Interaction of Cholera Toxin B-subunit with Human T-lymphocytes
E. V. Navolotskaya1*, V. B. Sadovnikov1, D. V. Zinchenko1, Y. A. Zolotarev2, V. M. Lipkin3, and V. P. Zav’yalov4
1Branch of Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: navolotskaya@fibkh.serpukhov.su, elenanavolots@gmail.com2Institute of Molecular Genetics, Russian Academy of Sciences, 123182 Moscow, Russia
3Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia
4University of Turku, Turku, 20500, Finland
* To whom correspondence should be addressed.
Received May 4, 2017; Revision received June 2, 2017
In this work, 125I-labeled cholera toxin B-subunit (CT-B) (specific activity 98 Ci/mmol) was prepared, and its high-affinity binding to human blood T-lymphocytes (Kd = 3.3 nM) was determined. The binding of the 125I-labeled CT-B was inhibited by unlabeled interferon-α2 (IFN-α2), thymosin-α1 (TM-α1), and by the synthetic peptide LKEKK, which corresponds to sequences 16-20 of human TM-α1 and 131-135 of IFN-α2 (Ki 0.8, 1.2, and 1.6 nM, respectively), but was not inhibited by the unlabeled synthetic peptide KKEKL with inverted sequence (Ki > 1 µM). In the concentration range of 10-1000 nM, both CT-B and peptide LKEKK dose-dependently increased the activity of soluble guanylate cyclase (sGC) but did not affect the activity of membrane-bound guanylate cyclase. The KKEKL peptide tested in parallel did not affect sGC activity. Thus, the CT-B and peptide LKEKK binding to a common receptor on the surface of T-lymphocytes leads to an increase in sGC activity.
KEY WORDS: peptides, receptors, thymosin-α1, interferon-α, T-lymphocytesDOI: 10.1134/S0006297917090061
Abbreviations: cGMP, cyclic guanosine monophosphate; CT-B, cholera toxin B-subunit; HPLC, high performance liquid chromatography; IFN, interferon; IL, interleukin; iNOS, inducible NO-synthase; Kd, equilibrium dissociation constant; Ki, inhibition constant; mGC, membrane-bound guanylate cyclase; PMSF, phenylmethylsulfonyl fluoride; sGC, soluble guanylate cyclase; TM-α1, thymosin-α1.
During structural-functional studies on interferons-α
(IFNs-α), the octapeptide LKEKKYSP was earlier prepared, which is
fragment 131-138 of human IFN-α2, and it bound with
high affinity to mouse thymocytes and human fibroblasts [1, 2]. Unlabeled
IFN-α2 and thymosin-α1
(TM-α1) competitively inhibited the binding of the
labeled octapeptide. Comparison of amino acid sequences of the
octapeptide and TM-α1 revealed that they contained an
identical five-membered fragment LKEKK, which corresponds to sequences
16-20 of TM-α1 and 131-135 of IFN-α2.
We supposed that this fragment could be responsible for binding of
TM-α1 and IFN-α2 to various cell
types, and that the corresponding synthetic peptide could be capable of
binding and having biological activity.
We recently synthesized peptide LKEKK, which corresponds to sequences 16-20 of human TM-α1 and 131-135 of IFN-α2, prepared [3H]LKEKK, and showed that it binds with high affinity to T-lymphocytes isolated from donor blood (Kd = 3.7 nM) [3]. Binding of the labeled peptide was competitively inhibited by unlabeled TM-α1, IFN-α2, and cholera toxin B-subunit (CT-B) with Ki equal to 2.0, 2.2, and 3.6 nM, respectively. The KKEKL peptide with inverted sequence did not inhibit the binding of [3H]LKEKK (Ki > 10 µM), which indicated high specificity of the labeled peptide receptor. Treatment of the cells with proteases did not influence the binding, which indicated non-protein nature of the receptor. Based on these findings, we concluded that peptide LKEKK, TM-α1, IFN-α2, and CT-B should have a common non-protein receptor on T-lymphocytes. Moreover, we hypothesized that this receptor could be represented by the cholera toxin receptor GM1-ganglioside.
