Antiinflammatory Effect of Rosiglitazone via Modulation of mRNA Stability of Interleukin 10 and Cyclooxygenase 2 in Astrocytes
E. V. Pankevich1, A. A. Astakhova2, D. V. Chistyakov2,3*, and M. G. Sergeeva2
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119234 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; E-mail: Chistyakof@gmail.com
3Pirogov Russian National Research Medical University, 117997 Moscow, Russia
* To whom correspondence should be addressed.
Received June 30, 2017; Revision received August 7, 2017
Investigation of molecular mechanisms of proinflammatory stimuli signaling in astrocytes is important for understanding their role in pathogenesis of central nervous system diseases as well as in functioning of the innate immunity system in non-immune cells. Here we show that lipopolysaccharide (LPS) stimulation of primary rat astrocytes led to conventional inflammatory response: increase in both proinflammatory (tumor necrosis factor, TNFα; prostaglandin E2, PGE2) and antiinflammatory marker (interleukin 10, IL-10) levels. The protein level of cyclooxygenase 2 (COX-2) was also increased. Rosiglitazone strengthened LPS-induced mRNA expression of COX-2 and IL-10 but not TNFα. Rosiglitazone is an agonist of nuclear receptor PPARγ, but its impact on IL-10 expression was not influenced by a PPARγ antagonist, GW9662, suggesting PPARγ-independent effect of rosiglitazone. The degradation of mRNA is one of the steps of inflammation regulation and might be affected by small molecules. In experiments with actinomycin D, we found that mRNA half-lives of IL-10, COX-2, and TNFα in naive astrocytes were 70, 44, and 19 min, respectively. LPS stimulation caused 2-fold increase in IL-10 and COX-2 mRNA decay rates, whereas addition of rosiglitazone restored them to the initial level. TNFα decay rate was not changed by these stimulations. This suggests that mRNA decay rate could be regulated by small molecules. Moreover, rosiglitazone could be used as a substance stimulating the resolution of inflammation without influence on proinflammatory signals. These results open new perspectives in the search for inflammation resolution modulators.
KEY WORDS: rosiglitazone, inflammatory response, astrocytes, mRNA stabilityDOI: 10.1134/S0006297917110050
Abbreviations: CNS, central nervous system; COX-2, cyclooxygenase 2; IL-10, interleukin 10; LPS, lipopolysaccharide; PGE2, prostaglandin E2; TLR4, toll-like receptor 4; TNFα, tumor necrosis factor alpha.
Inflammation has been proved to be an important part of the pathogenesis
of many chronic nervous system diseases including brain tumors and
neurodegenerative states (Alzheimer’s disease, Parkinson’s
disease, multiple sclerosis, etc.) [1]. While
active search for antiinflammatory drugs is conducted, rosiglitazone,
an agonist of the nuclear receptor PPARγ, has been proposed as
one of them [2-5]. Treatment
with rosiglitazone was shown to lead to a decrease in expression of
CD14, a component of the CD14/TLR4/MD2 complex recognizing
lipopolysaccharide (LPS) that induces an inflammatory response in
monocytes [6]. There is also evidence of
effectiveness of rosiglitazone against Alzheimer’s disease [7]. However, a better understanding of the effects of
rosiglitazone at the molecular level is required for development of
therapy strategy using this drug.
Cellular models are typically used to elucidate molecular mechanisms of action of potential antiinflammatory drugs. An antiinflammatory effect of rosiglitazone has been shown in different cell types including immune system cells [8], where an inflammatory response plays a crucial role, as well as in microglia in a model of Parkinson’s disease [9]. Rosiglitazone was shown to downregulate cell production of tumor necrosis factor alpha (TNFα) and interleukins 1β and 6 (IL-1β, IL-6), affecting expression levels of corresponding genes [9, 10]. A common model of neuroinflammatory conditions in vitro involves stimulation of nervous system cells by an agonist of toll-like receptor 4 (TLR4), lipopolysaccharide (LPS) [11]. Previously, we demonstrated that stimulation of astrocytes by LPS leads to induction of a marker of inflammation cyclooxygenase 2 (COX-2) and that rosiglitazone treatment leads to an increase in COX-2 expression at the mRNA level [12, 13]. In this work, we studied a possible mechanism of this effect.
Inflammation at the cellular level is a process developing in time that is regulated at the levels of transcription and translation. It recently became clear that a posttranscriptional level of control is also actively involved in regulation of inflammation. For instance, modulation of mRNA degradation is important for regulation of inflammatory genes [14]. Such studies are mainly performed for immune system cells mediating inflammatory response beyond the central nervous system (CNS). It should be noted that cells of the immune system (leukocytes, macrophages, etc.) have myeloid or lymphoid development from mesodermal cells. The immunocompetent cells of the CNS – microglial cells and astrocytes – have different origins: microglia belong to the myeloid lineage while astrocytes have common precursors with neurons and have ectodermal origin. While the regulation at the mRNA degradation level has been well established for immune cells, the characterization of this process in astrocytes has been in the shadow until recently. Previously, we showed that LPS stimulation changes the rate of mRNA degradation of several genes in astrocytes [13]. Therefore, we supposed that one of the mechanisms of rosiglitazone action could be an impact on the mRNA degradation rate.
