Biochemical Variations in Cytolytic Activity of Ortho- and Paramyxoviruses in Human Lung Tumor Cell Culture
O. P. Zhirnov1,2
1Ivanovsky Institute of Virology, Gamaleya Microbiology and Epidemiology Research Center, 123098 Moscow, Russia; E-mail: zhirnov@inbox.ru2Educational Noncommercial Organization Russian–German Academy of Social and Medical Sciences, 109004 Moscow, Russia
Received May 29, 2017; Revision received June 15, 2017
Human lung cancer cells (Calu-3 line) were studied for the development of apoptosis, necrosis, and autophagy in response to infection with ortho- and paramyxoviruses. Biochemical pathways underlying various mechanisms of cell death differed for different viruses. When infected with murine Sendai paramyxovirus, Calu-3 cells demonstrated typical necrotic features such as cell swelling (but not shrinkage), lack of chromatin DNA laddering, of caspase 3 and 8 activation, and of apoptotic cleavage of poly(ADP-ribose) polymerase (PARP) protein; an activation of antiapoptotic protein kinase Akt was also revealed. In contrast, infection with avian influenza virus A/FPV/Rostock/34 (H7N1 subtype) or Newcastle disease virus (NDV, avian paramyxovirus) caused the development of typical apoptotic markers such as cell shrinkage, ladder-type chromosomal DNA fragmentation, caspase 3 and 8 activation, and proteolytic cleavage of PARP in the absence of Akt activation. Notably, no upregulation of p53 protein phosphorylation was observed in all infected cells, which indicates that p53 is not involved in the virus-induced death of Calu-3 cells. Cell death caused by the influenza virus was accompanied by overstimulation of autophagy, whereas no stimulation of autophagy was observed in the NDV-infected cells. Infection with Sendai virus caused moderate stimulation of autophagy, which suggests that the mechanism of the virus-induced cell death and the balance between autophagy and cell death in infected cancer cells depend on the virus type and might significantly differ even for closely related viruses. Therefore, an optimal strategy for oncolytic virus-mediated destruction of tumor cells in cancer patients requires selection of the most appropriate oncolytic virus based on the mechanism of its cytolytic action in a particular type of tumor.
KEY WORDS: Sendai virus, Newcastle disease virus, influenza virus, apoptosis, necrosis, autophagy, oncolytic virusesDOI: 10.1134/S0006297917090085
Infection of cells with cytolytic viruses usually leads to cell death. Cells counteract viral infection by committing to rapid cell death to prevent virus propagation, while the virus suppresses premature cell death to provide its own replication. Typically, cell death happens after the virus has propagated and formed the progeny. In most cases, viral infection develops in two stages: an early stage, when the virus stimulates cell metabolism (in particular, autophagy) to support its reproduction, and a late stage, when the cell enters programmed cell death that might occur via apoptosis and/or necrosis [1-3].
The two major types of cell death – apoptosis and necrosis – differ in the underlying molecular mechanisms and biochemical pathways, changes in cell morphology, and immune and inflammatory responses of adjacent noninfected cells and the entire organism [4]. Based on the biochemical pathways involved, there are two types of apoptosis: canonical and autophagy-induced. There are also seven subtypes of necrosis, the two major subtypes being necroptosis and pyroptosis [5]. It is commonly believed that cell death by apoptosis is a physiological process that does not induce inflammation or acute immune response, while necrosis causes inflammation and provokes strong immune response [5-7]. However, recent data suggest that apoptosis induced by viral infection might cause inflammation as a part of the antiviral immune response [6, 8].