The purpose of the present work was to test this hypothesis. To do this, we prepared 125I-labeled CT-B, characterize its binding to donor blood T-lymphocytes, tested the ability of unlabeled TM-α1, IFN-α2 and peptides LKEKK and KKEKL to inhibit the binding, and studied the influence of CT-B and peptides LKEKK and KKEKL on the activity of soluble and membrane-bound guanylate cyclase.
MATERIALS AND METHODS
The following reagents were used in this work: human thymosin-α1 (Immundiagnostik AG, Germany); phenylmethylsulfonyl fluoride (PMSF) and Tris (Fluka, USA); media for cell cultivation, fetal calf serum, and Hepes (ICN, USA); sucrose, BSA, EDTA, EGTA, and sodium aside (NaN3) (Serva, Germany); all other reagents were of domestic production and were used after due purification.
Peptides LKEKK and KKEKL were synthesized with automated synthesizers (model 430A, Vega Coupler, and model C250, Applied Biosystems (USA)) using the corresponding Boc/Bzl-tactics of peptide synthesis and purified using preparative reversed-phase chromatography (Gilson chromatograph, France; Waters SymmetryPrep C18 column (19 × 300 mm), Malva, Greece). The synthesized peptides were characterized by data of analytic reversed-phase HPLC using the Gilson chromatograph and XTerra RP18 column (Malva) and amino acid analysis (hydrolysis with 6 M HCl for 24 h at 110°C; 4151 Alpha Plus amino acid analyzer (LKB, Sweden)), and mass spectrometry (Finnigan mass spectrometer, USA).
Human recombinant interferon-α2 was obtained from State Research Institute of Special Purity Biopreparations (St. Petersburg, Russia).
CT-B (20 µg) was labeled with 125I by the method of Salacinski et al. [4] using Na125I (1 mCi) and iodogen. To isolate the iodinated protein, the reaction mixture was placed on a column (0.9 × 10 cm) with Sephadex G-25. The column was eluted with 50 mM phosphate buffer (pH 7.4) at 5 ml/h. The elution peak of 125I-labeled CT-B was determined in a control experiment with unlabeled CT-B. The radioactivity of the fractions was measured with a Mini-Gamma Counter (LKB, Sweden). Fractions with the maximal radioactivity with positions corresponding to the unlabeled protein peak in the control were combined, and the total and specific activities of the resulting preparation were determined.
Mononuclear cells were isolated from blood of healthy donors as described by Boyum et al. [5]. T-cells were isolated by the method of Patel et al. [6] based on dense polystyrene granules loaded with mouse antibodies against human CD3. This method resulted in a preparation of the target T-cell population (CD3+) with >95% purity.
The 125I-labeled CT-B was bound to T-lymphocytes according to the following scheme: the cells (106 cells/ml) were incubated with the labeled protein (10–11-10–7 M, three parallel samples for each concentration) in 1 ml of RPMI-1640 medium containing 10 mM Hepes, 20 mM NaN3, and 50 µM PMSF, pH 7.4, at 4°C for 40 min. After the incubation, the reaction mixture was filtered through GF/A glass-fiber filters (Whatman, England). The filters were washed three times with 5 ml of ice-cold saline containing 10 mM Hepes (pH 7.4). Radioactivity was determined on the filters with the Mini-Gamma Counter. The specific binding of 125I-labeled CT-B to the cells was determined by the difference between its total and nonspecific binding, and the nonspecific binding of 125I-labeled CT-B was determined in the presence of 10–4 M unlabeled CT-B (1000-fold surplus with respect to the highest concentration of the labeled protein of 10–7 M). To determine the equilibrium dissociation constant Kd, the dependence was plotted of the ratio of molar concentrations of bound (B) and free (F) 125I-labeled CT-B on the molar concentration of the bound 125I-labeled CT-B (B) [7].