In this work, we studied an impact of rosiglitazone on expression and production of the main cytokines – the markers of inflammation. To characterize an inflammatory process under LPS stimulation, we chose TNFα as a proinflammatory marker and interleukin 10 (IL-10) as a cytokine with antiinflammatory action. We also studied the effect of rosiglitazone on COX-2 expression as this protein is related to proinflammatory markers (as a key enzyme in prostaglandin synthesis) as well as to antiinflammatory markers (as a possible source of cyclopentenone prostaglandins and other antiinflammatory oxylipins) [15]. We compared the effect of rosiglitazone on stabilization and degradation of COX-2, IL-10, and TNFα mRNA during inflammatory response in primary rat astrocytes.
We found that rosiglitazone stabilizes IL-10 and COX-2 mRNA but not TNFα mRNA. Our data point to a new effect of rosiglitazone – not suppressing proinflammatory response but stimulating activation of antiinflammatory processes, important factors of which are IL-10 and COX-2.
MATERIALS AND METHODS
Reagents. LPS, SB203580, GW9662, actinomycin D, and antibodies against β-tubulin (Sigma-Aldrich, Germany); rosiglitazone (Cayman, Germany); streptomycin-penicillin, trypsin, EDTA, fetal bovine serum, and DMEM culture medium (PanEko, Russia); antibodies against COX-2 (D5H5), phospho-p38 (4511), p38 (9212) (Cell Signaling Technology, USA); Western Blotting Substrate ECL and ELISA kits for TNFα and IL-10 (Thermo Fisher Scientific, USA) were used in this study.
Cell culture and stimulations. Primary cultures of rat astrocytes were obtained from neonatal Wistar rats according to a protocol used previously [11, 12]. In brief, the animals were aseptically decapitated and the brains were isolated, washed in ice-cold Puck’s buffer (mM: 137 NaCl, 5.4 KCl, 0.2 KH2PO4, 0.17 Na2HPO4, 5 glucose, 58.4 sucrose, pH 7.4), and minced against meshes of 250- and 136-µm size, and then the tissue fragments were placed into culture flasks. The material was supplied with DMEM (1 g/liter D-glucose, 10% fetal bovine serum, 50 units/ml streptomycin, 50 µg/ml penicillin) and incubated at 37°C, 5% CO2, and 10% humidity. Five days later, the cultures were shaken to detach microglial cells and given fresh portions of media with the same composition. The cells were cultured for an additional six days with media being changed every two days. After monolayer formation, the cells were treated with trypsin, then the cells were plated into 6-well culture plates at 750,000 cells/well. These cells were used for experiments after two days.
Western blot analysis. Analysis of changes in COX-2 expression at the protein level was carried out using classical techniques for the preparation of cell lysates, determination of protein concentrations using Bradford reagent, separation of proteins with different molecular weights by SDS-PAGE with denaturing Laemmli buffer, and protein transfer to nitrocellulose membranes (0.2 µm). The membranes were blocked in TBS buffer with 5% fat-free milk and 0.05% Tween 20. The membranes were then treated with primary antibodies against COX-2 or phospho-p38. After treatment of the membranes with secondary antibodies, proteins were visualized using chemiluminescent substrate. Band intensity was measured using a Gel Doc™ XR+ System (Bio-Rad, USA) and Quantity One software (Bio-Rad 4.6.9) and normalized to the intensity of the respective bands obtained for β-tubulin.
ELISA. After the experiments, the supernatant was collected and stored at –70°C for further analysis. The levels of released prostaglandin E2 (PGE2), TNFα, and IL-10 were determined using commercial enzyme-linked immunoassay kits and a Synergy H4 plate reader (BioTek, USA) following the manufacturer’s instructions.
Measurement of relative mRNA level. Total RNA was isolated using PowerLyzer RNA Isolation (MO BIO, USA) following the manufacturer’s protocol. RNA concentration was determined spectrophotometrically. The first cDNA strand was prepared using reverse transcription with a First Strand cDNA Synthesis Kit (Thermo Scientific, USA). The reaction was performed in accordance with the manufacturer’s instructions using oligo(dT) primers. The relative mRNA of IL-10, COX-2, and TNFα were determined by real-time PCR using a DTlite 4 thermocycler (DNK-Tekhnologiya, Russia) and a qPCRmix-HS (Evrogen, Russia) kit. The oligonucleotide primers and TaqMan probes were used to develop fragments of genes with the following sequences (Table 1); annealing temperature of primers – 57°C. A single cDNA prepared from 70 ng RNA in a reverse transcription reaction was taken into one 25 µl reaction mixture. The relative level of mRNA was calculated using the ΔCt, 2–ΔCt method [16]. Levels of β-actin were used as the constitutive for normalization.
