REVIEW: Electron Transfer through the Acceptor Side of Photosystem I: Interaction with Exogenous Acceptors and Molecular Oxygen
D. A. Cherepanov1,2*, G. E. Milanovsky1, A. A. Petrova1, A. N. Tikhonov3*, and A. Yu. Semenov1*
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119992 Moscow, Russia; E-mail: tscherepanov@gmail.com, semenov@genebee.msu.ru2Semenov Institute of Chemical Physics, Russian Academy of Sciences, 119991 Moscow, Russia
3Lomonosov Moscow State University, Faculty of Physics, 119991 Moscow, Russia; E-mail: an_tikhonov@mail.ru
* To whom correspondence should be addressed.
Received June 29, 2017; Revision received August 1, 2017
This review considers the state-of-the-art on mechanisms and alternative pathways of electron transfer in photosynthetic electron transport chains of chloroplasts and cyanobacteria. The mechanisms of electron transport control between photosystems (PS) I and II and the Calvin–Benson cycle are considered. The redistribution of electron fluxes between the noncyclic, cyclic, and pseudocyclic pathways plays an important role in the regulation of photosynthesis. Mathematical modeling of light-induced electron transport processes is considered. Particular attention is given to the electron transfer reactions on the acceptor side of PS I and to interactions of PS I with exogenous acceptors, including molecular oxygen. A kinetic model of PS I and its interaction with exogenous electron acceptors has been developed. This model is based on experimental kinetics of charge recombination in isolated PS I. Kinetic and thermodynamic parameters of the electron transfer reactions in PS I are scrutinized. The free energies of electron transfer between quinone acceptors A1A/A1B in the symmetric redox cofactor branches of PS I and iron–sulfur clusters FX, FA, and FB have been estimated. The second-order rate constants of electron transfer from PS I to external acceptors have been determined. The data suggest that byproduct formation of superoxide radical in PS I due to the reduction of molecular oxygen in the A1 site (Mehler reaction) can exceed 0.3% of the total electron flux in PS I.
KEY WORDS: photosystem I, electron transport, kinetic modeling of electron transfer, exogenous electron acceptors, interaction with oxygenDOI: 10.1134/S0006297917110037
Abbreviations: A0A and A0B, dimers of chlorophyll a molecules that are primary electron acceptors in cofactor A and B branches; A1A and A1B, phylloquinone molecules that are secondary electron acceptors in A and B branches; Asc−⋅, monodehydroascorbate; AscH−, ascorbate anion (fully reduced state); CBC, Calvin–Benson cycle; Chl, chlorophyll; Cl2NQ, 2,3-dichloro-1,4-naphthoquinone; DCPIP, 2,6-dichlorophenolindophenol; DMF, dimethylformamide; Em, midpoint potential; Fc, ferrocene; Fc+, ferrocenium cation; Fd, ferredoxin; Fld, flavodoxin; FNR, ferredoxin:NADP oxidoreductase; FQR, ferredoxin:quinone reductase; FX, FA, and FB, iron–sulfur clusters; FX–core, complexes depleted of iron–sulfur FA/FB clusters; ΔG, free energy; Km, Michaelis constant; menB, mutant strain of Synechocystis sp. PCC 6803 cyanobacterium; MV, methyl viologen; NDH, NADH dehydrogenase; O2•−, superoxide anion radical; P700, special pair of chlorophyll a molecules in PS I; Pc, plastocyanin; PGR5, proton gradient regulation protein gene 5 (proton gradient regulation 5); PGRL1, PGR5-like protein 1 gene (PGR5-like protein 1); PhQ, phylloquinone; PQ, plastoquinone; PQH2, plastoquinol; PS I (II), photosystem I (II); PSA, photosynthetic apparatus; PsaA, PsaB, PsaC, PsaD and PsaE, subunits within photosystem I; ROS, reactive oxygen species; SCE, saturated calomel electrode; SHE, standard hydrogen electrode; WT, wild-type Synechocystis sp. PCC 6803 cyanobacterium.
ELECTRON TRANSPORT IN OXYGENIC PHOTOSYNTHETIC SYSTEMS
Alternative routes of electron transfer in photosynthetic systems of oxygenic type. In oxygenic photosynthetic systems (chloroplasts of higher plants, cyanobacteria, algae), two photosystems (PS) I and II under sequential light-driven functioning provide electron transfer from water, splitted by PS II, to NADP+, the terminal physiological acceptor of PS I (Fig. 1). The electron transport reactions are coupled to generation of the transmembrane difference in electrochemical potentials of hydrogen ions serving as an energy source for the operation of ATP synthase complexes providing the ATP synthesis from ADP and Pi. Products of the light stage of photosynthesis – NADPH and ATP – enable the assimilation of carbon dioxide and the synthesis of carbohydrates in the Calvin–Benson cycle (CBC). The optimal balance between ATP and NADPH molecules (ATP/NADPH = 3/2), necessary for operation of the CBC, is achieved by fine regulation of electron flux partitioning between alternative paths of electron transport (noncyclic, cyclic, and pseudocyclic [1-4]). The key role in the distribution of electrons is played by the acceptor side of PS I [5]. Under metabolic conditions, when the CBC is active, the main electron flux from PS I is directed from ferredoxin (Fd) to NADP+ through ferredoxin-NADPH reductase. However, during the initial stage of the induction period of photosynthesis, when the CBC enzymes are inactivated after dark adaptation, the outflow of electrons from PS I is slow due to low consumption of NADPH in the CBC. In this case, the contribution of alternative electron transport pathways – cyclic electron flow around PS I and pseudocyclic electron transfer – is known to increase (Fig. 2). Electron transfer along the “short” route from the acceptor site of PS I to the plastoquinone (PQ) pool occurs with the participation of ferredoxin-quinone reductase (FQR) and the cytochrome b6f complex [6-8]. The “long” route of cyclic electron transport around PS I is associated with electron transfer through NAD(P)H-dehydrogenase [9-12]. Genetic and biochemical studies demonstrated the participation of various proteins in the cyclic electron transport (see, for example, review [4]).
Fig. 1. Schemes of noncyclic and cyclic pathways of electron transport in chloroplasts.
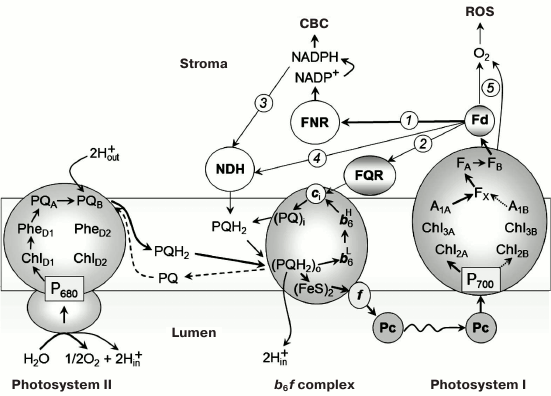
Fig. 2. Electron transport chain in oxygenic photosynthetic systems (Z-scheme of photosynthesis). APX, ascorbate-peroxidase; SOD, superoxide-dismutase.
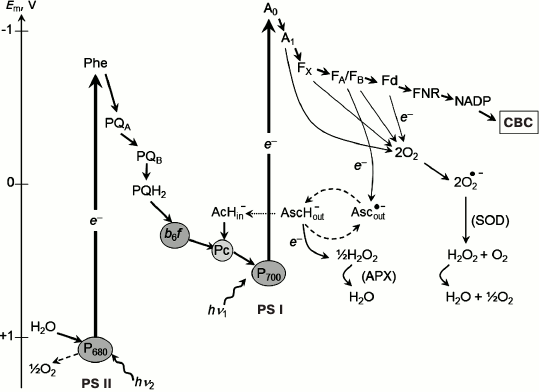
The concept of cyclic electron transport in photosynthetic systems was introduced in the literature in 1963, when Arnon et al. [13] demonstrated that Fd can serve as a cofactor of so-called “cyclic phosphorylation”. It is believed that competition for reduced Fd is one of the major mechanisms for redistributing electron fluxes on the PS I acceptor side [14-16]. More than half a century has passed since the publication of that article [13], but questions concerning the mechanisms of Fd-dependent cyclic electron transport are still a subject of scientific discussions. In 1995, Bendall and Manasse [17] suggested the existence of FQR, which provides a cyclic pathway of electron transfer around PS I: PS I → Fd → FQR → PQ → b6f → Pc → P700. Later, genes whose products provide for cyclic electron transport were found in Arabidopsis chloroplasts [4, 6, 18]. These genes are PGR5 (proton gradient regulation 5 protein gene) and PGRL1 (PGR5-like protein 1 gene). Expression of these genes is necessary for inducing nonphotochemical quenching of the excited state of chlorophyll (Chl) and for protecting the photosynthetic apparatus from photodegradation. It has been shown that the PGR5 and PGRL1 proteins can be involved in Fd-dependent electron transport around PS I [6, 7, 18-22]. This suggests that PGR5 and PGRL1 can be considered as the FQR components. However, the exact role of the PGR5/PGRL1 proteins (direct participation or regulatory functions) remained unclear, until it was established that PGRL1 can accept electrons from Fd and reduce PQ [20]. PGRL1 is a redox-regulated protein, its activity is associated with the presence of a Fe-containing cofactor and six cysteine residues. The PGR5 protein is required for transfer of electrons from Fd to PGRL1.
There is also a notion that the Fd-dependent cyclic transport pathway is associated with the reduction of the PQ molecule in the Qi site of the cytochrome b6f complex, oriented toward the stroma. It is assumed that the electrons accepted by the cytochrome complex from PS I come directly from ferredoxin:NADP oxidoreductase (FNR) (see, for example, the review [23]). The formation of the FNR–b6f supercomplex should facilitate this reaction.
FNR can be included in cyclic electron transport, either directly (PS I → Fd → FNR → PQ) or through the NADH dehydrogenase complex (NDH) (Fig. 2). This pathway is considered to be linked to NADPH (or NADH) oxidation. The presence of NDH has been demonstrated by chloroplast genome sequencing for Nicotiana tabacum and Marchantia polymorpha [24, 25]. NDH has also been found in cyanobacteria [26, 27]. Genetic and biochemical analysis convincingly showed involvement of chloroplast NDH in cyclic transport of electrons around PS I [12, 28, 29]. This is also indicated by NDH localization in stromal regions of thylakoid membranes enriched with PS I complexes [9]. There is evidence that NDH and PS I can form a supercomplex in plant chloroplasts [10, 11] and in cyanobacterial cells [30]. The nature of the electron donor for NDH is a subject of discussions [4]. It is believed that NADPH can serve as the electron donor for NDH (PS I → Fd → FNR → NADPH → NDH → PQ → b6f). However, different point of view was discussed in [31-33]. Johnson [31] suggested that Fd can donate electrons to the plastoquinone pool bypassing the NDH complex (Fig. 1). There are indications that in chloroplasts, NDH accepts electrons from reduced Fd, but not from NAD(P)H [32, 33]. Genetic and biochemical analysis demonstrated that NDH transfers electrons to PQ from stromal donors [34].
Redistribution of electron fluxes between alternative pathways can be controlled by the redox state of the chloroplast electron transport chain. Intensive reduction of thioredoxin occurs under excess of reducing agents on the acceptor side of PS I, thereby activating thioredoxin-dependent enzymes [35-37]. In particular, the light-induced activation of CBC enzymes contributes to an increase in the outflow of electrons from PS I to the CBC [38-40].
Besides the alternative ways of cyclic transport of electrons described above, the so-called pseudocyclic electron transport contributes to chloroplast functioning, in which water molecule oxidized by PS II serves as the direct electron donor. On the other hand, the water molecule is formed as the terminal product as a result of electron transfer from PS I to molecular oxygen O2 (H2O → PS II → PS I → O2 → H2O) (for more details, see below in the section “Pseudocyclic transport of electrons in chloroplasts (“water–water” cycle)”).