A new strategy for treatment of human cancer that includes targeted lysis of tumor cells with viruses (so-called viral oncolytics) has been extensively developed in the last years [9-12]. This approach requires comprehensive understanding of the molecular mechanisms that underlie the cytolytic activity of various families of oncolytic viruses. In this work, cell death pathways in human lung adenocarcinoma Calu-3 cell line after infection with two different strains of paramyxoviruses and the influenza virus (orthomyxovirus) are compared. The Calu-3 cells die by either apoptosis (when infected with avian influenza virus A/FPV/Rostock/34 or avian Newcastle disease paramyxovirus) or necrosis (when infected with murine Sendai paramyxovirus). Therefore, the mechanism of cell death is determined by the properties of the viral strain and might differ for viruses within the same viral family. This implies that successful tumor treatment depends on the tumor genotype and phenotype and will require the search for and selection of the optimal oncolytic virus(es) or targeted artificial modification of viral cytolytic properties.
MATERIALS AND METHODS
Cells. Human lung adenocarcinoma Calu-3 cell line was obtained from the cell culture collection of the Institute of Virology (Marburg, Germany) and was grown in a mixture of equal parts of minimal Dulbecco’s Modified Eagle Medium (DMEM) and DMEM-F12 supplemented with 10% fetal calf serum (Gibco BRL, USA) and antibiotics gentamycin (25 µg/ml), penicillin (50 U/ml), and streptomycin (50 µg/ml) [13, 14].
Viruses. Sendai virus (strain 960/ZH), Newcastle disease virus (NDV; vaccine strain LaSota), and avian influenza virus A/FPV/Rostock/34 (H7N1 subtype) were propagated in the allantoic cavity of 10-day-old leukosis-free chick embryos for 48-72 h at 36°C. The multiplicity of infection (MOI) was 104 focus-forming units (ffu) per embryo [13]. The allantoic fluid containing viral particles was collected and stored at –80°C.
Virus infectivity assay. Calu-3 cells were grown in a 24-well plate. The cells were incubated for 1 h at 37°C with 10-fold dilutions of the assayed viruses in DMEM. After 13-15 h, the cells were fixed with 4% paraformaldehyde and incubated with antibodies against Sendai virus, NDV, or influenza A virus and then with anti-specific antibodies conjugated with horseradish peroxidase. Infected cells (viral foci) were stained with tetramethylbenzidine substrate True Blue (KPL, USA). Stained foci were counted under a light microscope at ×75 magnification and expressed in ffu per ml of the initial virus dilution [3, 14].
Chromatin DNA ladder analysis. Cell pellets were suspended in 50 µl of PBS, mixed with 300 µl of buffer containing 0.6% SDS, 10 mM EDTA, and 15 mM Tris-HCl (pH 8.0), and then with 100 µl of 5 M NaCl. The lysates were incubated overnight at 4°C and then centrifuged at 14,000 rpm for 20 min to remove chromatin aggregates. The supernatants were subsequently treated with RNase A (0.1 mg/ml) at 37°C for 30 min and proteinase K (0.5 mg/ml) for another 30 min at 37°C. DNA was precipitated by adding three volumes of 96% ethanol and incubating the mixture at –20°C overnight. DNA precipitates were spun down by centrifugation at 14,000 rpm for 20 min, dissolved in 10 mM Tris-HCl (pH 8.0), and analyzed by electrophoresis in 1% agarose gel as described earlier [1].
Electrophoresis in polyacrylamide gel (PAGE) and Western blotting. Proteins from the infected cells were fractionated by SDS-PAGE and transferred on a Protran 0.45-µm nitrocellulose membrane (Schleicher & Schull, Germany) by the semi-dry method [3]. The membrane was washed in PBS, blocked for 2 h with 3% fat-free milk in PBS, and incubated overnight at 5°C in PBS containing 0.5% bovine serum albumin (Sigma, USA) and corresponding primary antibody. The antibodies used were against poly(ADP-ribose) polymerase (PARP; Santa Cruz, USA), phosphorylated forms of Akt protein kinase (Ser473-pho), p70S6 protein kinase (Ser371-pho), p53 (Ser15-pho), and S6 ribosomal protein S6 (Ser240/244-pho), activated caspase 3a (Cell Signaling, USA), autophagosomal microtubule-associated protein light chain 3 (LC3; Novus, Germany), caspase 8 (kindly provided by G. M. Cohen), and mammalian histone 3 (obtained in our lab by immunization of guinea pigs). The membrane was then washed with PBS, treated with species-specific secondary antibodies conjugated with horseradish peroxidase (Dako, USA), and developed using an enhanced chemiluminescence (ECL) kit with SuperSignal WB substrate (Pierce, USA) [2].
