Self-Organization of Recombinant Membrane Porin OmpF from Yersinia pseudotuberculosis in Aqueous Environments
E. V. Sidorin*, V. A. Khomenko, N. Yu. Kim, P. S. Dmitrenok, A. M. Stenkova, O. D. Novikova, and T. F. Solov’eva
Elyakov Pacific Institute of Bioorganic Chemistry, Far-Eastern Branch of the Russian Academy of Sciences, 690022 Vladivostok, Russia; E-mail: sev1972@mail.ru* To whom correspondence should be addressed.
Received July 17, 2017; Revision received August 17, 2017
Recombinant porin OmpF (an integral protein of bacterial outer membrane) from Yersinia pseudotuberculosis was synthesized in Escherichia coli cells as inclusion bodies. By combining the methods of anion-exchange and gel filtration chromatographies, recombinant OmpF (rOmpF) was isolated as an individual protein in its denatured state, and its characteristic properties (molecular mass, N-terminal amino acid sequence, and hydrodynamic radius of the protein in 8 M urea solution) were determined. According to the data of gel filtration, dynamic light scattering, optical spectroscopy, and binding of the hydrophobic fluorescent probe 8-anilino-1-naphthalenesulfonic acid, the rOmpF is fully unfolded in 8 M urea and exists in random coil conformation. In aqueous solutions, rOmpF undergoes conformational changes, reversible self-association, and aggregation. When transferred from 8 M urea into water, PBS (containing 0.15 M NaCl, pH 7.4), or buffer containing 0.8 M urea (pH 8.0), fully unfolded rOmpF forms relatively compact monomeric intermediates prone to self-association with formation of multimers. The oligomeric intermediates have high content of native protein-like secondary structure and pronounced tertiary structure. In acidic media (pH 5.0, close to the protein isoelectric point), rOmpF undergoes rapid irreversible aggregation. Therefore, we found that medium composition significantly affects both porin folding and processes of its self-association and aggregation.
KEY WORDS: Yersinia pseudotuberculosis, self-organization and aggregation of outer membrane protein, recombinant porin OmpF, dynamic light scatteringDOI: 10.1134/S0006297917110086
Abbreviations: AI, aggregation index; 8-ANS, 8-anilino-1-naphthalenesulfonic acid; DLS, dynamic light scattering; IBs, inclusion bodies; MALDI-TOF MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; PEG, polyethylene glycol; RH, hydrodynamic radius; rOmpF, recombinant OmpF.
The problem of polypeptide chain self-arrangement into a highly
organized functional structure has been in the center of scientific
attention since the 1960s, when it was postulated that the native
structure of a protein molecule is determined by the unique amino acid
sequence of the protein, as well as by its environment. The prevalent
approach to studying this problem is investigation of protein
folding/unfolding and characterization of partially and/or incorrectly
folded intermediates prone to association or aggregation. Under certain
conditions, self-association might serve as an additional structuring
factor [1]. However, aggregation of incorrectly
folded states can stabilize them and result in the formation of either
irregular amorphous aggregates or ordered structures, e.g. amyloid
fibrils that might contribute to the development of various
neurodegenerative disorders (Alzheimer’s disease,
Parkinson’s disease, prionic diseases, cystic fibrosis, etc.) [2]. Recent insights into the mechanisms of protein
folding and accompanying processes of protein aggregation are extremely
important not only for prediction of the protein spatial structure from
its amino acid sequence, but also for solving many practical problems,
such as production of recombinant proteins, design of artificial
proteins with predetermined properties, and development of medications
for diseases related to incorrect protein folding.
Protein folding has been mostly studied in small single-domain soluble proteins. However, the folding mechanisms of large multidomain and membrane proteins are of particular interest. It was shown that many β-barrel integral membrane proteins of bacterial and mitochondrial outer membranes spontaneously fold in detergent-containing micelles and lipid bilayers in vitro. The first protein of this type for which successful folding has been demonstrated was OmpA from Escherichia coli [3]. Later, another E. coli outer membrane protein, trimeric porin OmpF, was folded into its native structure from a fully unfolded state [4]. According to the body of evidence obtained so far, transmembrane proteins of this structural type fold in a more cooperative manner than the α-helical proteins [5]. Their self-assembly proceeds without formation of stable intermediates, whereas folding of α-helical proteins from a random coil into the native structure occurs through intermediate states that are similar to the molten globule of water-soluble — globular proteins.
The goal of our study was to investigate the behavior of unfolded OmpF porin from Yersinia pseudotuberculosis in different aqueous solutions. This study was possible because fully unfolded β-barrel proteins are soluble in water because of their low hydrophobicity. The polarity of porin calculated by the Capaldi method is 43% [6]. To observe folding intermediates and to follow protein states during prolonged periods of incubation in solutions, we performed experiments in the absence of compounds that model a membrane environment, i.e. excluded the final stage of protein self-assembly. We were able to collect information on the protein association that accompanied folding, as well as on the aggregation that competed with the folding from the random coil into the native structure.