The literature on the functional role of alternative pathways of electron transport is contradictory [3, 41-46]. It is considered, for example, that the fraction of pseudocyclic transport of electrons is small (~10%) compared to the total flux of electrons coming from PS II. The transfer of electrons along alternative paths is accompanied by the generation of a transmembrane difference in electrochemical potentials of hydrogen ions, which allows ATP synthesis during initial stages of the induction period of photosynthesis, when the CBC is inactive. With light-induced activation of the CBC, noncyclic (linear) electron flux increases, and the relative contributions of the alternative paths of electron outflow from PS I decrease. According to various estimates, even a relatively small branching of the electron flux to the cyclic electron transfer chain and oxygen, ~10-15% of the noncyclic flow to NADP+, can provide the required stoichiometry of ATP/NADPH [1, 47, 48].
Regulation of electron transport. Let us briefly consider some aspects of regulation of electron transport in chloroplasts. The ability of photosynthetic organisms to respond rapidly to changes in external conditions determines their productivity and survival under extreme conditions. Rapid changes in light intensity and temperature can damage the photosynthetic apparatus [48-56]. In plants, two strategies are present for protecting the photosynthetic apparatus (PSA) from light stress caused by excess solar energy: (i) rapid response of the PSA to light fluctuations and (ii) relatively slow plant response linked to changes in content of various PSA components during a decrease or amplification of light intensity or temperature and gas composition of the environment. Structural and functional reorganization of the PSA in response to changes in external conditions is achieved by various feedbacks; these processes occur in the time range from a few seconds to several tens of minutes (rapid PSA regulation) or several hours/days (slow PSA rearrangement).
Mechanisms of rapid regulation of the PSA are associated with the following processes: (i) pH-dependent control of electron transport due to photoinduced changes in pH inside the thylakoid and stroma, (ii) redox-dependent activation/inactivation of CBC enzymes, (iii) redistribution of absorbed light energy between PS II and PS I, (iv) redistribution of electron fluxes between the alternative pathways, and (v) chloroplast movements within the plant cell, shielding them from excessive light. Rapid mechanisms of regulation are seen most clearly when studying the induction phenomena of photosynthesis [37, 48, 50, 53, 54, 57-64].
Even though the molecular structure of the PSA has been rather thoroughly studied, questions about alternative electron transfer pathways in oxygenic photosynthetic systems are still contentious. These are, for instance, the question of the interaction of molecular oxygen (O2) with the electron transport chain of oxygenic photosynthetic organisms [42, 43, 65]. Below, we describe the electron transport reactions on the acceptor side of PS I, paying attention to the mechanisms of PS I interaction with exogenous electron acceptors, and with molecular oxygen (O2) in particular. Mathematical modeling is a useful tool for quantitative analysis of electron transport processes in photosynthetic systems, and it is given special attention in this review. Molecular oxygen is an acceptor with redox potential Em = –0.155 V relative to the normal hydrogen electrode at pH 7.0 and concentration of [O2] = 1 M [66], capable of accepting electrons from low-potential acceptors of PS I (Mehler reaction [67], Fig. 2). This results in formation of superoxide radical (O2•−) and other reactive oxygen species (ROS) [41-43, 65, 68]. Superoxide radicals O2•− dismutate, forming hydrogen peroxide H2O2 (2 O2•− → H2O2 + O2) [67, 69]. In intact chloroplasts, H2O2 is converted into water by interacting with ascorbate, catalyzed by peroxidase [41, 42, 70, 71]. The major electron donors for O2 in chloroplasts are electron carriers on the acceptor side of PS I. The sections “Mathematical modeling of electron transfer reactions in pigment–protein complex of photosystem I” and “Pseudocyclic electron transport in chloroplasts (“water–water” cycle)” of our review are devoted to experimental studies and mathematical modeling of the interaction of O2 with PS I.
General principles of mathematical modeling of electron transport processes of photosynthesis. Mathematical modeling is an effective tool for analyzing electron transport processes in photosynthetic systems (see, for example, [72, 73]). Below we briefly consider the basic principles for constructing mathematical models that describe the light stages of oxygenic photosynthesis.
Deterministic description of the processes of electron transport is based on the use of chemical kinetics equations that establish the relationship between the concentrations of interacting electron carriers by the law of mass action, sometimes supplemented by the Michaelis–Menten equations to describe the individual stages of electron transport. In the fast mixing approximation, the time-dependent concentrations of reagents can be considered as variables independent of spatial coordinates. In this case, a system of ordinary differential equations can be used to describe the interaction of metabolites (or electron carriers). The application of the law of mass action for describing the interaction between the reacting components is justified if the encounter of spatially separated reagents occurs by their rapid diffusion. In this case, the rates of individual reactions can be considered (in first approximation) proportional to the products of the reacting substances:
where kij is the apparent (effective) rate constant for interactions of reactants with concentrations of ci(r⃗, t) and cj(r⃗, t). We emphasize that the concentrations of the reacting components at different points in the system can depend on the spatial coordinate r⃗. Simulation of the electron and proton transport processes in chloroplasts has shown that such an approximation can be useful, for example, if rapid diffusion of PQ and plastocyanin (Pc) allows efficient interaction between spatially separated pigment–protein complexes of PS II, PS I, and cytochrome b6f complex [73, 74].
Equation (1) is known as the law of mass action. On its basis, a kinetic model has been developed that describes key reactions of electron transfer and transmembrane proton transfer in chloroplasts [73-75]. Quantitative analysis of the model revealed the key functional role of PS I in regulation of pH levels and ATP concentration in thylakoids, providing pH-dependent activation of the Calvin–Benson cycle and regulation of PS II activity. It has been shown that pH-dependent regulatory processes change the distribution of electron fluxes on the acceptor side of PS I and control the electron transfer rate between PS II and PS I. In general, light-induced activation of the Calvin–Benson cycle results in a significant increase in the electron flux from PS I to NADP+ and a decrease in the electron flux to molecular oxygen [75]. Therefore, the biochemical processes in thylakoid membranes and chloroplast stroma are regulated by complex systems of feedbacks, in which PS I is a key node; quantitative analysis of these reactions is impossible without a detailed kinetic model of PS I [73]. Recently, a hierarchical model of PS I was developed that describes the interaction of the pigment–protein complex with the native donor (Pc) and acceptors (plastoquinone-NAD(P)H reductase and ferredoxin-NADPH reductase) of electrons in detail [76]. Within the framework of this model, a qualitative description of PS I transition dynamics was performed. However, this model did not consider the interaction of PS I with oxygen; the kinetic parameters characterizing the interaction of iron–sulfur clusters were taken from an article by Santabarbara et al. [77], who inaccurately describe the thermodynamics of electron transfer reactions on the acceptor side of PS I [78].
To describe diffusion-controlled photosynthetic reactions in spatially heterogeneous photosynthetic systems (for example, PQ diffusion in thylakoid membrane or Pc diffusion in intrathylakoid space), mathematical tools are required that are based on partial differential equations. This is because diffusion restrictions can affect the kinetic characteristics of the system. Integral protein complexes can significantly retard PQ diffusion in the thylakoid membrane [79, 80]. Restricted diffusion of Pc in the narrow lumen space may slow down electron transfer from the cytochrome b6f complex to PS I [81]. The high concentration of buffering groups on the surface of thylakoid membranes significantly decelerates the proton diffusion in the near-surface layer [46, 82-86]. The slowing of ion diffusion in the near-surface layer of lipid membranes is caused, in part, by the presence of a barrier due to structuring of surface water layers near the charged interface [87, 88]. Correct description of diffusion-controlled electron transport reactions in chloroplasts requires considering the spatial heterogeneity of the lamellar system. Within the deterministic approach to finding concentrations of reagents in relation to their spatial coordinates ci(r⃗, t)top, the mathematical apparatus of partial differential equations is commonly used, and the mathematical task is related to integration of the system of diffusion-type equations (2):
where Di is the diffusion coefficient of the i-th component, and the function Fik characterizes its interaction with the other reagents of the system. As an example of modeling the diffusion-controlled processes of electron transport in chloroplasts, we refer to studies in which heterogeneous lateral pH profiles inside and outside the thylakoids were shown to form under conditions of intense ATP synthesis [74, 86]. The results of mathematical modeling are in good agreement with the experimental data on transmembrane pH difference (ΔpH) in chloroplasts, and they also suggest that alkalization of the space between closely adjacent thylakoids in grana can be one of the factors of attenuation of PS II activity [46, 82].
A more complete review on the utilization of the deterministic approach for description of electron and proton transport processes in chloroplasts can be found in recent articles [72-74].
Stochastic modeling of diffusion-controlled bioenergetic processes in the cell has recently become more relevant. The unusually complex topology of the spatially inhomogeneous lamellar system of chloroplasts greatly complicates the task of correct consideration of the diffusion-controlled stages of electron and proton transport within the framework of the deterministic approach. New perspectives are given by using the Monte Carlo method, based on considering spatial movement of diffusing metabolites or electron carriers by the random walk method. The productivity of this approach for describing photosynthetic processes in complex supramolecular bioenergetic systems is obvious, since the “scene” on which these processes are played may be as odd as possible. Without dwelling on the particulars of the description of photosynthetic systems by stochastic dynamics, we will only refer the reader to some original articles and reviews on this topic [72, 79, 89-94].
STRUCTURAL AND FUNCTIONAL ORGANIZATION OF PHOTOSYSTEM I
Molecular organization of photosystem I. Photosystem I is one of two key pigment–protein complexes of the electron transfer chain of oxygenic photosynthesis. This complex is localized in the thylakoid membranes of cyanobacteria, algae, and plants and carries out light-dependent electron transfer accompanied by oxidation of Pc (or cytochrome c6 in cyanobacteria) on the donor side and reduction of Fd or flavodoxin (Fld) on the acceptor side. PS I includes large light-harvesting antenna consisting of Chl and carotenoids molecules. The central part of the complex contains the photosynthetic reaction center catalyzing the formation of transmembrane difference of electrical potentials [95-97].
The X-ray crystallographic structure of the trimeric PS I complex with 2.5 Å resolution was first obtained for the cyanobacterium Synechococcus elongatus [98]. Recently, the PS I structure of plant origin [99] has also been deciphered. Each monomer of cyanobacterial PS I has molecular mass of ~320 kDa and consists of 12 subunits. The electron transfer cofactors and most of the chlorophyll (Chl) molecules and carotenoids of the light-harvesting antenna are localized within two large transmembrane subunits, PsaA and PsaB, and on a small external subunit PsaC. Ninety molecules of Chl and 22 molecules of carotenoids serve as an antenna. Six molecules of Chl a, two molecules of phylloquinone (PhQ), and three iron–sulfur clusters (Fe4S4) participate in the primary stages of electron transfer and subsequent reduction of the external electron acceptors – Fd or Fld. These cofactors implement electrogenic electron transfer through the coupling membrane.
The light-induced primary charge separation is associated with transfer of an electron from an excited primary electron donor, which is a dimer (a special pair) of Chl a molecules, designated P700, to the primary electron acceptor A0 (two pairs of Chl a molecules). Then, the electron is sequentially transferred from A0 to the phylloquinone acceptor A1 and further to Fe4S4 clusters FX, FA, and FB.
The photoreduced terminal cluster FB was shown to directly reduce Fd and Fld [100-103]. The electron transfer cofactors on the acceptor side of PS I are localized in the PsaA and PsaB protein subunits and are organized in the form of two symmetrical branches A and B. The symmetry axis extends from the P700 dimer on the lumen side to the FX cluster located on the stromal side. Each of the branches includes two Chl a molecules (A0A and A0B) and a PhQ molecule (A1A or A1B). Both branches were shown to participate in electron transfer [104] (see also the reviews [77, 105]) at room temperature at ratio ~3 : 1 in favor of branch A in cyanobacterial PS I complexes [106-108].
Kinetics and thermodynamics of electron transfer reactions in photosystem I. In study of primary stages of electron transfer in the reaction center, the use of laser technology that allows recording of fast electron transfer processes plays a crucial role. Upon laser flash excitation, charge separation occurs between P700 and the chlorophyll acceptor A0 to form the primary ion-radical pair P700+A0A– (and/or P700+A0B–) within <100 fs [109]. Then, the electron is sequentially transferred, first to the quinone acceptor A1A/A1B (30 ps), and then to the iron–sulfur cluster FX (200 ns), from which the electron is transferred to the terminal iron–sulfur clusters FA/FB (<1 µs). In the absence of exogenous electron acceptors, charge recombination between [FA/FB]– and P700+ occurs with a characteristic time τ ≈ 30-100 ms. In the absence of FA/FB, the recombination of the P700+FX– pair occurs in the 0.5-5 ms range. In PS I complex lacking all three Fe4S4 clusters, the reduced quinone acceptor A1– and the electron “vacancy” (“hole”) in P700+ recombine with a characteristic time of 20-200 µs [95, 105, 110].