Table 1. Sequences of primers and probes

Note: Fluorophore at the 5′-end and quencher at the 3′-prime
end are indicated in the probe sequences.
Statistics. The data are presented as mean ± standard error obtained from three independent experiments. The data were subjected to the ANOVA test. The difference was considered statistically significant at p-value < 0.05 (* p-value < 0.05 in case of one-way analysis, # 0.05 in case of multi-factor analysis for the treatment of cells with several compounds).
Determination of mRNA degradation rate. To determine the mRNA degradation rate, the levels of IL-10, COX-2, and TNFα mRNA were assessed in the presence of the transcriptional inhibitor actinomycin D in accordance with the classical technique [17]. It is known that after addition of actinomycin D only the level of the existing mRNA changes, which allows determination of the half-life of mRNA. Cells were stimulated with LPS (100 ng/ml) for 1 h, then de novo mRNA transcription was inhibited by addition of actinomycin D (5 µg/ml). Similar treatment was performed on cells without stimulation of LPS (untreated cells). Others stimulation remained unchanged. The level of mRNA in untreated cells prior to the addition of actinomycin D was taken as 100%. In LPS-stimulated cells, mRNA level was taken as 100% after 1 h of LPS stimulation before actinomycin D was added. The values of mRNA half-lives of the investigated genes under these conditions were calculated on the assumption that mRNA decay occurs in accordance with first-order kinetics [18].
RESULTS
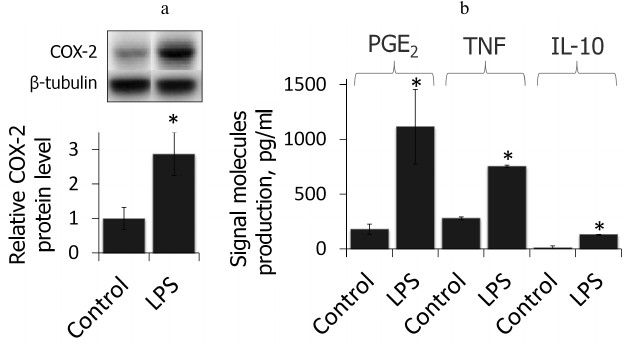
LPS induces inflammatory response in primary rat astrocytes. First, we showed that the cell model used in this study reflected proinflammatory state after LPS stimulation (4-h treatment, 100 ng/ml, time and concentration were chosen from previous studies [19]). We estimated the relative level of COX-2 protein and the primary product of its enzymatic activity, PGE2, as well as production of TNFα and IL-10 after treatment with LPS (Fig. 1). LPS was shown to cause an increase in COX-2 protein level accompanied by an increase in production of PGE2 as well as TNFα and IL-10, indicating the proinflammatory conditions of the model.
Fig. 1. COX-2 protein expressions and PGE2, TNFα, and IL-10 release after LPS treatment (100 ng/ml, 4 h) characterize the acute inflammatory conditions. a) Levels of COX-2 and β-tubulin proteins evaluated by Western blotting. Levels of COX-2 expression in untreated control astrocytes were taken for control. b) PGE2, TNFα, and IL-10 concentrations measured by ELISA (* p-value < 0.05 compared with unstimulated cells).
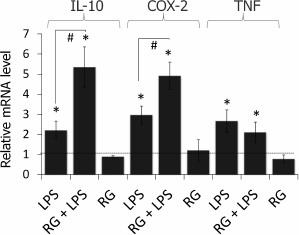
Rosiglitazone strengthens LPS-induced COX-2 and IL-10 but not TNFα mRNA expression in astrocytes. To estimate the effect of rosiglitazone on inflammatory response in astrocytes, we measured relative expression levels of IL-10, COX-2, and TNFα (Fig. 2). Cells were incubated with rosiglitazone for 30 min (10 µM, concentration and time were chosen from previous studies [19]), then incubated with no additional stimulation or stimulated with LPS (100 ng/ml) for 4 h. Untreated cells were used as a control; the expression level of the COX2 gene in control cells was taken as 1. LPS stimulation of astrocytes was found to cause 3-, 2.5-, and 2-fold increase in COX-2, IL-10, and TNFα expression level, respectively, in comparison to control. Rosiglitazone treatment increased LPS-induced expression of COX-2 and IL-10 1.5 and 2.5 times, respectively, but did not affect LPS-induced expression of TNFα. Rosiglitazone alone did not change expression levels of our genes of interest.
Fig. 2. Change in expression of IL-10, COX-2, and TNFα mRNA after LPS and rosiglitazone (RG) treatment in primary rat astrocytes. Cells were pretreated for 30 min with RG (10 µM) and subsequently stimulated with LPS (100 ng/ml, 4 h). Levels of mRNA were determined by real-time RT-PCR (* p-value < 0.05 compared with unstimulated cells, # p-value < 0.05).