Cell microscopy. Cells (live cell monolayer) were analyzed under an inverted Axiovert 35 microscope (Opton, Germany) at ×250 magnification. Photographs were taken through the objective with an additional ×4.5 magnification in a photographic chamber.
RESULTS
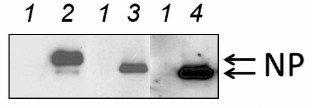
In this work, three viruses were studied – avian influenza virus (orthomyxovirus) and murine and avian paramyxoviruses. These viruses differ in their replication strategy and have different cell tropism profiles. The viruses were compared in their ability to infect cancer cells and cause their death by apoptosis, necrosis, or autophagy. Human lung adenocarcinoma Calu-3 cell line was used as a model. First, it was determined if the studied viruses could infect Calu-3 cells by analyzing accumulation of the major nucleocapsid NP protein and determining the virus yield in the culture liquid. Figure 1 shows that all three viruses efficiently infected Calu-3 cells and synthesized high amounts of NP protein. Calu-3 cells produced all the studied viruses with high yield of (1-5)·108 ffu/ml cultural liquid (data not shown), which correlates well with the results of our earlier studies [13, 14].
Fig. 1. Accumulation of nucleocapsid NP protein in infected Calu-3 cells. The cells were infected at MOI of 1 ffu/cell. The amount of NP protein was determined 27 h after infection by Western blotting using antibodies against corresponding viral NP proteins. Lanes: 1) control noninfected cells; 2-4) cells infected with Sendai virus, NDV, and influenza virus A/FPV/34, respectively.
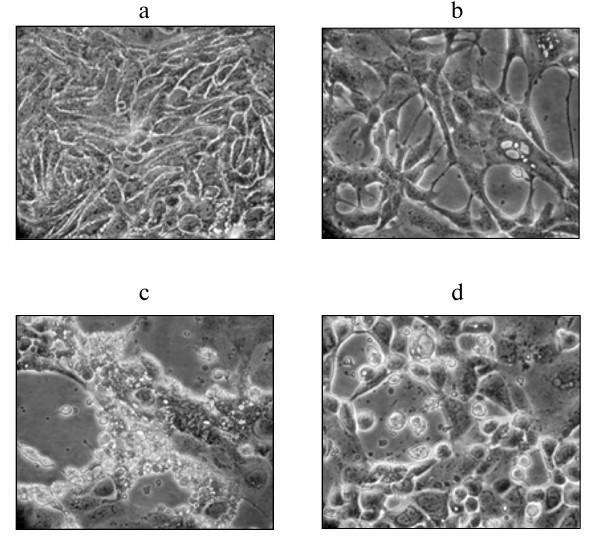
Next, morphological changes in the Calu-3 cell monolayer infected with the avian influenza virus A/FPV/34, avian NDV (strain LaSota), and murine Sendai virus (strain 960/ZH) at MOI of 1 ffu/cell were studied. Figure 2 shows that viral infection resulted in the development of clearly pronounced cell pathologies at the late stages of infection (i.e. 25-29 h after virus addition). Some cytolytic activity was observed at earlier stages in cell cultures infected with the influenza virus and NDV. When infected with Sendai virus, cells underwent pronounced cell lysis 35-45 h after infection. However, these viruses affected cell morphology differently. Infection with either NDV or influenza virus resulted in cell shrinkage and disintegration into fragments that detached from the flask surface. When infected with Sendai virus, the cells stayed attached to the surface but acquired round shape and formed lacunas in the cell monolayer (Fig. 2b). These observations suggest that different viruses induce different cell death mechanisms in human cancer cells – apoptosis in the case of influenza and NDV infection and necrosis in the case of Sendai virus infection (according to the cell morphology).