The free energy difference ΔG of electron transfer between A0 and A1 is more than 420 meV, while ΔG of the subsequent electron transfer from A1 to FA/FB is approximately 200 meV [77, 105, 111]. At room temperature, the direct transfer of an electron from A1 to FX occurs along the two alternative branches. Electron transfer along branch A (A1A → FX) is characterized by lifetime τ ≈ 300 ns and activation energy of 110 meV, and electron transfer along branch B (A1B → FX) occurs faster (τ ≈ 11-17 ns) and is almost temperature-independent [108, 112]. It was shown that charge recombination of the ion-radical pair P700+[FA/FB]– in the reaction P700+[FA/FB]– → P700[FA/FB] is characterized by activation energy ~220 meV [113], and charge recombination involving terminal cluster FX (P700+FX– → P700FX) is also temperature-dependent and occurs through intermediate state P700+A1– [95, 114].
MATHEMATICAL MODELING OF ELECTRON TRANSFER REACTIONS IN
PIGMENT–PROTEIN COMPLEX OF PHOTOSYSTEM I
Biochemical reactions in the thylakoid membranes of chloroplasts and in the coupling membranes of photosynthetic bacteria and algae are characterized by complex spatial structural organization and many regulatory cross couplings between enzyme complexes. To analyze quantitatively regulation of energy flows in chloroplasts, methods of chemical kinetics and mathematical modeling are widely used [73]. In their physical nature, electron transfer processes between cofactors of pigment–protein complexes differ from reactions in solutions by the absence of diffusion processes of reagents, and therefore their mathematical description is usually based on probability models whose dynamic properties can be characterized by a system of ordinary differential equations.
One of the key elements of the regulation of oxygenic photosynthesis that determines the direction of biochemical fluxes is the PS I complex; therefore, a considerable amount of both experimental [107, 112, 113, 115-120] and theoretical studies have been devoted to its mathematical modeling [76-78, 111, 114, 121-123]. PS I is one of the most intricately organized pigment–protein photosynthetic complexes (see section “Molecular organization of photosystem I”). Functionally, PS I is a molecular electric generator that converts light energy of the visible part of the sunlight spectral range into directed electron flux through the photosynthetic membrane. When a light quantum is absorbed, the primary processes of photoinduced charge separation proceed with participation of the special pair of Chl molecules P700 and four additional Chl molecules organized symmetrically around the rotation axis passing through the dimer P700 perpendicular to the membrane plane (Fig. 1). Electron transfer from P700 to quinone acceptors A1A and A1B occurs within ~25 ps, and the rate of these reactions does not decrease upon lowering temperature. Since this stage of electron transfer does not limit subsequent reactions, it is usually not included in the mathematical model when analyzing slower photoinduced processes.
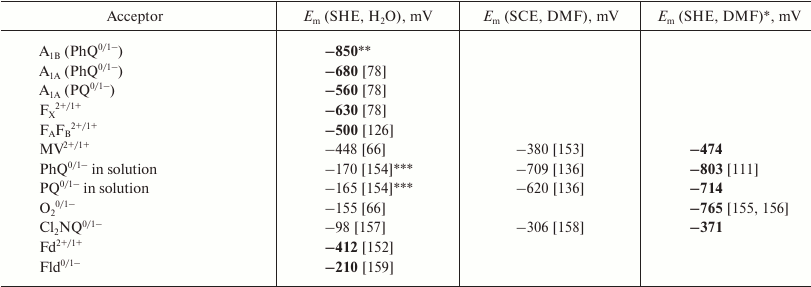
Redox potentials of the electron transfer cofactors in the PS I reaction center are significantly lower compared to cofactors of the similar PS II and the bacterial reaction center, which, on one hand, causes difficulties in experimental determination of their midpoint redox potentials, and, on the other hand, gives rise to incidental reactions in the process of functioning due to the escape of an electron to molecular oxygen with the formation of toxic superoxide oxygen radical. The reaction of molecular oxygen reduction to superoxide anion radical in water at 1 atm pressure of gaseous oxygen has potential –0.33 V relative to the standard hydrogen electrode (SHE) [124]. In air, the oxygen partial pressure is 0.2 atm, and the equilibrium concentration of dissolved oxygen in the water is ~250 µM [125]. The reduction of molecular oxygen to the superoxide anion usually occurs in thermodynamically nonequilibrium conditions, when the resulting superoxide rapidly interacts with other substances or is removed in vivo by superoxide dismutase. In these cases, to characterize the metabolic flux, it is important to know the probability of electron transfer during a bimolecular collision from the donor to molecular oxygen; this probability is more consistent with the redox potential of molecular oxygen calculated for conditions when its concentration in water is 1 M, and this value is given in Table 1.
Table 1. Midpoint redox potentials of
internal and exogenous electron acceptors of PS I in protein matrix,
aqueous environment, and dimethylformamide (DMF)
* Values of Em in DMF relative to aqueous SHE
were calculated by the method described by Ptushenko et al. [111].
** This value is based on Em estimate of quinone
A1A in Milanovsky et al. [78] and
difference of –170 mV between Em of
sites A1B and A1A, calculated by Ptushenko et al.
[111].
*** Data for water-soluble PhQ and PQ analogs.
In Table 1, the first column represents oxidation-reduction state pair of PS I cofactors and various electron acceptors interacting with PS I. The second column characterizes the midpoint potential (Em) of this compound relative to SHE, and the third column shows the Em values relative to the saturated calomel electrode (SCE) in dimethylformamide (DMF). It should be noted that the extremely low values of redox potentials of electron carriers of PS I makes their direct electrochemical determination possible only for terminal acceptors – iron-sulfur clusters FA and FB [126]. The redox potentials of FA and FB are close and almost indistinguishable upon titration at room temperature; therefore, in Table 1 they are denoted together as a joint FA/FB cluster. Redox potentials of other cofactors and their interaction with protein matrix can only be estimated on the basis of various electrostatic models and quantum chemical calculations (section “Redox potential calculation of electron transfer chain cofactors in photosystem I”), molecular dynamics modeling (section “Modeling of photosystem I cofactor interaction with protein environment using molecular dynamics”), and also on the basis of the kinetics of various electron transfer reactions within PS I complexes (section “Kinetic modeling of electron transfer in PS I”) and the interaction of PS I with exogenous electron acceptors (section “Interaction of photosystem I with exogenous electron acceptors”).
Redox potential calculations of electron transferring cofactors in photosystem I. Direct quantum-chemical calculation of redox potentials of PS I cofactors by ab initio methods [127] requires consideration of various electrostatic interactions, which are usually analyzed within the phenomenological Poisson–Boltzmann equation [128, 129]. However, several simplifying assumptions were made, so the total error of such calculations exceeded 0.5 V [111]. A potentially more accurate approach is semiempirical electrostatic calculations in which the midpoint potentials of cofactors in a protein are compared with known electrochemical potentials of these compounds in polar aprotic solvents, such as DMF or nitrobenzene [111, 130-132]. In terms of its physical-chemical properties, DMF is close to peptide groups of the protein core, so the interaction of the cofactor with DMF in solution is similar to its interaction with the protein environment in the binding site, which simplifies the analysis compared to ab initio calculations. However, the use of semiempirical models faces several methodological difficulties. First, in such calculations the values of redox potentials obtained in nonaqueous solvents must be brought to SHE in water, which cannot be done without special assumptions [111]. Second, semiempirical methods do not allow unambiguous determination of the value of the dielectric constant and require detailed conformational analysis of the microscopic structure of the protein [133-135].
To compare data obtained in different solvents, redox potentials of ferrocene and cobaltocene are used in electrochemistry, which are not sensitive to the chemical characteristics of solvent molecules. Conversion of potentials obtained in DMF relative to SCE to potentials in water relative to SHE can be carried out in two stages: first, the potential is calculated with respect to pair Fc/Fc+, and then transition to the potential relative to SHE in water is performed with the correction of +430 mV; this value corresponds to the Em value of the Fc/Fc+ pair relative to the SHE in water [111]. For example, the redox potential of phylloquinone PhQ0/1 relative to SCE in DMF is –709 mV, and Em of the Fc/Fc+ pair under the same conditions is +524 mV [136], so the potential of PhQ relative to Fc is –1233 mV. Thus, with a correction of +430 mV, the value of Em(PhQ0/1) relative to SHE in water is –803 mV. The third column in Table 1 shows the Em values in DMF determined relative to aqueous SHE. These values are closest to the redox potentials of the cofactors in the protein environment (the corresponding values are indicated in Table 1 by bold font).
In several studies [130, 132], the authors take the Em of PhQ in DMF relative to SHE equal to –405 or –455 mV, which is 0.4 V higher than the value obtained against the Fc/Fc+ pair; such a large error arises from ignoring the interfacial jump in the potential between water and DMF, which can be as high as several hundred mV. In a study by Ptushenko et al. [111], it was shown that a consistent calculation of the redox potentials of cofactors in the protein matrix of PS I also requires, in addition to the interfacial potential jump, consideration of heterogeneous distribution of the dielectric constant of the protein complex, which can be carried out by molecular modeling methods that are discussed in the next section of this review.
Modeling of photosystem I cofactor interaction with protein environment using molecular dynamics. Molecular dynamics approaches are widespread in biochemical and biophysical studies, and their use in the last decade has become almost universal. However, although the atomic structure of PS I has been known for more than fifteen years [98], molecular dynamics have not been practically used for the study of PS I until recently. Molecular dynamics modeling was used to study the interaction of trimeric PS I with a layer of detergent molecules of n-dodecyl-β-D-maltoside toroidally distributed around the complex [137]. Within the same model, the interaction of PS I and [Fe-Fe]-hydrogenase in a complex capable of photocatalytic generation of molecular hydrogen was studied [138]. The details of the molecular interaction of the PsaC and PsaD subunits in the stromal portion of PS I from rice chloroplasts (Oryza sativa L.) were studied using molecular dynamics and bioinformatics methods [139]. The molecular mechanism of primary reactions of photoinduced charge separation in PS I from the cyanobacterium Thermosynechococcus elongatus was analyzed by combined methods of molecular dynamics, continual electrostatics, and quantum chemistry [140]. Molecular dynamics modeling allowed us to follow in real time the conformational changes taking place in the protein during the electron transfer reactions along the two branches of the cofactors. A map of the structural mobility of PS I pigment–protein complex was calculated, which included a relatively rigid hydrophobic core and more mobile peripheral regions. The molecular dynamics model made it possible to calculate the dielectric response of the system accompanying the primary and secondary charge separation reactions, while the direct chemical interaction of the chlorophyll and quinone molecules at the A0 and A1 sites with the nearby protein environment was calculated ab initio by quantum chemistry methods. The calculation of the reorganization energy of charge separation reactions was carried out with a combination of molecular dynamics methods and continual electrostatics. Factors that could lead to asymmetry between the two branches of the electron carriers in PS I were analyzed. Based on the calculation of the redox potentials and the reorganization energy of the formation of the primary P700+A0– and secondary P700+A1– ion radical pairs, it was shown that a small difference in the redox potentials between the primary electron acceptors A0A and A0B in A and B branches leads to an asymmetry of electron transfer in ratio of 70 : 30 in favor of branch A [106]. The quinone reduction reactions in A1 sites are thermodynamically irreversible in the sub-microsecond range and are accompanied by additional increase in asymmetry between the cofactor branches of PS I. A comparison of redox potentials of cofactors in PS I pigment–protein complexes of cyanobacteria and plants was carried out by combined methods of molecular dynamics, continual electrostatics, and quantum chemistry [132]. The calculations showed that the protonated state of the carboxyl group of Asp-B575 can be different in plants and cyanobacteria, which should reduce the redox potentials of quinones at binding sites A1A and A1B in plant PS I compared to cyanobacteria.
Kinetic modeling of electron transfer in PS I upon flash excitation. The mathematical description of the regulation of electron flow in photosynthetic complexes is largely facilitated by the fact that electron transfer processes can be initiated by short light pulses, and subsequent reactions can be recorded by various physical methods, for example, using spectral measurements. The kinetic and thermodynamic parameters thus obtained are used in the construction of a general kinetic model for the regulation of photosynthetic electron flows.