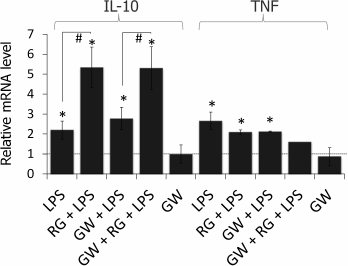
Rosiglitazone is known to mediate most of its effects (including enhancement of LPS-induced expression of COX-2) as an agonist of the nuclear receptor PPARγ [19]; however, there is evidence of its PPARγ-independent effects [20, 21]. To estimate the impact of PPARγ to the observed effect of rosiglitazone on IL-10 and TNFα expression, we used a selective antagonist of PPARγ, GW9662. Cells were preincubated with GW9662 for 30 min (1 µM, concentration and time were chosen from previous studies [19]). Then, the cells were stimulated with LPS and rosiglitazone (Fig. 3). Inhibition of PPARγ did not prevent strengthening effect of rosiglitazone on LPS-induced IL-10 expression. GW9662 at the concentration of 1 µM did not change the effect of rosiglitazone on IL-10 expression. Thus, the effect of rosiglitazone might not depend on the activation of PPARγ. Hence, the effect may be mediated through PPARγ-independent mechanisms, but further studies are required to support this hypothesis.
Fig. 3. Effect of GW9662 (GW, a PPARγ antagonist) on the effect of rosiglitazone (RG) on LPS-induced expression of IL-10 and TNFα mRNA in primary rat astrocytes. Cells were pretreated for 30 min with GW9662 (1 µM). Then the cells were treated with RG (10 µM, 30 min) and subsequently stimulated with LPS (100 ng/ml, 4 h). Levels of mRNA were determined by real-time RT-PCR (* p-value < 0.05 compared with unstimulated cells, # p-value < 0.05).
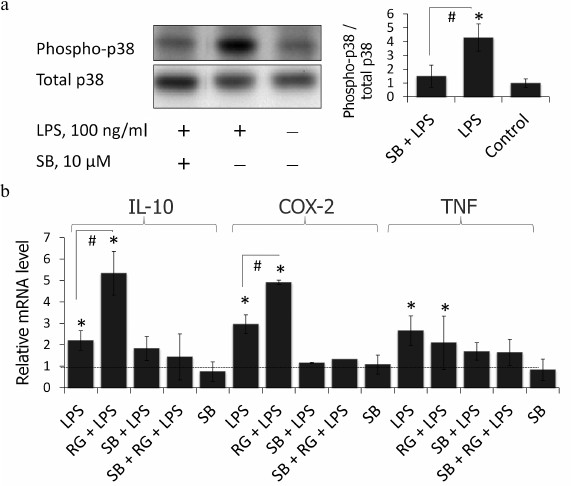
p38 MAPK inhibition blocks LPS-stimulated induction of COX-2, IL-10, and TNFα in astrocytes. Rosiglitazone was previously shown to activate p38 MAPK via a PPARγ-independent pathway in keratinocytes [22]. Therefore, we tested if p38 MAPK takes part in the rosiglitazone effect on expression of inflammatory genes in LPS-stimulated astrocytes. To do that, cells were preincubated with p38 MAPK inhibitor, SB203580 (10 µM), for 30 min (time and concentration were chosen from previous studies [13]). SB203580 inhibited phosphorylation of p38 MAPK under these conditions in astrocytes (Fig. 4a). This prevented induction of expression of all three genes after LPS stimulation (Fig. 4b). However, suppression of gene expression was observed also in the cells treated with LPS and rosiglitazone. These data indicate that rosiglitazone strengthens inflammatory response induced by LPS but not triggers it through any other TLR-4-independent mechanisms.
Fig. 4. a) Effect of SB203580 (SB, p38 inhibitor of MAPK) and LPS on the induction of phosphorylation of p38. Cells were pretreated for 30 min with SB203580 (10 µM) and subsequently kept for 4 h without any additional stimulation or with LPS (100 ng/ml). The p38 and phospho-p38 protein levels were evaluated by Western blotting. b) Influence of SB203580 on rosiglitazone (RG) effect on LPS-induced expression of IL-10, COX-2, and TNFα mRNA in primary rat astrocytes. Cells were pretreated for 30 min with SB203580 (10 µM). Then the cells were treated RG (10 µM, 30 min) and subsequently stimulated with LPS (100 ng/ml, 4 h). Levels of mRNA were determined by real-time RT-PCR (* p-value < 0.05 compared with unstimulated cells, # p-value < 0.05).