Fig. 2. Cell pathologies induced by viral infection of Calu-3 cells. The cells were infected at MOI of 1 ffu/cell and examined 27 h after infection under an Opton inverted light microscope (magnification, ×230) and in a photographic chamber (additional magnification, ×4.5). Panels (a)-(d) represent typical fields of observation: a) control noninfected cells; b-d) cells infected with Sendai virus, NDV, and influenza virus A/FPV/34, respectively.
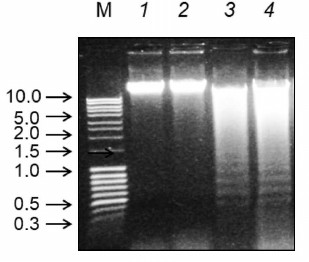
To confirm the necrotic and apoptotic types of cell death, the profiles of chromatin DNA degradation in the infected cells (Fig. 3) were studied. Cell cultures infected with the influenza virus and NDV exhibited characteristic “ladder” DNA pattern typical for apoptotic fragmentation of chromosomal DNA (Fig. 3, lanes 3 and 4). In cells infected with the Sendai virus, the DNA degradation pattern was diffuse, i.e. typical for the necrotic type of cell death. These results correlate with the data on the morphological changes in the infected cells.
Fig. 3. Chromatin DNA ladder profiles in Calu-3 cells infected with Sendai virus, NDV, and influenza virus A/FPV/34 (lanes 2-4, respectively); lane 1, control noninfected cells. The cells were infected at MOI of 1 ffu/cell; DNA was isolated 27 h after infection and analyzed by electrophoresis in 1% agarose gel with ethidium bromide staining. Lane M, DNA size markers (arrows left of the panel; DNA fragment sizes are shown in kb).
To elucidate virus-dependent mechanisms of Calu-3 cell death, the profiles of apoptotic, necrotic, and autophagic protein markers in the infected cells were compared using PAGE and Western blotting. Cell extracts were assayed for PARP, activated caspases 3 and 8, major autophagic protein LC3, and phosphorylated forms of p53 (Ser15-pho), Akt protein kinase (Ser473-pho), S6 ribosomal protein (Ser240/244-pho), and p70S6 (Ser371-pho) protein kinase (Fig. 4). Extracts from cells infected with the influenza virus or NDV contained typical for apoptosis proteolytic product (80 kDa) of PARP (110 kDa) and activated forms of caspase 3a (28 kDa) and caspase 8a (47, 32, and 18 kDa) (Fig. 4, lanes 3 and 4), but not the phosphorylated form of Akt protein kinase. In contrast, extracts from the cells infected with Sendai virus contained no 80-kDa product of PARP proteolysis even 45 h after infection or activated forms of caspases 3a and 8a; however, the levels of phosphorylated Akt (Ser473-pho) protein kinase were clearly increased (Fig. 4, lane 2). These data confirmed once again that infection with the influenza virus and NDV induces apoptosis, while infection with the Sendai paramyxovirus causes cell death by the necrotic mechanism.
Fig. 4. Marker proteins of apoptotic, necrotic, and autophagic pathways in Calu-3 cells infected with Sendai virus, NDV, and influenza virus A/FPV/34 (lanes 2-4, respectively); lane 1, control noninfected cells. The cells were infected at MOI of 1 ffu/cell. Equal protein amounts of cell extracts were loaded on a gel wells and fractionated by PAGE. Proteins of interest were identified by Western blotting using specific antibodies (shown to the left) and visualized by the ECL method using secondary horseradish peroxidase-conjugated antibodies.