In one of the first mathematical models of PS I, kinetics of P700 recombination was analyzed for complexes in which the site of the secondary electron acceptor A1 was occupied either by native PhQ or by PQ (mutant strain menB), and the complexes contained different numbers of iron–sulfur clusters FX, FA, and FB [121]. In later works, kinetic models considered the presence of two symmetrically arranged quinone binding sites A1A and A1B, whose reactions with nearby cofactors P700, A0, and FX proceeded at different rates [77, 78, 122, 123]. In the study of Milanovsky et al. [78], simulation of photoinduced electron transfer reactions in the acceptor part of PS I was carried out using the kinetic scheme shown in Fig. 3. In this scheme, the secondary ion-radical pair P700+A1– was considered as the initial state for kinetic modeling, with electron flux being distributed between branches A and B, reaching quinones A1A and A1B with probabilities δ and (1 – δ), respectively. The kinetics of electron transfer in the complex itself and its interaction with exogenous acceptors is described by the following system of ordinary differential equations:
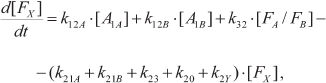 (5)
(5)
Fig. 3. Kinetic model of PS I used for interpretation of P700+ recombination kinetics. P700+ recombination pathways are marked by dashed lines. Reaction rate constants are noted above the corresponding arrows as ki. Direct interaction of FX with exogenous electron acceptors was only considered for FX–core complexes (dashed lines). Estimates of redox potentials of cofactors participating in different stages of charge separation and external acceptors are shown on the right; references to studies from which the values were taken are given in square brackets.
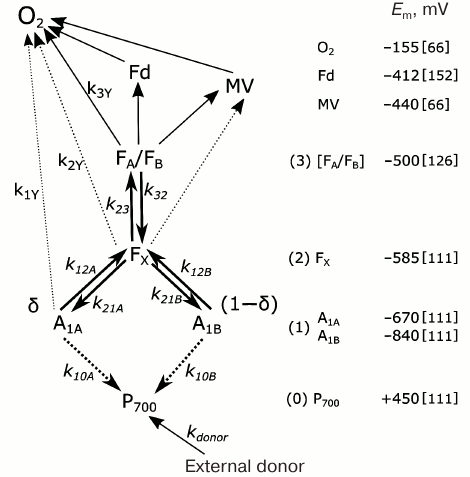
Here kij denote rate constants of monomolecular electron transfer reactions from cofactor i to cofactor j; indices 0, 1, 2, and 3 correspond to electron carriers P700, A1, FX, and [FA/FB]; kdonor is the rate constant of P700+ reduction by the external electron donor 2,6-dichlorophenolindophenol (DCPIP); kiY denote rate constants of electron transfer from various PS I cofactors to exogenous electron acceptors, in particular, molecular oxygen or methyl viologen (MV). Reactions with external acceptors were modeled using the Michaelis–Menten equation; in this approximation, the rate of electron transfer from PS I cofactor to external acceptor (kext) depends on its concentration [EA] nonlinearly:
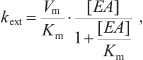 (8)
(8)where Vm is the maximum reaction rate, and Km is the Michaelis constant (equal to the dissociation constant of the external acceptor–PS I complex). If [EA] << Km, the reaction rate is a linear function of the concentration [EA]; therefore, the parameter ka = Vm/Km can be considered an effective bimolecular rate constant for the interaction of the exogenous acceptor with PS I.
The rate of electron transfer reactions between cofactors decreases exponentially as a function of distance between them; therefore, electron transfer between distant cofactors is usually carried out in steps with participation of intermediate carriers between which thermodynamic equilibrium is established. For example, the recombination reaction of the ion pair P700+[FA/FB]– occurs through activation reduction of acceptors FX and A1 [141], so experimentally observed recombination rate k30 can be expressed through rate constant k10A of direct recombination of the ion pair P700+A1A– and two equilibrium constants of intermediate reactions K23 and K12A:
In this case, only recombination through quinone in branch A is considered, since the midpoint potential of quinone A1A is at least 100 mV more positive than the potential of quinone A1B [111, 130], which makes the FX– → A1A– path more likely than intermediate reduction of quinone A1B (values of redox potentials of cofactors FX, A1A, and A1B are given in Fig. 3 and Table 1).
The main kinetic and thermodynamic parameters of PS I characterizing electron transfer reactions between cofactors A1A, A1B, FX, and FA/B, are given in Table 2. The rate of direct electron transfer between FX and FA/B clusters is not measured directly, but the data obtained by Diaz-Quintana et al. [100] indicate that direct electron transfer reactions in the acceptor part of PS I are limited by the A1A → FX stage, so rate constant k23 can be estimated as 107 s–1. The asymmetry factor δ was assumed to be 80% [107, 108].
Table 2. Kinetic and thermodynamic
parameters of electron transfer reactions between PS I cofactors
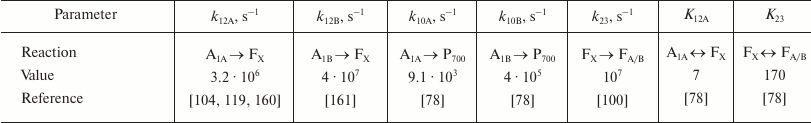
Note: Reaction rate constants kij are given in
accordance with the scheme in Fig. 3;
equilibrium constants Kij were calculated according
to Eqs. (3)-(7). The data are for room temperature.
Interaction of photosystem I with exogenous electron acceptors. Recently, we studied the interaction of various samples of PS I complexes from cyanobacterium Synechocystis sp. PCC 6803 with exogenous electron acceptors: MV, the high-potential derivative of naphthoquinone 2,3-dichloronaphthoquinone (Cl2NQ), and molecular oxygen [120]. The efficiency of the interaction of PS I with the acceptors was determined by measuring laser-induced absorption changes at wavelength 820 nm, which reflects the reduction kinetics of the photooxidized chlorophyll dimer P700. Four types of PS I samples were investigated: (i) intact complexes from wild-type cyanobacteria (WT) containing native PhQ and a full set of redox cofactors; (ii) FX–core-complexes devoid of iron–sulfur clusters FA/FB and external subunits PsaC, PsaD and PsaE; (iii) complexes from mutant cyanobacterium strain menB with PQ in the A1 binding site, and (iv) complexes from menB mutant with high-potential Cl2NQ in the A1 site.
Exogenous Cl2NQ was shown to efficiently accept electrons from reduced terminal iron–sulfur clusters FA/FB in PS I samples from the wild strain of Synechocystis sp. PCC 6803 and from the menB mutant, and less efficiently from the reduced FX cluster in FX–core samples. In all cases, the efficiency of interaction between Cl2NQ and PS I was significantly higher than that of the conventionally used artificial acceptor MV. In addition, the interaction of MV with PS I samples from menB containing Cl2NQ in the A1 site was studied. In this case, the effect of MV on the kinetics of charge recombination in PS I was significantly different from the effect observed in the other three types of samples.
The data were analyzed using a kinetic model considering electron transfer reactions between P700, quinone acceptor A1, iron–sulfur clusters, and exogenous electron donor and electron acceptors, including MV, Cl2NQ, and molecular oxygen [78]. The kinetic parameters of the forward and backward electron transfer reactions were obtained by deconvolution (decomposition into components) of the reduction kinetics of P700+. In accordance with the model, the acceleration of charge recombination in the reaction P700+[FA/FB]– → P700[FA/FB] observed upon substitution of PhQ by PQ in the A1 site indicates that the backward electron transfer from terminal acceptors to P700+ occurs through the transient reduction of quinone at the A1 site.
The results of kinetic modeling of electron transfer reactions in PS I at various concentrations of exogenous acceptor MV are shown in Fig. 4. The experimentally observed kinetics of reduction of photooxidized P700 is marked by black dots and simulated kinetics by thin black line (1). In the absence of external acceptor (Fig. 4a), two components can be discerned in the kinetics, which are associated with charge recombination from terminal acceptors of PS I (line 2) and P700+ reduction by external donor DCPIP (line 3), which compensates for electron transfer from the terminal acceptors of PS I to molecular oxygen dissolved in the incubation medium (line 4). Upon addition of exogenous acceptor, competing with oxygen for electron transfer from terminal acceptors (Fig. 4b, line 5), progressively fewer electrons recombine from intra-protein acceptors of PS I, and at 50 µM MV concentration the recombination kinetics of P700 becomes completely determined by electron transfer from reduced DCPIP (Fig. 4c).
Fig. 4. Effect of different concentrations of MV as exogenous acceptor on reduction of P700+ in wild-type PS I. The experimental reduction kinetics of P700 are shown by black dots, and the modeled kinetics is a solid black line (1); fractions of reduced P700 are shown by thick dashed lines (simple dashed line 2 for reduction by terminal acceptors and dot-dashed line 3 for external DCPIP donor); reduced external acceptors are shown by colored dot lines (red, 4 for molecular oxygen and blue, 5 for MV).
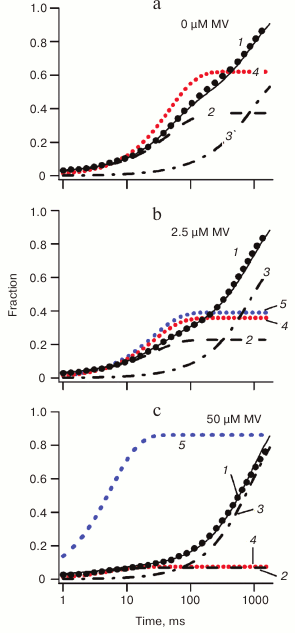
The kinetic model indicated that ΔG of electron transfer reaction FX → FA/FB for native PS I is –130 meV, and ΔG values of reaction A1 → FX are –50 and –220 meV for the branches A and B, respectively. In the case of PS I complex isolated from the menB mutant containing PQ molecule in the A1 binding site, the reaction A1 → FX was shown to be endergonic (ΔG0 = +75 meV). The interaction of PS I with exogenous electron acceptors MV and Cl2NQ was quantitatively described by Michaelis–Menten kinetics. The second order rate constants of the electron transfer from FA/FB and FX to Cl2NQ were calculated. The model indicates that the probability of incidental formation of superoxide anion radical in the A1 site due to reduction of molecular oxygen by PS I during the normal functioning of the complex (the Mehler reaction) may exceed 0.3% of the total electron flux. Since molecular oxygen is one of the main products of oxygenic photosynthesis, it is necessary to prevent its interaction with cofactors of the electron transport chain. Obviously, the high rate of electron transfer from PhQ in the A1 site to terminal Fe4S4 clusters (characteristic time <10–6 s) in the native system is necessary to prevent the interaction of PhQ with molecular oxygen.
The kinetic and thermodynamic parameters of reactions of PS I with exogenous acceptors are summarized in Table 3. The effective bimolecular rate constants ka and dissociation constants of protein–acceptor complexes Km, determined in accordance with Eq. (1), are shown. Comparison of the rates of interaction between exogenous acceptors and different types of PS I samples containing different quinones and different amounts of iron–sulfur clusters made it possible to determine that the interaction of PS I with native water-soluble Fd and Fld acceptors occurs on the surface of the PsaC subunit near iron–sulfur clusters FA and FB, whereas reactions with less polar molecular oxygen and artificial acceptor MV are more efficient within the A1 site itself but lower that the rate of transfer from A1 to Fx.[78]. Molecular oxygen and the oxidized form of ascorbic acid were previously shown to directly oxidize PQ and Cl2NQ in the A1 site under continuous illumination [68]. Analysis of PS I molecular structure [98] revealed two symmetrically arranged cavities partially filled with water near the A1A and A1B sites on the inwardly facing surface of the lipid bilayer of the PsaA/PsaB heterodimer [78]. These cavities are indicated in Fig. 5 with purple spheres. The cavities are connected with aqueous phase at the interface of PsaA/PsaE and PsaB/PsaC subunits, respectively, and they have sufficiently high volume to accommodate two benzene rings of the MV molecule ~0.6 nm from the oxygen atoms of the quinones in the A1A and A1B sites. Through the same channels, molecular oxygen from the aqueous phase can approach the A1A and A1B sites.