Rosiglitazone prevents destabilization of IL-10 and COX-2 mRNA during inflammatory response in astrocytes. Posttranscriptional regulation of mRNA has been actively studied recently. Genes involved in inflammatory response are of particular interest because their decay rates (including those of COX-2, TNFα, and IL-10 mRNA) changes in response to different stimuli [14]. However, to date the existence of expression regulation through alteration of mRNA decay rate has not been shown in astrocytes for these genes. Previously, we showed that COX-2 expression is regulated at the level of mRNA stability in astroglial rat tumor cell line C6 after treatment with LPS and rosiglitazone [23]. Therefore, we studied how rosiglitazone affects stabilization of mRNA of inflammatory response genes in astrocytes.
We determined changes in IL-10, COX-2, and TNFα mRNA levels in the presence of transcription inhibitor actinomycin D (according to the method described above) after stimulations with LPS and rosiglitazone (Fig. 5). We calculated mRNA half-lives of the studied genes (Table 2). As a control, we estimated the decay rate of β-tubulin mRNA, as a gene not involved directly in the signal pathway of toll-like receptors. LPS was found to destabilize COX-2 and IL-10 mRNA (Fig. 5). Pretreatment of cells with rosiglitazone prevented decrease in COX-2 and IL-10 mRNA half-lives induced by LPS. Therefore, the increase of LPS-induced expression of COX-2 and IL-10 caused by rosiglitazone may be mediated by mRNA stabilization. There was no significant change in TNFα mRNA stability after LPS stimulation (with and without preliminary rosiglitazone treatment) in comparison to naive cells. These data suggest that rosiglitazone takes part in posttranscriptional regulation of IL-10 and COX-2 expression in LPS-stimulated astrocytes and thus can increase availability of the mRNA of these genes for protein synthesis. This might lead to an increase in the level of antiinflammatory agents at the late stages of inflammatory response.
Our data suggest a new effect of rosiglitazone, not suppressing proinflammatory response (TNFα expression) but stimulating the activation of resolution of inflammation (by strengthening IL-10 and COX-2 expression).
Fig. 5. Effect of rosiglitazone (RG) and LPS on IL-10, COX-2, TNFα, and β-tubulin mRNA turnover. Cells were pretreated for 30 min with RG (10 µM) and subsequently stimulated with LPS (100 ng/ml, 1 h). Then the cells were incubated with actinomycin D (5 µg/ml) for 15, 30, 60, or 180 min. The levels of mRNA were determined by real-time RT-PCR; the values are normalized to the level of mRNA in the cells prior to addition of actinomycin D (* p-value < 0.05 compared with unstimulated cells (control)).
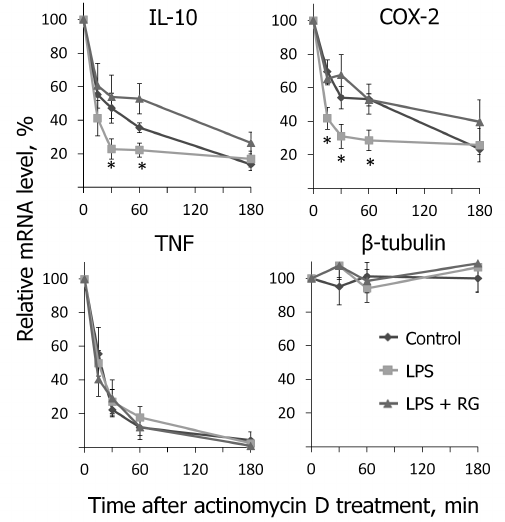
Table 2. Half-lives (τ1/2) of
IL-10, COX-2, and TNFα mRNA
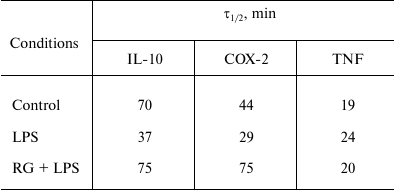
Note: Half-lives were defined as described before [18] (decay constant was calculated from linear
regression at 0, 15, 30, and 60-min time points).
DISCUSSION
The toll-like receptor (TLR) pathway is an important signaling cascade acting within innate immunity [24]. Although most research on this pathway on molecular level is related to cells of the immune system, there is no doubt that localization of TLR on cells of non-immune origin indicates involvement of those cells in various manifestations of innate immunity and inflammatory response. Astrocytes are of particular interest because they are cells of ectodermal (non-immune) origin. Thus, astrocytes were not considered as CNS located inflammatory response mediators for a long time. Just very recently this issue was reevaluated. Astrocytes were demonstrated to play an active role in inflammatory responses that accompany neurodegenerative disorders and various pathologies of the brain [1, 25]. Nevertheless, understanding of the characteristics of innate immune responses in astroglial cells is very limited, and this represents a significant gap in nervous tissue biology.