To evaluate the ability of TM-α1, IFN-α2, and of the LKEKK and KKEKL peptides to inhibit the specific binding of 125I-labeled CT-B to T-lymphocytes, the cells (106 cells/ml) were incubated with the labeled protein (3 nM) and one of the potential competitors (the concentration range was 10–10-10–5 M, three repeats for each concentration), as described above. The inhibition constant (Ki) was determined from the formula:
Ki = [IC]50/(1 + [L]/Kd) [8],
where [L] is molar concentration of 125I-labeled CT-B; Kd is the equilibrium dissociation constant of the 125I-labeled CT-B complex with the receptor; [IC]50 is the unlabeled ligand concentration that caused 50% inhibition of the specific binding of the labeled protein. The [IC]50 value was determined from the inhibition curve. The Kd value was determined earlier as described above.
To prepare subcellular fractions from T-lymphocytes, the cells were homogenized in 10 mM Tris-HCl (pH 7.4) on an ice bath, and the homogenate was centrifuged at 100,000g (10 min at 4°C). The supernatants were taken, and the precipitates were resuspended in equal volumes of the buffer solution. The supernatants and resuspended precipitates were used for determination of the activity of soluble guanylate cyclase (sGC) and membrane-bound guanylate cyclase (mGC), respectively [9]. The cells were preliminarily incubated with CT-B or with one of the peptides (concentration range 1-1000 nM) at 37°C for 30 min (increasing the incubation time neither increased the activity of sGC nor changed the activity of mGC). The guanylate cyclase activity was measured by monitoring the conversion of α-[³²P]GTP to [³²P]cGMP [10], and the product was isolated by precipitation with zinc carbonate and chromatography on a column with aluminum oxide [11]. The enzyme activity is expressed in pmol cGMP produced in 10 min per mg protein. The protein concentration was determined by the method of Lowry et al. [12] with BSA as a standard. Mean values were determined from data of three independent experiments. The significance of differences between the experimental and control data was assessed with Student’s t-test (p < 0.05).
RESULTS
The main characteristics of the peptides (purity, data of amino acid analysis, molecular weight) are presented in Table 1. The specific activity of 125I-labeled CT-B was 98 Ci/mmol.
Table 1. Main characteristics of
peptides
Binding of 125I-labeled CT-B to T-lymphocytes. The experiments revealed that under our conditions 125I-labeled CT-B specifically bound to donor blood T-lymphocytes. Figure 1 shows the dependence of the 125I-labeled CT-B specific binding to T-lymphocytes at 4°C on the incubation time. Note that dynamic equilibrium in the “labeled peptide–receptor” system was established after approximately 40 min. Therefore, to determine the Kd value, the binding of 125I-labeled CT-B to the cells was conducted for 40 min. The nonspecific binding of 125I-labeled CT-B to T-lymphocytes under these conditions was 9.2 ± 0.7% of its total binding.
Fig. 1. Dependence of total (1), specific (2), and nonspecific (3) binding of 125I-labeled CT-B to human blood T-lymphocytes on incubation time. Specific binding of labeled protein was determined as the difference between its total and nonspecific binding.
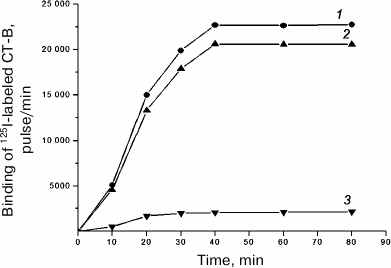
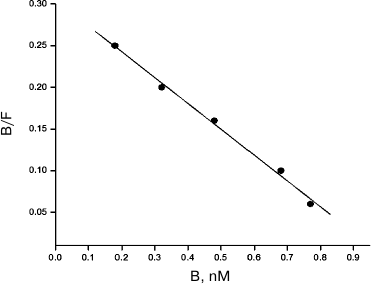
Figure 2 presents the Scatchard plot that describes the specific binding of 125I-labeled CT-B to T-lymphocytes. The linearity of the plot indicates that the T-cells have one receptor type to CT-B and the value Kd = 3.3 ± 0.2 nM shows high affinity of the protein to the receptor.