Table 3. Parameters of interaction of PS I
with external cofactors according to the kinetic model

Note: Definitions of constants are provided in “Mathematical
modeling of electron transfer reactions in pigment–protein
complex of photosystem I”. Data for Fd and Fld are from Setif [162]; data for MV, Cl2NQ, and
O2 are from Milanovsky et al. [78].
Fig. 5. Structure of acceptor side of PS I. Phylloquinone molecules A1A and A1B are shown in blue (oxygen atoms are red); Fe and S atoms of iron–sulfur clusters FX and FA are shown in blue and yellow, respectively. The intra-protein cavities that are connected with the aqueous phase in the interface between the PsaA/PsaE and PsaB/PsaC subunits are indicated in purple. Secondary structure of the protein is shown by gray spirals.
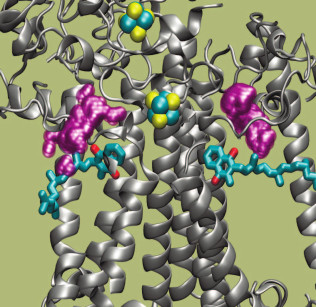
Taking into account the second-order rate constant of electron transfer from Cl2NQ in the A1 site of PS I to oxygen (Table 3), the oxygen concentration in water, the significantly more negative value of the midpoint redox potential of PhQ as compared to Cl2NQ (Table 1), and the rate constant of electron transfer from A1 to FX [112], it is possible to estimate the probability of oxygen reduction in the A1 site (bypassing the iron–sulfur clusters FX, FA, and FB) with the formation of superoxide radical, which is >0.3% under experimental conditions. A direct indication of the possibility of oxygen reduction directly by phylloquinone A1 was demonstrated on isolated PS I complexes [142]. Oxygen is one of the main products of photosynthesis; therefore, it is necessary to prevent its interaction with the redox cofactors of PS I for effective functioning of the complex. Apparently, this factor is responsible for the evolutionary significance of the high rate of electron transfer from phylloquinone A1 to terminal iron–sulfur clusters FA/FB and further to native exogenous acceptors, Fd and Fld (see Table 3). However, it should be noted that with deficit of exogenous electron acceptors and with high illumination (when the PS I acceptor pool is partially reduced), the flux of electrons to molecular oxygen with formation of superoxide radical increases [143]. Under these conditions, to prevent damage to the photosynthetic apparatus by reactive oxygen species, a pseudocyclic pathway of electron transfer involving superoxide dismutase is activated (Fig. 2). The mechanism and features of this path of electron transfer are discussed in the next section.
PSEUDOCYCLIC ELECTRON TRANSPORT IN CHLOROPLASTS
(“WATER–WATER” CYCLE)
The problem of interaction of molecular oxygen (O2) with the electron transport chain of chloroplasts has been the subject of discussions for many decades since the discovery of the Mehler reaction [41-43, 67]. As mentioned above, molecular oxygen is an effective electron acceptor (Em = –155 mV) that can accept an electron from low-potential acceptors of PS I. As a result of this reaction, superoxide radicals (O2•–) are formed, which then transform into other ROS [41-43, 65]. Superoxide radicals O2•– dismutate with the formation of H2O2 (2 O2•– → H2O2 + O2) [67, 69]. This reaction is catalyzed by the superoxide dismutase bound to the thylakoid membrane [41]. In intact chloroplasts, H2O2 molecules are converted to water by ascorbate reducing H2O2 to water. This reaction is catalyzed by ascorbate peroxidase: AscH– + ½ H2O2 → Asc⋅– + H2O, where AscH– and Asc⋅– denote ascorbate and monodehydroascorbate [41]. Regeneration of AscH– in chloroplasts can occur due to the reduction of Asc⋅– by low-potential PS I acceptors [144-148]. In addition to ascorbate, an important role in the protection of plants from oxidative stress, which is initiated by the ROS formation, is played by the glutathione pathway of ROS inactivation [41, 42, 70, 71].
Considering the complete balance of O2 production and O2 consumption reactions, associated with electron transfer from water to PS I and then to O2, the total number of O2 released and absorbed molecules are equal. Two electrons, donated by one water molecule splitted in PS II, are transferred to the O2 molecule reduced by PS I. Eventually, one molecule of water is formed. This flow of electrons is sometimes called a water–water cycle: H2O → PS II (½ O2 ↑) → (2e–) → PS I → O2 → H2O (½ O2 ↓). This pathway of electron transport has been termed in the literature as the “pseudocyclic electron transport” or “water–water” cycle [41]. Estimates of the relative contribution of pseudocyclic electron transport available in the literature are widely dispersed. It is generally believed that the contribution of this cycle in plants does not exceed 10-15% of the noncyclic electron flux [3, 41-44]. However, there is some evidence that the electron flux from PS I to O2 can be much higher. In higher plants, under certain conditions, it reaches approximately 30% of the total flux of electrons donated by PS II to the electron transport chain [41, 75, 149]. In other oxygenic photosynthetic organisms, electron flow from PS I to O2 can reach 40-50% of the total electron flux [48, 143].
The nature of electron donors in the Mehler reaction remains a matter of debates in the literature. A recent review [143] considers the hypothesis that PhQ in the A1 site of PS I from plant thylakoids is the main electron donor for molecular oxygen.
The question of the physiological significance of pseudocyclic electron transport involving O2 is debatable [3, 41-46]. This cycle is not associated with the formation of NADPH. However, its functioning, as in the case of cyclic electron transport around PS I, is connected with the operation of proton pumps generating a transmembrane difference in the electrochemical potentials of hydrogen ions (ΔµH+), which is the energy source for the operation of ATP synthase complexes. This allows synthesis of additional ATP molecules, providing the appropriate ATP/NADPH ratio [3, 41]. Simple estimates show that even a relatively small contribution of pseudocyclic electron transport (10%) to the total electron flux makes it possible to maintain the stoichiometry of the products of the light stage of photosynthesis ATP/NADPH = 3/2 which is necessary for optimal CBC functioning [47]. There are indications that the water–water cycle can play the role of a “starter” that triggers energy-dependent stages of photosynthesis in the initial stages of chloroplast illumination [41, 42]. It is also assumed that the outflow of electrons from PS I to molecular oxygen avoids the accumulation of an excessive number of reduced carriers on the acceptor side of PS I [44].
The flow of electrons from PS I to O2 is a dynamic characteristic of chloroplasts; its value can vary during the induction period observed after beginning of illumination of chloroplasts adapted to darkness. After a sufficiently long adaptation of the PSA to darkness (usually this time is ≥10-20 min), key CBC enzymes become inactive [36, 40, 64, 150, 151], and therefore a rapid accumulation of excess reduced NADPH occurs. Accordingly, the outflow of electrons from PS I to the CBC slows down. For effective CBC operation, regeneration of NADP+ – the physiological acceptor of PS I – is required. Pseudocyclic transport of electrons, ensuring generation of ΔµH+, should promote synthesis of ATP, thereby creating conditions for CBC functioning. For these reasons, at the beginning of the induction period, the contribution of pseudocyclic electron transport can be significantly higher than in the steady state, when the CBC enzymes are active and noncyclic electron flux (PS I → CBC) dominates over alternative pathways. Mathematical modeling of such flow redistribution has been described earlier (see reviews [73, 74]). As noted above, pseudocyclic electron transport can be a link in the regulation of electron transport, which, along with cyclic electron flow around the PS I, allows optimal balance between the products of the light stage of photosynthesis, ATP and NADPH. The electron flux from PS I to O2 can serve as a kind of bypass that draws away excess electrons from the acceptor site of PS I to oxygen, thereby protecting the photosynthetic apparatus from possible damage under stress conditions (dehydration, excess light [44]).
Charge transfer through electron transport chains of oxygenic photosynthetic organisms can be carried out through noncyclic, cyclic, and pseudocyclic modes of transport. This review describes the key role of the interaction of PS I with water-soluble electron acceptors in the regulation of these flows. The electron transfer reactions between redox cofactors of PS I and its interaction with native and exogenous donors and acceptors of electrons have been discussed. We have shown that kinetic modeling of PS I during pulsed excitation of the complex makes it possible to determine the kinetic and thermodynamic parameters of charge transfer reactions and to estimate the rate of reactive oxygen species generation in PS I complexes under different environmental conditions.
Acknowledgments
The authors are grateful to M. D. Mamedov and B. V. Trubitsyn for productive discussion of the results.
This study was supported by the Russian Science Foundation (project No. 17-14-01323). Analysis of the molecular dynamics simulation data, shown in Fig. 5 and in the section “Modeling of photosystem I cofactor interaction with protein environment using molecular dynamics”, had partial financial support from the Russian Science Foundation (project No. 14-50-00029).
REFERENCES
1.Allen, J. F. (2003) Cyclic, pseudocyclic and
noncyclic photophosphorylation: new links in the chain, Trends Plant
Sci., 8, 15-19.
2.Joliot, P., and Joliot, A. (2006) Cyclic electron
flow in C3 plants, Biochim. Biophys. Acta, 1757,
362-368.
3.Miyake, C. (2010) Alternative electron flows
(water–water cycle and cyclic electron flow around PSI) in
photosynthesis: molecular mechanisms and physiological functions,
Plant Cell Physiol., 51, 1951-1963.
4.Shikanai, T. (2007) Cyclic electron transport
around photosystem I: genetic approaches, Annu. Rev. Plant
Biol., 58, 199-217.
5.Breyton, C., Nandha, B., Johnson, G. N., Joliot,
P., and Finazzi, G. (2006) Redox modulation of cyclic electron flow
around photosystem I in C3 plants, Biochemistry, 45,
13465-13475.
6.Munekage, Y., Hashimoto, M., Miyake, C., Tomizawa,
K.-I., Endo, T., Tasaka, M., and Shikanai, T. (2004) Cyclic electron
flow around photosystem I is essential for photosynthesis,
Nature, 429, 579-582.
7.Iwai, M., Takizawa, K., Tokutsu, R., Okamuro, A.,
Takahashi, Y., and Minagawa, J. (2010) Isolation of the elusive
supercomplex that drives cyclic electron flow in photosynthesis,
Nature, 464, 1210-1213.
8.Strand, D. D., Fisher, N., and Kramer, D. M. (2016)
Distinct energetics and regulatory functions of the two major cyclic
electron flow pathways in chloroplasts, in Chloroplasts: Current
Research and Future Trends (Kirchhoff, H., ed.) Caister Academic
Press, pp. 89-100.
9.Sazanov, L. A., Burrows, P., and Nixon, P. J.
(1996) Detection and characterization of a complex I-like NADH-specific
dehydrogenase from pea thylakoids, Biochem. Soc. Trans.,
24, 739-743.
10.Peng, L., Shimizu, H., and Shikanai, T. (2008)
The chloroplast NAD(P)H dehydrogenase complex interacts with
photosystem I in Arabidopsis, J. Biol. Chem., 283,
34873-34879.
11.Peng, L., Fukao, Y., Fujiwara, M., Takami, T.,
and Shikanai, T. (2009) Efficient operation of NAD(P)H dehydrogenase
requires supercomplex formation with photosystem I via minor LHCI in
Arabidopsis, Plant Cell, 21, 3623-3640.
12.Shikanai, T., Endo, T., Hashimoto, T., Yamada,
Y., Asada, K., and Yokota, A. (1998) Directed disruption of the tobacco
ndhB gene impairs cyclic electron flow around photosystem I,
Proc. Natl. Acad. Sci. USA, 95, 9705-9709.
13.Tagawa, K., Tsujimoto, H. Y., and Arnon, D. I.
(1963) Role of chloroplast ferredoxin in the energy conversion process
of photosynthesis, Proc. Natl. Acad. Sci. USA, 49,
567-572.
14.Yamamoto, H., Kato, H., Shinzaki, Y., Horiguchi,
S., Shikanai, T., Hase, T., Endo, T., Nishioka, M., Makino, A.,
Tomizawa, K.-I., and Miyake, C. (2006) Ferredoxin limits cyclic
electron flow around PSI (CEF-PSI) in higher plants –
stimulation of CEF-PSI enhances non-photochemical quenching of Chl
fluorescence in transplastomic tobacco, Plant Cell Physiol.,
47, 1355-1371.