In the present research, a model of inflammatory response caused by stimulation of TLR with LPS was used. Previously we showed that LPS activated TLR-4 mediated response in astroglial cultures [11, 12]. This activation was accompanied with increase in COX-2 protein expression and PGE2 production. Additionally, increased release of TNFα and IL-10 into the cell culture medium was observed. Thus, classic characteristics of inflammatory response were observed, and proinflammatory (TNFα, PGE2) as well as antiinflammatory (IL-10) markers were detected. Increase in COX-2 production can be observed as a proinflammatory response component (this is a key enzyme involved into synthesis of the proinflammatory mediator PGE2 and other primary prostaglandins), as well as an antiinflammatory mediator (primary prostaglandins serve as a source for production of cyclopentenone prostaglandins that are referred to as signaling lipids of resolution of inflammatory responses [26, 27]).
We demonstrated that rosiglitazone enhances LPS-stimulated expression of COX-2 and IL-10 mRNA, but not TNFα mRNA. Previously our group reported that preincubation of primary rat astrocytes with rosiglitazone (30 min, 20 µM) resulted in increase in LPS-stimulated expression of the COX-2 gene 6-fold [11, 19]. Thus, our current data come into agreement with previously obtained results. There are no data on the effect of rosiglitazone on the IL-10 and TNFα genes in astrocytes. Our results support a hypothesis of antiinflammatory properties of rosiglitazone. Moreover, targeted activation of just IL-10 suggests that rosiglitazone might be a modulator of inflammatory response resolution. Indeed, currently not only pro- and antiinflammatory stages of inflammation, but resolution of the inflammation phase is recognized and this process is being actively studied as a complex of mechanisms directed toward elimination of consequences of inflammatory stimulus [28, 29]. Resolution of inflammation is linked to action of specific oxylipins that might be produced by COX-2 [30].
The mechanism of action of rosiglitazone is of great interest. Previously, we [19] and other groups reported that effects of rosiglitazone are related to its activity as a nuclear receptor PPARγ agonist. Nevertheless, IL-10 mRNA upregulation was provided in a PPARγ-independent fashion as indicated by our experiments with the PPARγ antagonist GW9662. PPARγ-independent effects of rosiglitazone, including its direct action on p38 MAPK were discussed in literature [22]. Still, we could not support or refute this interaction because within our model application of SB203580, a p38 MAPK inhibitor, turned out to suppress LPS-induced upregulation of genes, and the stimulatory effect of rosiglitazone was not revealed on that background. Probably, p38 MAPK is placed upstream to a rosiglitazone target within the signaling pathway, but this issue requires additional studies. Moreover, for cells of immune origin it has been demonstrated that rosiglitazone might downregulate production of TNFα and other proinflammatory cytokines, influencing corresponding gene expression levels [10]. In our case, rosiglitazone did not influence proinflammatory cytokines but induced IL-10, which is listed among cytokines of inflammation resolution.
Study of mRNA turnover of inflammatory response markers and rosiglitazone involvement into this process is a field of great interest. Indeed, posttranscriptional control of gene expression plays an active role in regulation of inflammation. It has been demonstrated that modulation of mRNA turnover rates is very important for genes of inflammatory response. Of interest was an observation that TLR-mediated LPS stimulation results in mRNA decay downregulation for multiple participants of inflammatory response [14]. We observed increase in mRNA decay rates for nuclear receptors of the PPAR family under LPS challenge [11, 13]. Previously, we demonstrated that COX-2 mRNA half-life corresponds to 75 min in naïve cells, while LPS challenge increases the half-life up to 120 min, and rosiglitazone treatment abolishes the LPS-induced effect and returns the mRNA decay rate to its control levels [23]. In similar experiments of the present study, we found that COX-2 mRNA decays twice slower in astrocytes, and LPS treatment reduces the mRNA half-life, but still rosiglitazone returns the rate to its initial level. It is not clear if the observed effect is related to peculiarities of astrocyte-intrinsic mechanisms of mRNA decay control, or if it results from a specific role that COX-2 plays within these cells. Indeed, the pattern of COX-2 mRNA turnover alteration under LPS stimulation in glioma cells did not distinguish those cells from immune cells (in both cases downregulation of degradation after TLR activation was observed). It is noteworthy that half-life values for mRNA of the studied genes after LPS treatment were lower than those reported elsewhere (more than 40 min for macrophages of variable origins [26, 31, 32], more than 2 h for cells of lung tumor [33], breast tumor [34], and C6 rat astroglioma [23]). At the same time, β-tubulin mRNA turnover time remained stable, corresponding to data found in literature. Our results indicate that a system of mRNA decay is activated in astrocytes and regulates inflammatory gene expression. This system might be of high importance for fine-tuning processes within the brain, especially inflammatory response.
Our results with rosiglitazone indicate that mRNA degradation can be controlled with low molecular weight ligands. Another important observation is that rosiglitazone might be a substance potent to stimulate processes relevant to resolution (in this case IL-10 release), without activating proinflammatory responses (TNFα release). This opens the field for the search for low molecular weight modulators of resolution.