Fig. 2. Analysis in Scatchard coordinates of 125I-labeled CT-B specific binding to human blood T-lymphocytes. B and F are molar concentrations of the bound and free protein, respectively.
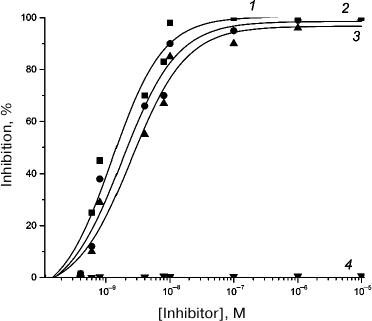
Evaluation of 125I-labeled CT-B binding to T-lymphocytes. To characterize the specificity of 125I-labeled CT-B binding to T-lymphocytes, unlabeled IFN-α2, TM-α1 and the LKEKK and KKEKL peptides were tested as potential competitors of the labeled protein. Table 2 presents the Ki values equal to 0.8 ± 0.2, 1.2 ± 0.2, and 1.6 ± 0.2 nM, respectively, and these values indicate that TM-α1, IFN-α2 and peptide LKEKK possess high inhibitory ability, whereas peptide KKEKL with the inverted sequence does not have such ability, its Ki > 1 µM (inhibition curves are presented in Fig. 3). The inability of peptide KKEKL to inhibit the binding indicates high specificity of the receptor.
Table 2. Inhibition by unlabeled ligands of
125I-labeled CT-B specific binding to human blood
T-lymphocytes
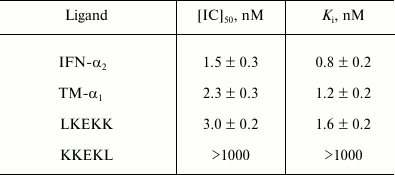
Fig. 3. Inhibition of 125I-labeled CT-B specific binding to rat small intestine epithelium membranes by unlabeled IFN-α2 (1), TM-α1 (2), and by synthetic peptides LKEKK (3) and KKEKL (4).
Influence of CT-B and peptides LKEKK and KKEKL on activity of sGC and mGC of T-lymphocytes. Data presented in Table 3 show that in the concentration range of 10-1000 nM both CT-B and peptide LKEKK have increased dose-dependently the activity of sGC and did not influence the activity of mGC. Peptide KKEKL with the inverted sequence, which was tested in parallel, did not influence the sGC activity, indicating high specificity of the action of CT-B and peptide LKEKK. Thus, CT-B and peptide LKEKK binding to the common receptor on the T-lymphocyte surface resulted in increase in sGC activity.
Table 3. Influence of CT-B and peptides
LKEKK and KKEKL on activity of soluble (sGC) and membrane-bound (mGC)
guanylate cyclase from human blood T-lymphocytes
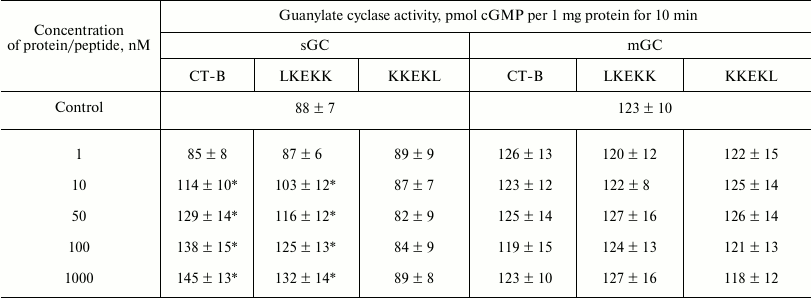
* p < 0.05.