15.Hald, S., Nandha, B., Gallois, P., and Johnson,
G. N. (2008) Feedback regulation of photosynthetic electron transport
by NADP(H) redox poise, Biochim. Biophys. Acta, 1777,
433-440.
16.Hald, S., Pribil, M., Leister, D., Gallois, P.,
and Johnson, G. N. (2008) Competition between linear and cyclic
electron flow in plants deficient in photosystem I, Biochim.
Biophys. Acta, 1777, 1173-1183.
17.Bendall, D. S., and Manasse, R. S. (1995) Cyclic
photophosphorylation and electron transport, Biochim. Biophys.
Acta, 1229, 23-38.
18.Munekage, Y., Hojo, M., Meurer, J., Endo, T.,
Tasaka, M., and Shikanai, T. (2002) PGR5 is involved in cyclic electron
flow around photosystem I and is essential for photoprotection in
Arabidopsis, Cell, 110, 361-371.
19.DalCorso, G., Pesaresi, P., Masiero, S., Aseeva,
E., Schunemann, D., Finazzi, G., Joliot, P., Barbato, R., and Leister,
D. (2008) A complex containing PGRL1 and PGR5 is involved in the switch
between linear and cyclic electron flow in Arabidopsis,
Cell, 132, 273-285.
20.Hertle, A. P., Blunder, T., Wunder, T., Pesaresi,
P., Pribil, M., Armbruster, U., and Leister, D. (2013) PGRL1 is the
elusive ferredoxin-plastoquinone reductase in photosynthetic cyclic
electron flow, Mol. Cell, 49, 511-523.
21.Munekage, Y. N., Genty, B., and Peltier, G.
(2008) Effect of PGR5 impairment on photosynthesis and growth in
Arabidopsis thaliana, Plant Cell. Physiol., 49,
1688-1698.
22.Suorsa, M., Grieco, M., Nurmi, M., Pietrzykowska,
M., Rantala, M., Paakkarinen, V., Tikkanen, M., Jansson, S., Aro, E.,
Jarvi, S., Grieco, M., Nurmi, M., Pietrzykowska, M., Rantala, M.,
Kangasjarvi, S., Paakkarinen, V., Tikkanen, M., Jansson, S., and Aro,
E. (2012) Proton gradient regulation 5 is essential for proper
acclimation of Arabidopsis photosystem I to naturally and
artificially fluctuating light conditions, Plant Cell,
24, 2934-2948.
23.Benz, J. P., Lintala, M., Soll, J., Mulo, P., and
Bolter, B. (2010) A new concept for ferredoxin-NADP(H) oxidoreductase
binding to plant thylakoids, Trends Plant Sci., 15,
608-613.
24.Ohyama, K., Fukuzawa, H., Kohchi, T., Shirai, H.,
Sano, T., Sano, S., Umesono, K., Shiki, Y., Takeuchi, M., Chang, Z.,
Aota, S., Inokuchi, H., and Ozeki, H. (1986) Chloroplast gene
organization deduced from complete sequence of liverwort marchantia
polymorpha chloroplast DNA, Nature, 322, 572-574.
25.Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi,
T., Hayashida, N., Matsubayashi, T., Zaita, N., Chunwongse, J.,
Obokata, J., Yamaguchi-Shinozaki, K., Ohto, C., Torazawa, K., Meng, B.
Y., Sugita, M., Deno, H., Kamogashira, T., Yamada, K., Kusuda, J.,
Takaiwa, F., Kato, A., Tohdoh, N., Shimada, H., and Sugiura, M. (1986)
The complete nucleotide sequence of the tobacco chloroplast genome: its
gene organization and expression, EMBO J., 5,
2043-2049.
26.Battchikova, N., Eisenhut, M., and Aro, E.-M.
(2011) Cyanobacterial NDH-1 complexes: novel insights and remaining
puzzles, Biochim. Biophys. Acta, 1807, 935-944.
27.Ogawa, T., and Mi, H. (2007) Cyanobacterial NADPH
dehydrogenase complexes, Photosynth. Res., 93, 69-77.
28.Endo, T., Shikanai, T., Sato, F., and Asada, K.
(1998) NAD(P)H dehydrogenase-dependent, antimycin A-sensitive electron
donation to plastoquinone in tobacco chloroplasts, Plant Cell
Physiol., 39, 1226-1231.
29.Joet, T., Cournac, L., Horvath, E. M., Medgyesy,
P., and Peltier, G. (2001) Increased sensitivity of photosynthesis to
antimycin A induced by inactivation of the chloroplast ndhB
gene. Evidence for a participation of the NADH-dehydrogenase complex to
cyclic electron flow around photosystem I, Plant Physiol.,
125, 1919-1929.
30.Kubota, H., Sakurai, I., Katayama, K., Mizusawa,
N., Ohashi, S., Kobayashi, M., Zhang, P., Aro, E.-M., and Wada, H.
(2010) Purification and characterization of photosystem I complex from
Synechocystis sp. PCC 6803 by expressing histidine-tagged
subunits, Biochim. Biophys. Acta, 1797, 98-105.
31.Johnson, G. N. (2011) Physiology of PSI cyclic
electron transport in higher plants, Biochim. Biophys. Acta,
1807, 384-389.
32.Yamamoto, H., Peng, L., Fukao, Y., and Shikanai,
T. (2011) An Src homology 3 domain-like fold protein forms a ferredoxin
binding site for the chloroplast NADH dehydrogenase-like complex in
Arabidopsis, Plant Cell, 23, 1480-1493.
33.Leister, D., and Shikanai, T. (2013) Complexities
and protein complexes in the antimycin A-sensitive pathway of cyclic
electron flow in plants, Front. Plant Sci., 4, 161.
34.Burrows, P. A., Sazanov, L. A., Svab, Z., Maliga,
P., and Nixon, P. J. (1998) Identification of a functional respiratory
complex in chloroplasts through analysis of tobacco mutants containing
disrupted plastid Ndh genes, EMBO J., 17,
868-876.
35.Motohashi, K., and Hisabori, T. (2006) HCF164
receives reducing equivalents from stromal thioredoxin across the
thylakoid membrane and mediates reduction of target proteins in the
thylakoid lumen, J. Biol. Chem., 281, 35039-35047.
36.Balsera, M., Schurmann, P., and Buchanan, B. B.
(2016) Redox regulation in chloroplasts, in Chloroplasts. Current
Research and Future Trends (Kirchhoff, H., ed.) Caister Academic
Press, pp. 187-208.
37.Dietz, K.-J., and Pfannschmidt, T. (2011) Novel
regulators in photosynthetic redox control of plant metabolism and gene
expression, Plant Physiol., 155, 1477-1485.
38.Woodrow, I. E., and Berry, J. A. (1988) Enzymatic
regulation of photosynthetic CO2, fixation in C3 plants,
Ann. Rev. Plant Physiol. Plant Mol. Biol., 39,
533-594.
39.Andersson, I. (2007) Catalysis and regulation in
rubisco, J. Exp. Bot., 59, 1555-1568.
40.Michelet, L., Zaffagnini, M., Morisse, S.,
Sparla, F., Perez-Perez, M. E., Francia, F., Danon, A., Marchand, C.
H., Fermani, S., Trost, P., and Lemaire, S. D. (2013) Redox regulation
of the Calvin–Benson cycle: something old, something new,
Front. Plant Sci., 4, 470.
41.Asada, K. (1999) The water–water cycle in
chloroplasts: scavenging of active oxygens and dissipation of excess
photons, Ann. Rev. Plant Physiol. Plant Mol. Biol., 50,
601-639.
42.Asada, K. (2006) Production and scavenging of
reactive oxygen species in chloroplasts and their functions, Plant
Physiol., 141, 391-396.
43.Badger, M. R., Von Caemmerer, S., Ruuska, S., and
Nakano, H. (2000) Electron flow to oxygen in higher plants and algae:
rates and control of direct photoreduction (Mehler reaction) and
rubisco oxygenase, Philos. Trans. R. Soc. London, 355,
1433-1446.
44.Ort, D. R., and Baker, N. R. (2002) A
photoprotective role for O2 as an alternative electron sink
in photosynthesis? Curr. Opin. Plant. Biol., 5,
193-198.
45.Egorova, E. A., and Bukhov, N. G. (2006)
Mechanisms and functions of photosystem I-related alternative electron
transport pathways in chloroplasts, Russ. J. Plant Physiol.,
53, 571-582.
46.Kuvykin, I. V., Ptushenko, V. V., Vershubskii, A.
V., and Tikhonov, A. N. (2011) Regulation of electron transport in C3
plant chloroplasts in situ and in silico: short-term
effects of atmospheric CO2 and O2, Biochim.
Biophys. Acta, 1807, 336-347.
47.Kramer, D. M., Avenson, T. J., and Edwards, G. E.
(2004) Dynamic flexibility in the light reactions of photosynthesis
governed by both electron and proton transfer reactions, Trends
Plant Sci., 9, 349-357.
48.Eberhard, S., Finazzi, G., and Wollman, F.-A.
(2008) The dynamics of photosynthesis, Annu. Rev. Genet.,
42, 463-515.
49.Allakhverdiev, S. I., and Murata, N. (2004)
Environmental stress inhibits the synthesis de novo of
proteins involved in the photodamage-repair cycle of photosystem II in
Synechocystis sp. PCC 6803, Biochim. Biophys. Acta,
1657, 23-32.
50.Demmig-Adams, B., Cohu, C. M., Muller, O., and
Adams, W. W. (2012) Modulation of photosynthetic energy conversion
efficiency in nature: from seconds to seasons, Photosynth. Res.,
113, 75-88.
51.Kasahara, M., Kagawa, T., Oikawa, K., Suetsugu,
N., Miyao, M., and Wada, M. (2002) Chloroplast avoidance movement
reduces photodamage in plants, Nature, 420, 829-832.
52.Murata, N., Allakhverdiev, S. I., and Nishiyama,
Y. (2012) The mechanism of photoinhibition in vivo:
re-evaluation of the roles of catalase, alpha-tocopherol,
non-photochemical quenching, and electron transport, Biochim.
Biophys. Acta, 1817, 1127-1133.
53.Foyer, C. H., Neukermans, J., Queval, G., Noctor,
G., and Harbinson, J. (2012) Photosynthetic control of electron
transport and the regulation of gene expression, J. Exp. Bot.,
63, 1637-1661.
54.Tikkanen, M., Grieco, M., Nurmi, M., Rantala, M.,
Suorsa, M., and Aro, E.-M. (2012) Regulation of the photosynthetic
apparatus under fluctuating growth light, Philos. Trans. R. Soc.
Lond. Biol., 367, 3486-3493.
55.Tikkanen, M., Mekala, N. R., and Aro, E.-M.
(2014) Photosystem II photoinhibition-repair cycle protects photosystem
I from irreversible damage, Biochim. Biophys. Acta, 1837,
210-215.
56.Li, Z., Wakao, S., Fischer, B. B., and Niyogi, K.
K. (2009) Sensing and responding to excess light, Annu. Rev. Plant
Biol., 60, 239-260.
57.Govindjee (1995) Sixty-three years since Kautsky:
chlorophyll a fluorescence, Aust. J. Plant Physiol.,
22, 131-160.
58.Stirbet, A., and Govindjee (2011) On the relation
between the Kautsky effect (chlorophyll a fluorescence
induction) and photosystem II: basics and applications of the OJIP
fluorescence transient, J. Photochem. Photobiol. B, 104,
236-257.
59.Stirbet, A., and Govindjee (2012) Chlorophyll
a fluorescence induction: a personal perspective of the thermal
phase, the J-I-P rise, Photosynth. Res., 113, 15-61.
60.Rochaix, J.-D. (2014) Regulation and dynamics of
the light-harvesting system, Annu. Rev. Plant Biol., 65,
287-309.
61.Tikhonov, A. N. (2013) pH-dependent regulation of
electron transport and ATP synthesis in chloroplasts, Photosynth.
Res., 116, 511-534.
62.Tikhonov, A. N. (2015) Induction events and
short-term regulation of electron transport in chloroplasts: an
overview, Photosynth. Res., 125, 65-94.