Acknowledgments
This study was supported by the Russian Science Foundation (project No. 16-15-10298; Figs. 1-4) and by the Russian Foundation for Basic Research (research project No. 16-34-01085 mol_a; Fig. 5).
REFERENCES
1.Amor, S., Peferoen, L. A., Vogel, D. Y., Breur, M.,
Van der Valk, P., Baker, D., and Van Noort, J. M. (2014) Inflammation
in neurodegenerative diseases – an update,
Immunology, 142, 151-166.
2.Carta, A. R. (2013) PPAR-γ: therapeutic
prospects in Parkinson’s disease, Curr. Drug Targets,
14, 743-751.
3.Heneka, M. T., and Landreth, G. E. (2007) PPARs in
the brain, Biochim. Biophys. Acta, 1771, 1031-1045.
4.Rainsford, K. D. (2007) Anti-inflammatory drugs in
the 21st century, Subcell. Biochem., 42, 3-27.
5.Rolland, B., Deguil, J., Jardri, R., Cottencin, O.,
Thomas, P., and Bordet, R. (2013) Therapeutic prospects of PPARs in
psychiatric disorders: a comprehensive review, Curr. Drug.
Targets, 14, 724-732.
6.Stulc, T., Svobodova, H., Krupickova, Z.,
Dolezalova, R., Marinov, I., and Ceska, R. (2014) Rosiglitazone
influences the expression of leukocyte adhesion molecules and CD14
receptor in type 2 diabetes mellitus patients, Physiol. Res.,
63 (Suppl. 2), S293-298.
7.Landreth, G., Jiang, Q., Mandrekar, S., and Heneka,
M. (2008) PPARγ agonists as therapeutics for the treatment of
Alzheimer’s disease, Neurotherapeutics, 5,
481-489.
8.Croasdell, A., Duffney, P. F., Kim, N., Lacy, S.
H., Sime, P. J., and Phipps, R. P. (2015) PPARγ and the innate
immune system mediate the resolution of inflammation, PPAR Res.,
2015, 549691.
9.Pisanu, A., Lecca, D., Mulas, G., Wardas, J.,
Simbula, G., Spiga, S., and Carta, A. R. (2014) Dynamic changes in pro-
and anti-inflammatory cytokines in microglia after PPAR-γ agonist
neuroprotective treatment in the MPTPp mouse model of progressive
Parkinson’s disease, Neurobiol. Dis., 71,
280-291.
10.Li, X., Xu, B., Wang, Y., and Wei, L. (2014)
Anti-inflammatory effect of peroxisome proliferator-activated
receptor-γ (PPAR-γ) on non-obese diabetic mice with
Sjogren’s syndrome, Int. J. Clin. Exp. Pathol., 7,
4886-4894.
11.Chistyakov, D. V., Aleshin, S., Sergeeva, M. G.,
and Reiser, G. (2014) Regulation of peroxisome proliferator-activated
receptor β/δ expression and activity levels by toll-like
receptor agonists and MAP kinase inhibitors in rat astrocytes, J.
Neurochem., 130, 563-574.
12.Astakhova, A. A., Chistyakov, D. V., Pankevich,
E. V., and Sergeeva, M. G. (2015) Regulation of cyclooxygenase 2
expression by agonists of PPAR nuclear receptors in the model of
endotoxin tolerance in astrocytes, Biochemistry (Moscow),
80, 1262-1270.
13.Chistyakov, D. V., Aleshin, S. E., Astakhova, A.
A., Sergeeva, M. G., and Reiser, G. (2015) Regulation of peroxisome
proliferator-activated receptors (PPAR) α and -γ of rat
brain astrocytes in the course of activation by toll-like receptor
agonists, J. Neurochem., 134, 113-124.
14.Anderson, P. (2010) Post-transcriptional regulons
coordinate the initiation and resolution of inflammation, Nat. Rev.
Immunol., 10, 24-35.
15.Basil, M. C., and Levy, B. D. (2016) Specialized
pro-resolving mediators: endogenous regulators of infection and
inflammation, Nat. Rev. Immunol., 16, 51-67.
16.Livak, K. J., and Schmittgen, T. D. (2001)
Analysis of relative gene expression data using real-time quantitative
PCR and the 2(–ΔΔC(T)) method, Methods,
25, 402-408.
17.Ross, J. (1995) mRNA stability in mammalian
cells, Microbiol. Rev., 59, 423-450.
18.Chen, C. A., Ezzeddine, N., and Shyu, A. (2008)
Chapter 17. Messenger RNA half-life measurements in mammalian cells, in
RNA Turnover in Eukaryotes: Nucleases, Pathways and Analysis of mRNA
Decay, Elsevier, pp. 335-357.
19.Aleshin, S., Grabeklis, S., Hanck, T., Sergeeva,
M., and Reiser, G. (2009) Peroxisome proliferator-activated receptor
(PPAR)-γ positively controls and PPARα negatively controls
cyclooxygenase-2 expression in rat brain astrocytes through a
convergence on PPARβ/δ via mutual control of PPAR expression
levels, Mol. Pharmacol., 76, 414-424.