DISCUSSION
Cholera toxin is a soluble hexameric protein produced by the gram-negative bacterium Vibrio cholerae. This protein consists of one A-subunit (CT-A) and five B-subunits (CT-B) [13]. In CT-A, there are two domains bound with a disulfide bond: CT-A1 is an enzyme that joins ADP-ribose to the regulatory Gsα-component of adenylate cyclase; CT-A2 is an α-helix localized inside a tunnel formed by CT-B [14]. CT-B forms a ring-shaped structure consisting of five monomers, each of which interacts with two neighboring molecules via hydrogen bonds. CT-B is responsible for toxin binding with monosialotetrahexosylganglioside (GM1a, Galβ3GalNAcβ4(Neu5Acα3)Galβ4GlcCer [15]) on the target cell surface [16, 17], and upon the binding the toxin enters the cell via endocytosis. CT-B is not toxic, and its receptor is present on the surface of most cells; therefore, this molecule is good as a carrier [18]. Moreover, according to many studies, CT-B is a good adjuvant, stimulates humoral immunity, and induces antiinflammatory mechanisms in vivo [19, 20]. However, pathways of the signal transmission from CT-B into the cells responsible for these effects are unknown.
Our studies have shown that 125I-labeled CT-B binds to donor blood T-lymphocytes with high affinity (Kd = 3.3 nM; Fig. 1). To characterize the binding specificity, unlabeled IFN-α2, TM-α1, peptide LKEKK, and peptide KKEKL with the inverted sequence were tested as potential competitors of 125I-labeled CT-B. Data presented in Table 2 demonstrate that IFN-α2, TM-α1 and peptide LKEKK have high inhibitory ability (the Ki values are, respectively, 0.8, 1.2, and 1.6 nM; the inhibition curves are presented in Fig. 3). The inability of peptide KKEKL to inhibit the binding (Ki > 1 µM) indicates that the receptor is highly specific. Our previous studies have shown that 3H-labeled synthetic peptide LKEKK binds with high affinity to a non-protein receptor on donor blood T-lymphocytes [3] and on membranes of rat small intestine epithelial cells [21]. In both cases, the binding was competitively inhibited by unlabeled IFN-α2, TM-α1, and CT-B. However, in the present work unlabeled IFN-α2, TM-α1, and peptide LKEKK were shown to inhibit the binding of 125I-labeled CT-B to its receptor on T-lymphocytes. These findings, as well as the cholera toxin lacking other receptors besides GM1-ganglioside [16, 17], indicate rather certainly that just GM1-ganglioside is the common receptor for CT-B, IFN-α2, TM-α1, and peptide LKEKK. Moreover, the ability of peptide LKEKK to bind to the receptor with high affinity and specificity indicates that the sequences 16-20 of TM-α1 and 131-135 of IFN-α2 are involved in the binding.
We found earlier that CT-B did not change the activity of adenylate cyclase [21]. The influence of CT-B on guanylate cyclase activity was not investigated. Mammalian cells are known to possess two enzymes synthesizing cGMP: soluble guanylate cyclase (sGC) and membrane-bound guanylate cyclase (mGC). sGC is a heteromer consisting of α- and β-subunits that is activated by direct interaction of NO with the β-subunit heme [22]. The genome of mammals encodes seven mGCs (from mGC-A to mGC-G), and they all are similar in topology: an extracellular ligand-binding domain, a short transmembrane region, and an intracellular domain with a catalytic site on the C-end [23].
The results of the present work revealed that in the concentration range of 10-1000 nM CT-B and peptide LKEKK enhanced dose-dependently the activity of sGC, but did not influence the activity of mGC (Table 3). Peptide KKEKL with the inverted sequence, which was tested in parallel, did not influence the activity of sGC, and this evidenced high specificity of the action of CT-B and peptide LKEKK on the cells. Thus, binding of CT-B and peptide LKEKK to a common receptor on the T-lymphocyte surface enhances the activity of sGC.