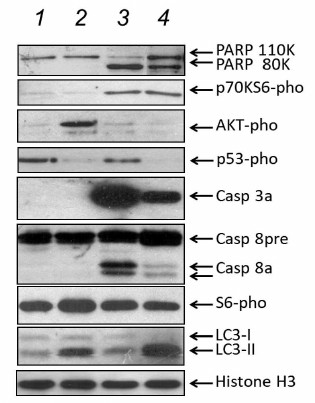
Notably, the content of phosphorylated p53 protein (Ser15-pho) in the control noninfected cells (Fig. 4, lane 1) was considerably higher than in all the infected cells (Fig. 4, lanes 2-4). The lowest content of phosphorylated p53 was observed in the cells infected with the Sendai virus and the influenza virus (Fig. 4, lanes 2 and 4, respectively), which indicates that infection of Calu-3 cells with viruses does result in p53 activation. This might be related to a defect in the p53 protein in Calu-3 cells (see ATCC® Cell Lines by Gene Mutation Guide; https://www.atcc.org/~/media/PDFs/Culture%20Guides/Cell_Lines_by_Gene_Mutation.ashx), that eliminates p53 from the processes of virus-induced death. It is well known that mutations in the p53 gene, that suppress its proapoptotic function, are typical events in many types of human lung non-small cell adenocarcinomas [15].
To investigate the relationship between cell death and intracellular autophagy, the content of the microtubule-associated protein 1A/1B light chain 3 (LC3), the main marker of intracellular autophagy, was studied. Infected cells were also assayed for phosphorylation of p70S6 (Ser371-pho) protein kinase, which serves as an indicator of the activity of the mTOR pathway known to be involved in autophagy inhibition [16-18]. Activation conversion of the LC3/I (electrophoretic mobility, 17 kDa) into its lipid-conjugated LC3/II form (electrophoretic mobility, 15 kDa) indicates autophagy overstimulation, and this was observed in Calu-3 cells infected with the influenza virus (Fig. 4, lane 4). Less pronounced increase in the LC3/II content was found in Calu-3 cells infected with the Sendai virus (Fig. 4, lane 2). When the cells were infected with NDV, the content of LC3/II was similar to that in the noninfected cells (Fig. 4, lanes 3 and 1, respectively). Infection of Calu-3 cells with the influenza virus and NDV, but not with Sendai virus, upregulated phosphorylation of p70S6 kinase, a phosphorylation substrate for the mTOR (Fig. 4, lanes 2-4), which suggests that Sendai virus suppresses the mTOR signaling pathway responsible for the inhibition of intracellular autophagy. Hence, infection of Calu-3 cells with Sendai virus resulted in autophagy suppression and necrosis. Infection of the same cells with the two other viruses either caused no autophagy stimulation (NDV) or activated autophagy (influenza virus). In both cases, the cell death was apoptotic.
DISCUSSION
The Calu-3 cell line was isolated from the pleural metastasis of a lung cancer patient and is a non-small cell adenocarcinoma of the epithelial type [19, 20]. Calu-3 cells bear mutations in the K-RAS (G13D) and p53 genes (M237I) (ATCC® Cell Lines by Gene Mutation Guide; https://www.atcc.org/~/media/PDFs/Culture%20Guides/Cell_Lines_by_Gene_Mutation.ashx), which is typical for this type of tumors [15]. Here, we studied the ability of various cytolytic viruses from the ortho- and paramyxovirus families to cause death of Calu-3 cells and investigated the mechanism of cell death based on the presence of apoptotic, necrotic, and autophagic markers in the infected cells. It was found that the type of cell death differs for different (even closely related) viruses. Thus, the influenza virus and avian NDV induced cell death by the apoptotic mechanism, while murine Sendai virus caused necrosis. These observations imply that different viruses use different cell pathways for replication, which in combination with other viral factors, promotes cell death via a particular mechanism. Our results suggest the possibility of directional manipulation of the cell death mechanism in cancer cells by either using specific viruses or by creating artificial (recombinant) viruses with a particular type of cytolytic activity. This approach might be promising for choosing optimal strategies for cancer treatment with oncolytic viruses.