63.Kangasjarvi, S., Tikkanen, M., Durian, G., and
Aro, E.-M. (2014) Photosynthetic light reactions – an
adjustable Hub in basic production and plant immunity signaling,
Plant Physiol. Biochem., 81, 128-134.
64.Edwards, D., and Walker, D. (1986)
Photosynthesis in C3- and C4-Plants: Mechanism and Regulation
[Russian translation], Mir, Moscow.
65.Ivanov, B., and Khorobrykh, S. (2003)
Participation of photosynthetic electron transport in production and
scavenging of reactive oxygen species, Antioxid. Redox Signal.,
5, 43-53.
66.Wardman, P. (1989) Reduction potentials of
one-electron couples involving free radicals in aqueous solution, J.
Phys. Chem. Ref. Data, 18, 1637.
67.Mehler, A. H. (1951) Studies on reactions of
illuminated chloroplasts: I. Mechanism of the reduction of oxygen and
other hill reagents, Arch. Biochem. Biophys., 33,
65-77.
68.Trubitsin, B. V., Mamedov, M. D., Semenov, A. Y.,
and Tikhonov, A. N. (2014) Interaction of ascorbate with photosystem I,
Photosynth. Res., 122, 215-231.
69.Asada, K., Kiso, K., and Yoshikawa, K. (1974)
Univalent reduction of molecular oxygen by spinach chloroplasts on
illumination, J. Biol. Chem., 249, 2175-2181.
70.Noctor, G., and Foyer, C. H. (1998) Ascorbate and
glutatione: keeping active oxygen under control, Annu. Rev. Plant
Physiol. Plant Mol. Biol., 49, 249-279.
71.Foyer, C. H., and Noctor, G. (2011) Ascorbate and
glutathione: the heart of the redox hub, Plant Physiol.,
155, 2-18.
72.Rubin, A., and Riznichenko, G. (2014)
Mathematical Biophysics, Springer, Boston.
73.Tikhonov, A. N. (2016) Modeling electron and
proton transport in chloroplasts, in Chloroplasts: Current Research
and Future Trends (Kirchhoff, H., ed.) Caister Academic Press,
Norfolk, UK, pp. 101-134.
74.Tikhonov, A. N., and Vershubskii, A. V. (2014)
Computer modeling of electron and proton transport in chloroplasts,
Biosystems, 121, 1-21.
75.Kuvykin, I. V., Vershubskii, A. V.,
Ptushenko, V. V., and Tikhonov, A. N. (2008) Oxygen as an alternative
electron acceptor in the photosynthetic electron transport chain of C3
plants, Biochemistry (Moscow), 73, 1063-1075.
76.Matsuoka, T., Tanaka, S., and Ebina, K. (2016)
Reduced minimum model for the photosynthetic induction processes in
photosystem I, J. Photochem. Photobiol. B, 160,
364-375.
77.Santabarbara, S., Heathcote, P., and Evans, M. C.
W. (2005) Modelling of the electron transfer reactions in photosystem I
by electron tunnelling theory: the phylloquinones bound to the PsaA and
the PsaB reaction centre subunits of PS I are almost isoenergetic to
the iron-sulfur cluster FX, Biochim. Biophys. Acta, 1708,
283-310.
78.Milanovsky, G. E., Petrova, A. A., Cherepanov, D.
A., and Semenov, A. Y. (2017) Kinetic modeling of electron transfer
reactions in photosystem I complexes of various structures with
substituted quinone acceptors, Photosynth. Res., 133,
185-199.
79.Kirchhoff, H. (2008) Significance of protein
crowding, order and mobility for photosynthetic membrane functions,
Biochem. Soc. Trans., 36, 967-970.
80.Kirchhoff, H. (2014) Diffusion of molecules and
macromolecules in thylakoid membranes, Biochim. Biophys. Acta,
1837, 495-502.
81.Kirchhoff, H., Hall, C., Wood, M., Herbstova, M.,
Tsabari, O., Nevo, R., Charuvi, D., Shimoni, E., and Reich, Z. (2011)
Dynamic control of protein diffusion within the granal thylakoid lumen,
Proc. Natl. Acad. Sci. USA, 108, 20248-20253.
82.Polle, A., and Junge, W. (1986) The slow rise of
the flash-light-induced alkalization by photosystem II of the
suspending medium of thylakoids is reversibly related to thylakoid
stacking, Biochim. Biophys. Acta, 848, 257-264.
83.Junge, W., and Polle, A. (1986) Theory of proton
flow along appressed thylakoid membranes under both non-stationary and
stationary conditions, Biochim. Biophys. Acta, 848,
265-273.
84.Cherepanov, D. A., Junge, W., and Mulkidjanian,
A. Y. (2004) Proton transfer dynamics at the membrane/water interface:
dependence on the fixed and mobile pH buffers, on the size and form of
membrane particles, and on the interfacial potential barrier,
Biophys. J., 86, 665-680.
85.Springer, A., Hagen, V., Cherepanov, D. A.,
Antonenko, Y. N., and Pohl, P. (2011) Protons migrate along interfacial
water without significant contributions from jumps between ionizable
groups on the membrane surface, Proc. Natl. Acad. Sci. USA,
108, 14461-14466.
86.Vershubskii, A. V., Trubitsin, B. V.,
Priklonskii, V. I., and Tikhonov, A. N. (2017) Lateral heterogeneity of
the proton potential along the thylakoid membranes of chloroplasts,
Biochim. Biophys. Acta, 1859, 388-401.
87.Cherepanov, D. A., Feniouk, B. A., Junge, W., and
Mulkidjanian, A. Y. (2003) Low dielectric permittivity of water at the
membrane interface: effect on the energy coupling mechanism in
biological membranes, Biophys. J., 85, 1307-1316.
88.Cherepanov, D. A. (2004) Force oscillations and
dielectric overscreening of interfacial water, Phys. Rev. Lett.,
93, 266104.
89.Blackwell, M. F., and Whitmarsh, J. (1990) Effect
of integral membrane proteins on the lateral mobility of plastoquinone
in phosphatidylcholine proteoliposomes, Biophys. J., 58,
1259-1271.
90.Drepper, F., Carlberg, I., Andersson, B., and
Haehnel, W. (1993) Lateral diffusion of an integral membrane protein:
Monte Carlo analysis of the migration of phosphorylated
light-harvesting complex II in the thylakoid membrane,
Biochemistry, 32, 11915-11922.
91.Blackwell, M., Gibas, C., Gygax, S., Roman, D.,
and Wagner, B. (1994) The plastoquinone diffusion coefficient in
chloroplasts and its mechanistic implications, Biochim. Biophys.
Acta, 1183, 533-543.
92.Kirchhoff, H., Horstmann, S., and Weis, E. (2000)
Control of the photosynthetic electron transport by PQ diffusion
microdomains in thylakoids of higher plants, Biochim. Biophys.
Acta, 1459, 148-168.
93.Kirchhoff, H., Mukherjee, U., and Galla, H.-J.
(2002) Molecular architecture of the thylakoid membrane: lipid
diffusion space for plastoquinone, Biochemistry, 41,
4872-4882.
94.Kovalenko, I. B., Abaturova, A. M., Riznichenko,
G. Y., and Rubin, A. B. (2011) Computer simulation of interaction of
photosystem I with plastocyanin and ferredoxin, Bio Systems,
103, 180-187.
95.Brettel, K., and Leibl, W. (2001) Electron
transfer in photosystem I, Biochim. Biophys. Acta, 1507,
100-114.
96.Fromme, P., and Mathis, P. (2004) Unraveling the
photosystem I reaction center: a history, or the sum of many efforts,
Photosynth. Res., 80, 109-124.
97.Mamedov, M., Govindjee, Nadtochenko, V., and
Semenov, A. (2015) Primary electron transfer processes in
photosynthetic reaction centers from oxygenic organisms, Photosynth.
Res., 125, 51-63.
98.Jordan, P., Fromme, P., Witt, H. T., Klukas, O.,
Saenger, W., and Krauss, N. (2001) Three-dimensional structure of
cyanobacterial photosystem I at 2.5 Å resolution, Nature,
411, 909-917.
99.Mazor, Y., Borovikova, A., and Nelson, N. (2017)
The structure of plant photosystem I super-complex at 2.8 Å
resolution, Nat. Plants, 3, 17014.
100.Diaz-Quintana, A., Leibl, W., Bottin, H., and
Setif, P. (1998) Electron transfer in photosystem I reaction centers
follows a linear pathway in which iron-sulfur cluster FB is the
immediate electron donor to soluble ferredoxin, Biochemistry,
37, 3429-3439.
101.Vassiliev, I. R., Jung, Y. S., Yang, F., and
Golbeck, J. H. (1998) PsaC subunit of photosystem I is oriented with
iron-sulfur cluster F(B) as the immediate electron donor to ferredoxin
and flavodoxin, Biophys. J., 74, 2029-2035.
102.Golbeck, J. H. (1999) A comparative analysis of
the spin state distribution of in vitro and in vivo
mutants of PsaC. A biochemical argument for the sequence of electron
transfer in photosystem I as
FX→FA→FB→ferredoxin/flavodoxin,
Photosynth. Res., 61, 107-144.
103.Mamedova, A. A., Mamedov, M. D., Gourovskaya,
K. N., Vassiliev, I. R., Golbeck, J. H., and Semenov, A. Y. (1999)
Electrometrical study of electron transfer from the terminal
FA/FB iron-sulfur clusters to external acceptors
in photosystem I, FEBS Lett., 462, 421-424.
104.Guergova-Kuras, M., Boudreaux, B., Joliot, A.,
Joliot, P., and Redding, K. (2001) Evidence for two active branches for
electron transfer in photosystem I, Proc. Natl. Acad. Sci. USA,
98, 4437-4442.
105.Srinivasan, N., and Golbeck, J. H. (2009)
Protein–cofactor interactions in bioenergetic complexes: the role
of the A1A and A1B phylloquinones in photosystem
I, Biochim. Biophys. Acta, 1787, 1057-1088.
106.Milanovsky, G. E., Ptushenko, V. V.,
Cherepanov, D. A., and Semenov, A. Y. (2014) Mechanism of primary and
secondary ion-radical pair formation in photosystem I complexes,
Biochemistry (Moscow), 79, 221-226.
107.Makita, H., and Hastings, G. (2015)
Directionality of electron transfer in cyanobacterial photosystem I at
298 and 77K, FEBS Lett., 589, 1412-1417.
108.Sun, J., Hao, S., Radle, M., Xu, W., Shelaev,
I., Nadtochenko, V., Shuvalov, V., Semenov, A., Gordon, H., Van der
Est, A., and Golbeck, J. H. (2014) Evidence that histidine forms a
coordination bond to the A0A and A0B chlorophylls
and a second H-bond to the A1A and A1B
phylloquinones in M688HPsaA and M668HPsaB variants of
Synechocystis sp. PCC 6803, Biochim. Biophys. Acta,
1837, 1362-1375.
109.Shelaev, I. V., Gostev, F. E., Mamedov, M. D.,
Sarkisov, O. M., Nadtochenko, V. A., Shuvalov, V. A., and Semenov, A.
Y. (2010) Femtosecond primary charge separation in Synechocystis
sp. PCC 6803 photosystem I, Biochim. Biophys. Acta, 1797,
1410-1420.
110.Vassiliev, I. R., Jung, Y. S., Mamedov, M. D.,
Semenov, A. Y., and Golbeck, J. H. (1997) Near-IR absorbance changes
and electrogenic reactions in the microsecond-to-second time domain in
photosystem I, Biophys. J., 72, 301-315.
111.Ptushenko, V. V., Cherepanov, D. A.,
Krishtalik, L. I., and Semenov, A. Y. (2008) Semi-continuum
electrostatic calculations of redox potentials in photosystem I,
Photosynth. Res., 97, 55-74.
112.Agalarov, R., and Brettel, K. (2003)
Temperature dependence of biphasic forward electron transfer from the
phylloquinone( s) A1 in photosystem I: only the slower
phase is activated, Biochim. Biophys. Acta, 1604,
7-12.
113.Jordan, R., Nessau, U., and Schlodder, E.
(1998) Charge recombination between the reduced iron-sulphur clusters
and P700+, in Photosynthesis: Mechanisms and Effects:
Volume I Proc. of the XIth Int. Congr. on Photosynthesis (Garab,
G., ed.) Budapest, Hungary, August 17-22, 1998, Springer Netherlands,
Dordrecht, pp. 663-666.