20Park, E. J., Park, S. Y., Joe, E.-H., and Jou, I. (2003)
15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT
inflammatory signaling through induction of suppressor of cytokine
signaling 1 (SOCS1) and SOCS3 in glia, J. Biol. Chem.,
278, 14747-14752.
21.Luna-Medina, R., Cortes-Canteli, M., Alonso, M.,
Santos, A., Martinez, A., and Perez-Castillo, A. (2005) Regulation of
inflammatory response in neural cells in vitro by
thiadiazolidinones derivatives through peroxisome
proliferator-activated receptor γ activation, J. Biol.
Chem., 280, 21453-21462.
22.Schiefelbein, D., Seitz, O., Goren, I., Dissmann,
J. P., Schmidt, H., Bachmann, M., Sader, R., Geisslinger, G.,
Pfeilschifter, J., and Frank, S. (2008) Keratinocyte-derived vascular
endothelial growth factor biosynthesis represents a pleiotropic side
effect of peroxisome proliferator-activated receptor-agonist
troglitazone but not rosiglitazone and involves activation of p38
mitogen-activated protein kinase: implications for diabetes-impaired
skin repair, Mol. Pharmacol., 74, 952-963.
23.Pankevich, E. V., Chistyakov, D.
V., Astakhova, A. A., Strelkova, O. S., and Sergeeva, M. G.
(2015) Regulation of cyclooxygenase 2 mRNA degradation by rosiglitazone
in C6 glioma cells in the presence of inflammation inductors, Biol.
Membr., 9, 337-341.
24.Takeda, K., and Akira, S. (2005) Toll-like
receptors in innate immunity, Int. Immunol., 17,
1-14.
25.Heales, S. J. R., Lam, A. A. J., Duncan, A. J.,
and Land, J. M. (2004) Neurodegeneration or neuroprotection: the
pivotal role of astrocytes, Neurochem. Res., 29,
513-519.
26.Font-Nieves, M., Sans-Fons, M. G., Gorina, R.,
Bonfill-Teixidor, E., Salas-Perdomo, A., Marquez-Kisinousky, L.,
Santalucia, T., and Planas, A. M. (2012) Induction of COX-2 enzyme and
down-regulation of COX-1 expression by lipopolysaccharide (LPS) control
prostaglandin E2 production in astrocytes, J. Biol. Chem.,
287, 6454-6468.
27.Sergeeva, M. G., and Varfolomeeva, A. T. (2006)
The Cascade of Arachidonic Acid [in Russian], Narodnoe
Obrazovanie, Moscow.
28.Ortega-Gomez, A., Perretti, M., and Soehnlein, O.
(2013) Resolution of inflammation: an integrated view, EMBO Mol.
Med., 5, 661-674.
29.Sugimoto, M. A., Sousa, L. P., Pinho, V.,
Perretti, M., and Teixeira, M. M. (2016) Resolution of inflammation:
what controls its onset? Front. Immunol., 7, 160.
30.Serhan, C. N., Chiang, N., Dalli, J., and Levy,
B. D. (2014) Lipid mediators in the resolution of inflammation, Cold
Spring Harb. Perspect. Biol., 7, a016311.
31.MacKenzie, K. F., Van Den Bosch, M. W. M., Naqvi,
S., Elcombea, S. E., McGuirea, V. A., Reithb, A. D., Blackshearc, P.
J., Deand, J. L. E., and Arthura, J. S. C. (2013) MSK1 and MSK2 inhibit
lipopolysaccharide-induced prostaglandin production via an
interleukin-10 feedback loop, Mol. Cell. Biol., 33,
1456-1467.
32.Bollmann, F., Wu, Z., Oelze, M., Siuda, D., Xia,
N., Henke, J., Daiber, A., Li, H., Stumpo, D. J., Blackshear, P. J.,
Kleinert, H., and Pautz, A. (2014) Endothelial dysfunction in
tristetraprolin-deficient mice is not caused by enhanced tumor necrosis
factor-α expression, J. Biol. Chem., 289,
15653-15665.
33.Li, H., Pan, G.-F., Jiang, Z.-Z., Yang, J., Sun,
L.-X., and Zhang, L.-Y. (2015) Triptolide inhibits human breast cancer
MCF-7 cell growth via downregulation of the ERα-mediated
signaling pathway, Acta Pharmacol. Sin., 36, 606-613.
34.Subbaram, S., Lyons, S. P., Svenson, K. B.,
Hammond, S. L., McCabe, L. G., Chittur, S. V., and DiPersio, C. M.
(2014) Integrin α3β1 controls mRNA splicing that determines
Cox-2 mRNA stability in breast cancer cells, J. Cell. Sci.,
127, 1179-1189.