., and Zhang, M. (2010) Activation of p38 MAPK
participates in brain ischemic tolerance induced by limb ischemic
preconditioning by up-regulating HSP 70, Exp. Neurol.,
224, 347-355.
38.Plumier, J. C., Krueger, A. M., Currie, R. W.,
Kontoyiannis, D., Kollias, G., and Pagoulatos, G. N. (1997) Transgenic
mice expressing the human inducible Hsp70 have hippocampal neurons
resistant to ischemic injury, Cell Stress Chaperones, 2,
162-167.
39.Kirino, T., Tsujita, Y., and Tamura, A. (1991)
Induced tolerance to ischemia in gerbil hippocampal neurons, J.
Cereb. Blood Flow Metab., 11, 299-307.
40.Aoki, M., Abe, K., Kawagoe, J., Nakamura, S., and
Kogure, K. (1993) Acceleration of HSP70 and HSC70 heat shock gene
expression following transient ischemia in the preconditioned gerbil
hippocampus, J. Cereb. Blood Flow Metab., 13,
781-788.
41.Glazier, S. S., O’Rourke, D. M., Graham, D.
I., and Welsh, F. A. (1994) Induction of ischemic tolerance following
brief focal ischemia in rat brain, J. Cereb. Blood Flow Metab.,
14, 545-553.
42.Juhaszova, M., Zorov, D. B., Kim, S. H., Pepe,
S., Fu, Q., Fishbein, K. W., Ziman, B. D., Wang, S., Ytrehus, K.,
Antos, C. L., Olson, E. N., and Sollott, S. J. (2004) Glycogen synthase
kinase-3β mediates convergence of protection signaling to inhibit
the mitochondrial permeability transition pore, J. Clin.
Invest., 113, 1535-1549.
43.Liu, T., Fang, Y., Liu, S., Yu, X., Zhang, H.,
Liang, M., and Ding, X. (2015) Limb ischemic preconditioning protects
against contrast-induced acute kidney injury in rats via
phosphorylation of GSK-3β, Free Radic. Biol. Med.,
81, 170-182.
44.Plotnikov, E. Y., Chupyrkina, A. A., Jankauskas,
S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P., and Zorov, D.
B. (2011) Mechanisms of nephroprotective effect of
mitochondria-targeted antioxidants under rhabdomyolysis and
ischemia/reperfusion, Biochim. Biophys. Acta, 1812,
77-86.
45.Oba, T., Yasukawa, H., Nagata, T., Kyogoku, S.,
Minami, T., Nishihara, M., Ohshima, H., Mawatari, K., Nohara, S.,
Takahashi, J., Sugi, Y., Igata, S., Iwamoto, Y., Kai, H., Matsuoka, H.,
Takano, M., Aoki, H., Fukumoto, Y., and Imaizumi, T. (2015) Renal
nerve-mediated erythropoietin release confers cardioprotection during
remote ischemic preconditioning, Circ. J., 79,
1557-1567.
46.Malhotra, S., Naggar, I., Stewart, M., and
Rosenbaum, D. M. (2011) Neurogenic pathway mediated remote
preconditioning protects the brain from transient focal ischemic
injury, Brain Res., 1386, 184-190.