114.Shinkarev, V. (2006) Functional modeling of
electron transfer in photosynthetic reaction centers, in Photosystem
I: the Light-Driven Plastocyanin:Ferredoxin Oxidoreductase
(Golbeck, J. H., ed.) Springer Netherlands, Dordrecht, pp. 611-637.
115.Palsson, L.-O. O., Flemming, C., Gobets, B.,
Van Grondelle, R., Dekker, J. P., and Schlodder, E. (1998) Energy
transfer and charge separation in photosystem I: P700 oxidation upon
selective excitation of the long-wavelength antenna chlorophylls of
Synechococcus elongatus, Biophys. J., 74,
2611-2622.
116.Shinkarev, V. P., Vassiliev, I. R., and
Golbeck, J. H. (2000) A kinetic assessment of the sequence of electron
transfer from FX to FA and further to
FB in photosystem I: the value of the equilibrium constant
between FX and FA, Biophys. J., 78, 363-372.
117.Shen, G., Zhao, J., Reimer, S. K., Antonkine,
M. L., Cai, Q., Weiland, S. M., Golbeck, J. H., and Bryant, D. A.
(2002) Assembly of photosystem I. I. Inactivation of the rubA
gene encoding a membrane-associated rubredoxin in the cyanobacterium
Synechococcus sp. PCC 7002 causes a loss of photosystem I
activity, J. Biol. Chem., 277, 20343-20354.
118.Mula, S., Savitsky, A., Mobius, K., Lubitz, W.,
Golbeck, J. H., Mamedov, M. D., Semenov, A. Y., and Van der Est, A.
(2012) Incorporation of a high potential quinone reveals that electron
transfer in photosystem I becomes highly asymmetric at low temperature,
Photochem. Photobiol. Sci., 11, 946-956.
119.Makita, H., Zhao, N., and Hastings, G. (2015)
Time-resolved visible and infrared difference spectroscopy for the
study of photosystem I with different quinones incorporated into the A1
binding site, Biochim. Biophys. Acta, 1847, 343-354.
120.Petrova, A. A., Boskhomdzhieva, B. K.,
Milanovsky, G. E., Koksharova, O. A., Mamedov, M. D., Cherepanov, D.
A., and Semenov, A. Y. (2017) Interaction of various types of
photosystem I complexes with exogenous electron acceptors,
Photosynth. Res., 133, 175-184.
121.Shinkarev, V. P., Zybailov, B., Vassiliev, I.
R., and Golbeck, J. H. (2002) Modeling of the P700+ charge
recombination kinetics with phylloquinone and plastoquinone-9 in the
A1 site of photosystem I, Biophys. J., 83,
2885-2897.
122.Makita, H., and Hastings, G. (2016) Modeling
electron transfer in photosystem I, Biochim. Biophys. Acta,
1857, 723-733.
123.Santabarbara, S., and Zucchelli, G. (2016)
Comparative kinetic and energetic modelling of phyllosemiquinone
oxidation in photosystem I, Phys. Chem. Chem. Phys., 18,
9687-9701.
124.Ilan, Y. A., Meisel, D., and Czapski, G. (1974)
The redox potential of the
O2–O2− system in aqueous
media, Israel J. Chem., 12, 891-895.
125.Battino, R., Rettich, T. R., and Tominaga, T.
(1983) The solubility of oxygen and ozone in liquids, J. Phys. Chem.
Ref. Data, 12, 163-178.
126.Golbeck, J. H., Parrett, K. G., and McDermott,
A. E. (1987) Photosystem I charge separation in the absence of center A
and B. III. Biochemical characterization of a reaction center particle
containing P-700 and FX, Biochim. Biophys. Acta,
893, 149-160.
127.Torres, R. A., Lovell, T., Noodleman, L., and
Case, D. A. (2003) Density functional and reduction potential
calculations of Fe4S4 clusters, J. Am. Chem.
Soc., 125, 1923-1936.
128.Bashford, D. (1997) An Object-Oriented
Programming Suite for Electrostatic Effects in Biological Molecules: An
Experience Report on the MEAD project. Lecture Notes in Computer
Science (Including Subseries Lecture Notes in Artificial Intelligence
and Lecture Notes in Bioinformatics), Springer, Berlin-Heidelberg,
pp. 233-240.
129.Witham, S., Takano, K., Schwartz, C., and
Alexov, E. (2011) A missense mutation in CLIC2 associated with
intellectual disability is predicted by in silico modeling to
affect protein stability and dynamics, Proteins Struct. Funct.
Bioinform., 79, 2444-2454.
130.Ishikita, H., and Knapp, E.-W. (2003) Redox
potential of quinones in both electron transfer branches of photosystem
I, J. Biol. Chem., 278, 52002-52011.
131.Ishikita, H., Saenger, W., Biesiadka, J., Loll,
B., and Knapp, E.-W. (2006) How photosynthetic reaction centers control
oxidation power in chlorophyll pairs P680, P700, and P870, Proc.
Natl. Acad. Sci. USA, 103, 9855-9860.
132.Kawashima, K., and Ishikita, H. (2017)
Structural factors that alter the redox potential of quinones in
cyanobacterial and plant photosystem I, Biochemistry, 56,
3019-3028.
133.Parson, W. W., and Warshel, A. (2008)
Calculations of electrostatic energies in proteins using microscopic,
semimicroscopic and macroscopic models and free-energy perturbation
approaches, in Biophysical Techniques in Photosynthesis
(Aartsma, T. J., and Matysik, J., eds.) Springer, Netherlands,
Dordrecht, pp. 401-420.
134.Mitra, R., Shyam, R., Mitra, I., Miteva, M.,
Alexov, E., Reihle, T., Loladze, V., Makhatadze, G., Guo, H., and Ha,
S. (2008) Calculating the protonation states of proteins and small
molecules: implications to ligand–receptor interactions, Curr.
Comp. Aided Drug Design, 4, 169-179.
135.Ptushenko, V. V., Cherepanov, D. A., and
Krishtalik, L. I. (2015) Electrostatics of the photosynthetic bacterial
reaction center. protonation of Glu L 212 and Asp L 213 – a
new method of calculation, Biochim. Biophys. Acta, 1847,
1495-1508.
136.Prince, R. C., Leslie Dutton, P., and Malcolm
Bruce, J. (1983) Electrochemistry of ubiquinones. Menaquinones and
plastoquinones in aprotic solvents, FEBS Lett., 160,
273-276.
137.Harris, B. J., Cheng, X., and Frymier, P.
(2014) All-atom molecular dynamics simulation of a photosystem
I/detergent complex, J. Phys. Chem. B, 118,
11633-11645.
138.Harris, B. J., Cheng, X., and Frymier, P.
(2016) Structure and function of photosystem I-[FeFe] hydrogenase
protein fusions: an all-atom molecular dynamics study, J. Phys.
Chem. B, 120, 599-609.
139.Zhang, Y., and Ding, Y. (2016) Molecular
dynamics simulation and bioinformatics study on chloroplast stromal
ridge complex from rice (Oryza sativa L.), BMC
Bioinformatics, 17, 1-12.
140.Milanovsky, G. E., Ptushenko, V. V., Golbeck,
J. H., Semenov, A. Y., and Cherepanov, D. A. (2014) Molecular dynamics
study of the primary charge separation reactions in photosystem I:
effect of the replacement of the axial ligands to the electron acceptor
A0, Biochim. Biophys. Acta, 1837, 1472-1483.
141.Brettel, K., and Golbeck, J. H. (1995) Spectral
and kinetic characterization of electron acceptor A1 in a photosystem I
core devoid of iron-sulfur centers FX, FB and
FA, Photosynth. Res., 45, 183-193.
142.Kozuleva, M. A., Petrova, A. A., Mamedov, M.
D., Semenov, A. Y., and Ivanov, B. N. (2014) O2 reduction by
photosystem I involves phylloquinone under steady-state illumination,
FEBS Lett., 588, 4364-4368.
143.Kozuleva, M. A., and Ivanov, B. N. (2016) The
mechanisms of oxygen reduction in the terminal reducing segment of the
chloroplast photosynthetic electron transport chain, Plant Cell
Physiol., 57, 1397-1404.
144.Forti, G., and Ehrenheim, A. M. (1993) The role
of ascorbic acid in photosynthetic electron transport, Biochim.
Biophys. Acta, 1183, 408-412.
145.Miyake, C., and Asada, K. (1994)
Ferredoxin-dependent photoreduction of the monodehydroascorbate radical
in spinach thylakoids, Plant Cell Physiol., 35,
539-549.
146.Grace, S., Pace, R., and Wydrzynski, T. (1995)
Formation and decay of monodehydroascorbate radicals in illuminated
thylakoids as determined by EPR spectroscopy, Biochim. Biophys.
Acta, 1229, 155-165.
147.Ivanov, B. (2000) The competition between
methyl viologen and monodehydroascorbate radical as electron acceptors
in spinach thylakoids and intact chloroplasts, Free Radic. Res.,
33, 217-227.
148.Ivanov, B. N. (2014) Role of ascorbic acid in
photosynthesis, Biochemistry (Moscow), 79, 282-289.
149.Miyake, C., and Yokota, A. (2000) Determination
of the rate of photoreduction of O2 in the water–water
cycle in watermelon leaves and enhancement of the rate by limitation of
photosynthesis, Plant Cell Physiol., 41, 335-343.
150.Buchanan, B. B. (1980) Role of light in the
regulation of chloroplast enzymes, Annu. Rev. Plant Physiol.,
31, 341-374.
151.Buchanan, B. B. (1991) Regulation of
CO2 assimilation in oxygenic photosynthesis: the
ferredoxin/thioredoxin system. Perspective on its discovery, present
status, and future development, Arch. Biochem. Biophys.,
288, 1-9.
152.Bottin, H., and Lagoutte, B. (1992) Ferrodoxin
and flavodoxin from the cyanobacterium Synechocystis sp. PCC
6803, Biochim. Biophys. Acta, 1101, 48-56.
153.Bird, C. L., and Kuhn, A. T. (1981)
Electrochemistry of the viologens, Chem. Soc. Rev. R. Soc.
Chem., 10, 49.
154.Ilan, Y. A., Czapski, G., and Meisel, D. (1976)
The one-electron transfer redox potentials of free radicals. I. The
oxygen/superoxide system, Biochim. Biophys. Acta, 430,
209-224.
155.Vasudevan, D., and Wendt, H. (1995)
Electroreduction of oxygen in aprotic media, J. Electroanal.
Chem., 392, 69-74.
156.Sawyer, D., Chiericato, G., and Angelis, C. A.
(1982) Effects of media and electrode materials on the electrochemical
reduction of dioxygen, Anal. Chem., 2, 1720-1724.
157.Currie, D. J., and Holmes, H. L. (1966)
Polarographic half-wave potentials of some 1,4-naphthoquinones, Can.
J. Chem., 44, 1027-1029.
158.Shalev, H., and Evans, D. H. (1989) Solvation
of anion radicals: GAS phase vs solution, J. Am. Chem. Soc.,
111, 2667-2674.
159.Pueyo, J. J., Gomez-Moreno, C., and Mayhew, S.
G. (1991) Oxidation-reduction potentials of ferredoxin-NADP+
reductase and flavodoxin from anabaena PCC 7119 and their electrostatic
and covalent complexes, Eur. J. Biochem., 202,
1065-1071.
160.Setif, P., and Brettel, K. (1993) Forward
electron transfer from phylloquinone A1 to iron-sulfur centers in
spinach photosystem I, Biochemistry, 32, 7846-7854.
161.Bautista, J. A., Rappaport, F., Guergova-Kuras,
M., Cohen, R. O., Golbeck, J. H., Wang, J. Y., Beal, D., and Diner, B.
A. (2005) Biochemical and biophysical characterization of photosystem I
from phytoene desaturase and ζ-carotene desaturase deletion
mutants of Synechocystis sp. PCC 6803: evidence for PsaA- and
PsaB-side electron transport in cyanobacteria, J. Biol. Chem.,
280, 20030-20041.
162.Setif, P. (2001) Ferredoxin and flavodoxin
reduction by photosystem I, Biochim. Biophys. Acta, 1507,
161-179.