Both NO and sGC are known to play an important role in the regulation of the activity of T-cells [24]. There are data indicating that T-cellular inducible NO-synthase (iNOS) and NO play a crucial suppressing role in the control of T-helper differentiation [25-27]. NO was shown to inhibit T-cells in the G1-phase and to induce apoptosis through activation of the sGC-dependent protein kinase G [28]. However, NO was found to display a suppressing effect only at high concentrations (>100 µM), whereas its low concentrations (5-25 µM) selectively increased the differentiation of Th1-cells and did not influence the differentiation of Th2-cells [29, 30]. The authors demonstrated that the activating effect of low concentrations of NO was mediated through cGMP and was manifested selectively on Th1-cells: NO activated sGC, which led to an increase in the level of cGMP that activated expression of the IL-12 receptor β2-subunit but did not influence the IL-4 receptor. Since IL-12 and IL-4 are key cytokines in the induction of Th1- and Th2-cells, respectively, they are responsible for the selective action of NO on the differentiation of T-cells. Thus, low doses of NO promote the differentiation of Th1-cells by the selective induction of IL-12Rβ2 via the sGC-cGMP-dependent pathway. Note that after infections, in iNOS-deficient mice an increased Th1-response is developing, which is accompanied by an increase in the level of IFN-γ and a decrease in the level of IL-4 [31-33]. These data show that NO selectively suppresses the expansion of Th1-cells through negative feedback that can be realized due to inhibition of IL-12 synthesis by activated macrophages [34]. This mechanism might be very useful in inflammatory diseases mainly mediated through Th1-cells. By contrast, a strong Th1-cell response is very desirable for the effective protection of the organism against intracellular pathogens.
As mentioned above, CT-B is now considered as a promising immunomodulating agent. Therefore, establishing the molecular mechanism of action of this protein is an important problem that must be solved for introduction into medical practice. The ability of CT-B and peptide LKEKK to enhance the activity of T-cellular sGC makes reasonable further study in detail of the influence of each of them on the sGC-cGMP pathway of signal transmission and activities mediated through it.
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (project No. 14-04-00177) and by the Russian Academy of Sciences Presidium Program “Molecular and Cell Biology” (the manager is V. M. Lipkin).
REFERENCES
1.Zav’yalov, V. P., Navolotskaya, E. V.,
Abramov, V. M., Galaktionov, V. G., Isaev, I. S., Kaurov, O. A.,
Kozhich, A. T., Maiorov, V. A., Prusakov, A. N., Vasilenko, R. N., and
Volodina, E. Y. (1991) The octapeptide corresponding to the region of
the highest homology between α-interferon and
thymosin-α1 effectively competes with both cytokines
for common high-affinity receptors on murine thymocytes, FEBS
Lett., 278, 187-189.
2.Zav’yalov, V. P., Navolotskaya, E. V.,
Vasilenko, R. N., Abramov, V. M., Volodina, E. Y., Roslovtseva, O. A.,
Prusakov, A. N., and Kaurov, O. A. (1995) The sequence 130-137 of human
interferon-α2 is involved in the competition of interferon,
prothymosin α and cholera toxin B subunit for common receptors on
human fibroblasts, Mol. Immunol., 32, 425-431.
3.Navolotskaya, E. V., Zinchenko, D. V., Zolotarev,
Y. A., Kolobov, A. A., and Lipkin, V. M. (2016) Binding of synthetic
LKEKK peptide to human T-lymphocytes, Biochemistry (Moscow),
81, 871-875.
4.Salacinski, P. R., McLean, C., Sykes, J. E.,
Clement-Jones, V. V., and Lowry, P. J. (1981) Iodination of proteins,
glycoproteins, and peptides using a solid-phase oxidizing agent,
1,3,4,6-tetrachloro-3 alpha,6 alpha-diphenyl glycoluril (Iodogen),
Anal. Biochem., 117, 136-146.