In this study, correlations between the type of cell death and intracellular autophagy in Calu-3 cells infected with different viruses were found. In particular, autophagy stimulation was accompanied by suppression of apoptosis. Thus, no apoptosis was observed in cells infected with the Sendai virus, and the cells exhibited high levels of autophagy. In contrast, cells infected with NDV underwent apoptosis in the absence of autophagy stimulation. When infected with the influenza virus, cells entered an intermediate state – they demonstrated autophagy overstimulation, but developed several apoptotic markers at the same time. The logical explanation is that autophagy suppresses apoptosis and promotes necrotic death in the infected cells. Moreover, excessive autophagy stimulation can induce apoptosis [21, 22], which might have taken place in the cells infected with the avian influenza virus A/FPV/34 (H7N1) (Figs. 3 and 4).
The intimate molecular mechanism of the cross-talk between autophagy induction and apoptosis in viral infection is not fully understood. It can only be hypothesized that partial suppression of autophagy in Calu-3 cells under Sendai virus infection, in comparison with influenza virus superstimulation, might be due to the downregulation of the mTOR signaling pathway, which is known to inhibit intracellular autophagy [19, 20]. This is confirmed by increased levels of the activated (phosphorylated) form of the anti-mTOR protein kinase Akt and its substrate (phosphorylated S6 ribosomal protein) and the absence of mTOR-mediated phosphorylation of p70S6 kinase (Ser371-pho) (Fig. 4, lane 2). Cells infected with the influenza virus and NDV showed no Akt activation and had increased levels of p70S6 kinase phosphorylation, which correlates with the induction of the autophagy-suppressing mTOR pathway and explains the absence of autophagy stimulation in the NDV-infected cells (Fig. 4, lane 3). However, the phenomenon of autophagy upregulation in the influenza-infected Calu-3 cells that exhibited high levels of LC3/II, but whose profile of apoptotic/necrotic markers (Akt-pho, p70S6K-pho, and S6-pho) was close to that of apoptotic cells (Fig. 4, lane 4), requires further investigation. Perhaps the key role in the intracellular autophagy stimulation in this case belongs to the viral channel-forming protein M2, which supposedly acts through downstream autophagic mediators and interacts with the cell protein Beclin [23].
The cytolytic activity of paramyxoviruses and influenza viruses usually occurs via the apoptotic mechanism in both healthy and tumor cells [9, 24-26]. Here, for the first time it is demonstrated that Sendai virus causes necrosis in the culture of Calu-3 cancer cells. Earlier, similar necrotic activity of the influenza virus was found in the human colon cancer Caco-2 cell line [1]. It should be noted that necrosis in the Calu-3 cell culture developed in the absence of changes in the state of cell caspases, e.g. caspases 3 and 8 (Fig. 4), which contradicts earlier data on necrosis induced by inactivated Sendai virus in human neuroblastoma CK-N-SH and SK-N-AS cell lines [27]. However, those authors [27] used replication-defective (UV-inactivated) Sendai virus, and, unlike Calu-3 cells, used neuroblastoma cells lines that had mutated caspase 8 gene and could not prevent necrosis development via caspase 8 activation or induce caspase-mediated apoptosis.
The important observation is that, unlike the influenza virus and NDV, Sendai virus caused no activation of caspase 8 in infected cells. This correlates well with our data that Sendai virus induces cell death by the necrotic mechanism. Moreover, this observation is an accord with the earlier published results of other studies that caspase 8 activation inhibits necrosis through the cleavage of major necrosis mediators, such as RIPK1, RIPK3, and CYLD [28-30]. Perhaps the activation of caspase 8 by the influenza virus and NDV suppresses necrosis. Further studies should focus on (i) searches for the viral factors (proteins) that induce apoptosis and, at the same time, stimulate autophagy and induce necrosis in Calu-3 cells (similarly to Sendai virus) and (ii) identification of proapoptotic viral factor(s) or proteins(s) in the influenza virus and NDV. Identification of such factors (genes) will allow development of artificial recombinant viruses with the targeted cytolytic activity against cancer cells of different origin.
Acknowledgments
This work is dedicated to the memory of my mother, Nadezhda Vasilyevna Zhirnova, a World War II veteran. I am immensely grateful for her invaluable and noble help and support in all aspects of my work.