5.Boyum, A., Berg, T., and Blomhoff, R. (1983) in
Iodinated Density Gradient Media – A Practical
Approach (Rickwood, D., ed.) Oxford, pp. 147-170.
6.Patel, D., Rubbi, C. P., and Rickwood, D. (1995)
Separation of T- and B-lymphocytes from human peripheral blood
mononuclear cells using density perturbation methods, Clin. Chim.
Acta, 240, 187-193.
7.Pennock, B. E. (1973) A calculator for finding
binding parameters from a Scatchard plot, Anal. Biochem.,
56, 306-309.
8.Cheng, Y. C., and Prusoff, W. (1973) Relationship
between the inhibition constant (Ki) and the
concentration of inhibitor which causes 50% inhibition
(IC50) of an enzymatic reaction, Biochem. Pharmacol.,
22, 3099-3108.
9.Carpentieri, U., Minguell, J. J., and Gardner, F.
H. (1981) Adenylate cyclase and guanylate cyclase activity in normal
and leukemic human lymphocytes, Blood, 57, 975-978.
10.Schultz, G., and Bohme, E. (1984) in Methods
of Enzymatic Analysis, Verlag Chemie, Weinheim, Germany, pp.
379-389.
11.Southam, E. (2001) Measurement of cGMP and
soluble guanylyl cyclase activity, Curr. Protoc. Toxicol.,
10, 10.5.
12.Lowry, O. H., Rosebrough, N. J., Farr, O. L., and
Randal, R. J. (1951) Protein measurement with the Folin phenol reagent,
J. Biol. Chem., 193, 265-275.
13.Lonnroth, I., and Holmgren, J. (1973) Subunit
structure of cholera toxin, J. Gen. Microbiol., 76,
417-427.
14.Merritt, E. A., Sarfaty, S., Akker, F. V. D.,
L’Hoir, C., Martial, J. A., and Hol, W. G. J. (1994) Crystal
structure of cholera toxin B-pentamer bound to receptor GM1
pentasaccharide, Protein Sci., 3, 166-175.
15.Chester, M. A. (1998) IUPAC-IUB joint commission
on biochemical nomenclature (JCBN). Nomenclature of glycolipids –
recommendations 1997, Eur. J. Biochem., 257, 293-298.
16.Holmgren, J., Lonnroth, I., and Svennerholm, L.
(1973) Tissue receptor for cholera exotoxin: postulated structure from
studies with GM ganglioside and related glycolipids, Infect.
Immun., 8, 208-214.
17.Schoen, A., and Freire, E. (1989) Thermodynamics
of intersubunit interactions in cholera toxin upon binding to the
oligosaccharide portion of its cell surface receptor, ganglioside GM1,
Biochemistry, 8, 5019-5024.
18.Kozireski-Chuback, D., Wu, G., and Ledeen, R. W.
(1999) Developmental appearance of nuclear GM1 in neurons of the
central and peripheral nervous systems, Dev. Brain Res.,
115, 201-208.
19.Baldauf, K. J., Royal, J. M., Hamorsky, K. T.,
and Matoba, N. (2015) Cholera toxin B: one subunit with many
pharmaceutical applications, Toxins, 7, 974-996.
20.Stratmann, T. (2015) Cholera toxin subunit B as
adjuvant – an accelerator in protective immunity and a break in
autoimmunity, Vaccines (Basel), 3, 579-596.
21.Navolotskaya, E. V., Sadovnikov, V. B.,
Zinchenko, D. V., Vladimirov, V. I., Zolotarev, Y. A., and Kolobov, A.
A. (2016) The LKEKK synthetic peptide as a ligand of rat intestinal
epithelial cell membranes, Russ. J. Bioorg. Chem., 42,
479-483.
22.Kots, A. Y., Martin, E., Sharina, I. G., and
Murad, F. (2009) A short history of cGMP, guanylyl cyclases, and
cGMP-dependent protein kinases, Handb. Exp. Pharmacol.,
191, 1-14.