I also thank E. Böttcher-Friebertshäuser and H. D. Klenk (Institute of Virology, Philipps University, Marburg, Germany) for kindly providing the Calu-3 cell culture, S. Ludwig (Institute of Molecular Virology, Westfälische Wilhelms University of Münster, Münster, Germany) for valuable discussion of the research topics, and G. M. Cohen (University of Leicester, United Kingdom) for kindly providing anti-caspase-8 antibodies.
This work was supported by the Ministry of Education and Science of the Russian Federation (project No. RFMEFI60721X0014).
I thank Veronique Zh. for help in preparing this report.
REFERENCES
1.Zhirnov, O., and Klenk, H. D. (2003) Human
influenza A viruses are proteolytically activated and do not induce
apoptosis in CACO-2 cells, Virology, 313, 198-212;
Erratum in: Virology (2003) 317, 383.
2.Zhirnov, O. P., and Klenk, H. D. (2007) Control of
apoptosis in influenza virus-infected cells by up-regulation of Akt and
p53 signaling, Apoptosis, 12, 1419-1432.
3.Zhirnov, O. P., and Klenk, H. D. (2013) Influenza A
virus proteins NS1 and hemagglutinin along with M2 are involved in
stimulation of autophagy in infected cells, J. Virol.,
87, 13107-13014.
4.Kondylis, V., Kumari, S., Vlantis, K., and
Pasparakis, M. (2017) The interplay of IKK, NF-κB and RIPK1
signaling in the regulation of cell death, tissue homeostasis and
inflammation, Immunol. Rev., 277, 113-127.
5.Pasparakis, M., and Vandenabeele, P. (2015)
Necroptosis and its role in inflammation, Nature, 517,
311-320.
6.Davidovich, P., Kearney, C. J., and Martin, S. J.
(2014) Inflammatory outcomes of apoptosis, necrosis and necroptosis,
Biol. Chem., 395, 1163-1171.
7.Creagh, E. M. (2014) Caspase crosstalk: integration
of apoptotic and innate immune signaling pathways, Trends
Immunol., 35, 631-640.
8.Man, S. M., and Kanneganti, T. D. (2016) Converging
roles of caspases in inflammasome activation, cell death and innate
immunity, Nat. Rev. Immunol., 16, 7-21.
9.Cuadrado-Castano, S., Sanchez-Aparicio, M. T.,
Garcia-Sastre, A., and Villar, E. (2015) The therapeutic effect of
death: Newcastle disease virus and its antitumor potential,
Virus Res., 209, 56-66.
10.Saga, K., and Kaneda, Y. (2015) Oncolytic Sendai
virus-based virotherapy for cancer: recent advances, Oncolyt.
Virother., 4, 141-147.
11.Matveeva, O. V., Guo, Z. S., Shabalina, S. A.,
and Chumakov, P. M. (2015) Oncolysis by paramyxoviruses: multiple
mechanisms contribute to therapeutic efficiency, Mol. Ther.
Oncolyt., 2, 15011-15021.
12.Chen, D., Yu, J., and Zhang, L. (2016)
Necroptosis: an alternative cell death program defending against
cancer, Biochim. Biophys. Acta, 1865, 228-236.
13.Zhirnov, O. P. (2017) Paramyxoviruses activation
by host proteases in cultures of normal and cancer cells, Vopr.
Virusol., 62, 65-72.
14.Zhirnov, O. P., Vorob’eva, I. V., Safonova,
O. A., Malyshev, N. A., Schwalm, F., and Klenk, H.-D. (2013) Pathogenic
effect of pandemic influenza virus H1N1 under replication in cultures
of human cells, Vopr. Virusol., 58, 20-28.
15.Liu, G., Pei, F., Yang, F., Li, L., Amin, A. D.,
Liu, S., Buchan, J. R., and Cho, W. C. (2017) Role of autophagy and
apoptosis in non-small-cell lung cancer, Int. J. Mol. Sci.,
18, E367.
16.Saitoh, M., Pullen, N., Brennan, P., Cantrell,
D., Dennis, P. B., and Thomas, G. (2002) Regulation of an activated S6
kinase 1 variant reveals a novel mammalian target of rapamycin
phosphorylation site, J. Biol. Chem., 277,
20104-20112.
17.Fumarola, C., Bonelli, M. A., Petronini, P. G.,
and Alfieri, R. R. (2014) Targeting PI3K/AKT/mTOR pathway in non-small
cell lung cancer, Biochem. Pharmacol., 90, 197-207.
18.Shimobayashi, M., and Hall, M. N. (2014) Making
new contacts: the mTOR network in metabolism and signaling crosstalk,
Nat. Rev. Mol. Cell Biol., 15, 155-162.
19.Fogh, J., Fogh, J. M., and Orfeo, T. (1977) One
hundred and twenty-seven cultured human tumor cell lines producing
tumors in nude mice, J. Natl. Cancer Inst., 59,
221-226.
20.Shen, B. Q., Finkbeiner, W. E., Wine, J. J.,
Mrsny, R. J., and Widdicombe, J. H. (1994) Calu-3: a human airway
epithelial cell line that shows cAMP-dependent Cl−
secretion, Am. J. Physiol. Lung Cell Mol. Physiol., 266,
L493-L501.
21.Levine, B., and Kroemer, G. (2008) Autophagy in
the pathogenesis of disease, Cell, 132, 27-42.
22.Marino, G., Niso-Santano, M., Baehrecke, E. H.,
and Kroemer, G. (2014) Self-consumption: the interplay of autophagy and
apoptosis, Nat. Rev. Mol. Cell Biol., 15, 81-94.
23.Rossman, J. S., and Lamb, R. A. (2009) Autophagy,
apoptosis, and the influenza virus M2 protein, Cell Host
Microbe, 6, 299-300.
24.Wei, B., Cui, Y., Huang, Y., Liu, H., Li, L., Li,
M., Ruan, K. C., Zhou, Q., and Wang, C. (2015) Tom70 mediates Sendai
virus-induced apoptosis on mitochondria, J. Virol., 89,
3804-3818.
25.Chattopadhyay, S., Fensterl, V., Zhang, Y.,
Veleeparambil, M., Yamashita, M., and Sen, G. C. (2013) Role of
interferon regulatory factor 3-mediated apoptosis in the establishment
and maintenance of persistent infection by Sendai virus, J.
Virol., 87, 16-24.
26.Koks, C. A., Garg, A. D., Ehrhardt, M., Riva, M.,
Vandenberk, L., Boon, L., De Vleeschouwer, S., Agostinis, P., Graf, N.,
and Van Gool, S. W. (2015) Newcastle disease virotherapy induces
long-term survival and tumor-specific immune memory in orthotopic
glioma through the induction of immunogenic cell death, Int. J.
Cancer, 136, E313-325.
27.Nomura, M., Ueno, A., Saga, K., Fukuzawa, M., and
Kaneda, Y. (2014) Accumulation of cytosolic calcium induces necroptotic
cell death in human neuroblastoma, Cancer Res., 74,
1056-1066.
28.O’Donnell, M. A., Perez-Jimenez, E.,
Oberst, A., Ng, A., Massoumi, R., Xavier, R., Green, D. R., and Ting,
A. T. (2011) Caspase 8 inhibits programmed necrosis by processing CYLD,
Nat. Cell Biol., 13, 1437-1442.
29.Feng, S., Yang, Y., Mei, Y., Ma, L., Zhu, D. E.,
Hoti, N., Castanares, M., and Wu, M. (2007) Cleavage of RIP3
inactivates its caspase-independent apoptosis pathway by removal of
kinase domain, Cell Signal., 19, 2056-2067.
30.Man, S. M., Karki, R., and Kanneganti, T. D.
(2017) Molecular mechanisms and functions of pyroptosis, inflammatory
caspases and inflammasomes in infectious diseases, Immunol.
Rev., 277, 61-75.