23.Kuhn, M. (2016) Molecular physiology of membrane
guanylyl cyclase receptors, Physiol. Rev., 96,
751-804.
24.Niedbala, W., Cai, B., and Liew, F. Y. (2006)
Role of nitric oxide in the regulation of T-cell functions, Ann.
Rheumatic Dis., 65, 37-40.
25.Nath, N., Morinaga, O., and Singh, I. (2010)
S-Nitrosoglutathione a physiologic nitric oxide carrier attenuates
experimental autoimmune encephalomyelitis, J. Neuroimmune
Pharmacol., 5, 240-251.
26.Lee, S. W., Choi, H., Eun, S. Y., Fukuyama, S.,
and Croft, M. (2011) Nitric oxide modulates TGF-β-directive
signals to suppress Foxp3+ regulatory T cell differentiation
and potentiate Th1 development, J. Immunol., 186,
6972-6980.
27.Yang, J., Zhang, R., Lu, G., Shen, Y., Peng, L.,
Zhu, C., Cui, M., Wang, W., Arnaboldi, P., Tang, M., Gupta, M., Qi, C.
F., Jayaraman, P., Zhu, H., Jiang, B., Chen, S. H., He, J. C., Ting, A.
T., Zhou, M. M., Kuchroo, V. K., Morse, H. C., Ozato, K., Sikora, A.
G., and Xiong, H. (2013) T-cell-derived inducible nitric oxide synthase
switches off Th17 cell differentiation, J. Exp. Med.,
210, 1447-1462.
28.Valenti, L., Mathieu, J., Chancerelle, Y.,
Levacher, M., Chanaud, B., De Sousa, M., Strzalko, S., Dinh-Xuan, A.
T., Giroud, J. P., and Florentin, I. (2003) Nitric oxide inhibits
spleen cell proliferative response after burn injury by inducing
cytostasis, apoptosis, and necrosis of activated T-lymphocytes: role of
the guanylate cyclase, Cell. Immunol., 221, 50-63.
29.Niedbala, W., Wei, X. Q., Piedrafita, D., Xu, D.,
and Liew, F. Y. (1999) Effects of nitric oxide on the induction and
differentiation of Th1 cells, Eur. J. Immunol., 29,
2498-2505.
30.Niedbala, W., Wei, X. Q., Campbell, C., Thomson,
D., Komai-Koma, M., and Liew, F. Y. (2002) Nitric oxide preferentially
induces type 1 T-cell differentiation by selectively up-regulating
IL-12 receptor β2 expression via cGMP, Proc. Natl. Acad. Sci.
USA, 99, 16186-16191.
31.Wei, X. Q., Charles, I. G., Smith, A., Ure, J.,
Feng, G. J., Huang, F. P., Xu, D., Muller, W., Moncada, S., and Liew,
F. Y. (1995) Altered immune responses in mice lacking inducible nitric
oxide synthase, Nature, 375, 408-411.
32.McInnes, I. B., Leung, B., Wei, X. Q., Gemmell,
C. C., and Liew, F. Y. (1998) Septic arthritis following
Staphylococcus aureus infection in mice lacking inducible nitric
oxide synthase, J. Immunol., 160, 308-315.
33.MacLean, A., Wei, X. Q., Huang, F. P., Al-Alem,
U. A., Chan, W. L., and Liew, F. Y. (1998) Mice lacking inducible
nitric-oxide synthase are more susceptible to herpes simplex
virus infection despite enhanced Th1 cell responses, J. Gen.
Virol., 79, 825-830.
34.Huang, F. P., Niedbala, W., Wei, X. Q., Xu, D.,
Feng, G. J., Robinson, J. H., Lam, C., and Liew, F. Y. (1998) Nitric
oxide regulates Th1 cell development through the inhibition of IL-12
synthesis by macrophages, Eur. J. Immunol., 28,
4062-4070.