MATERIALS AND METHODS
Escherichia coli strains TOP10 and BL21(DE3) (Invitrogen, USA), chromatography columns Source 15 Q PE 4.6/100 (Pharmacia Biotech, Sweden), HiTrap Desalting (Amersham Biosciences, Sweden), Superdex 200 10/300 GL (GE Healthcare, USA), Amicon Ultra (3-kDa cut-off) concentrating centrifuge tubes (Merck Millipore, Germany), cellulose membrane filters (0.45 µm) (Agilent, USA), acrylamide (Serva, Germany), sodium dodecyl sulfate (SDS) (Bio-Rad, USA), phenylmethanesulfonyl fluoride (PMSF) and polyethylene glycol (PEG) 20 kDa (Merck, Germany), 3,5-dimethoxy-4-hydroxycinnamic (sinapinic) acid (Bruker Daltonics, Germany), acetonitrile (quality 2; Cryochrom, Russia), 8-anilino-1-naphthalenesulfonic acid (8-ANS) and trifluoroacetic acid (TFA) (Sigma, USA), PolySorp 96-well plates (Nunc, USA), and pH 7.4 phosphate buffered saline (PBS) (Helicon, Russia) were used. All other reagents were of pure grade (Reakhim, Russia) and used without additional purification.
Buffers used were: TEN (50 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl, pH 8.0); PBS (10 mM NaH2PO4, 137 mM NaCl, 2 mM KCl, pH 7.4); buffer A (10 mM NaH2PO4/NaOH, pH 8.0, 8 M urea); buffer B (50/100 mM CH3COONa/CH3COOH (50/100 mM CH3COONa (200 ml), 17.4 M CH3COOH (0.33/0.66 ml), pH 5.0)); buffer C (100 mM NaH2PO4/NaOH, pH 8.0); buffer D (buffer A (20 ml), 50 mM buffer B (46.7 ml), buffer C (133.3 ml), pH 8.0); buffer E (50 mM buffer B (46.7 ml), buffer C (133.3 ml), pH 8.0).
Purification of Y. pseudotuberculosis rOmpF. Cloning and expression of rOmpF and isolation of inclusion bodies (IBs) from E. coli cells were performed as described in [7]. The IBs were washed twice with TEN buffer, dissolved in cold TEN buffer containing 8 M urea and 0.1 mM PMSF, and sonicated on ice for 20 min at 44 Hz. Insoluble pellet was removed by centrifugation at 12,000g for 10 min at 4°C. Before loading on the anion-exchange column, the supernatant was diluted 16 times with buffer A and filtered through a 0.45-µm membrane. Ion-exchange chromatography was performed on a Source 15Q column equilibrated with buffer A using an FPLC system (Amersham Pharmacia Biotech, USA). After loading the sample, the column was washed with 10 ml of buffer A to remove weakly bound proteins. The adsorbed proteins were then washed off at 0.5 ml/min by isocratic elution and with a continuous 0.2-1 M gradient of NaCl in buffer A (5 ml). Isocratic elution was performed in two steps with buffer A containing 0.2 and 2 M NaCl (7 and 5 ml, respectively). Collected fractions (0.5 ml) were analyzed by SDS-PAGE; fractions containing rOmpF were combined and concentrated using centrifuge concentrators with 3-kDa cut-off. The concentrated protein solutions were subjected to a second round of chromatography on a Source 15 Q column under identical conditions. The rechromatographed protein was concentrated and purified by FPLC gel filtration on a Superdex 200 column equilibrated with buffer A. Before loading on the column, SDS and NaCl were added to the sample to a final concentration of 0.035 M (1% w/v) and 1 M, respectively, and the sample (0.25 ml) was filtered through a 0.45-µm membrane. The elution rate was 0.5 ml/min; protein fractions (0.5 ml) were collected based on their absorbance at 280 nm. The major protein peak was eluted within 9.6 to 12.1 ml. Fractions containing purified rOmpF (shown by SDS-PAGE) were combined and concentrated as described above.
Gel filtration of homogenous denatured rOmpF. Gel filtration on a Superdex 200 10/300 GL column was used to investigate processes resulting in changes in size of the denatured porin particles (i.e. folding and self-association). The column was equilibrated with either PBS or buffer D. Samples (0.25 ml) of completely denatured porin (0.18 mg/ml) in buffer A were loaded on the column and then eluted at 0.5 ml/min.
Before mass spectrometry, dynamic light scattering, and optical spectrometric analysis, protein samples were desalted by gel filtration on a HiTrap Desalting column. Depending on the experimental task, the column was equilibrated with deionized water or PBS. Porin samples (0.1 ml, 1.8 mg/ml) in buffer A were loaded on the column and eluted at 1 ml/min. The elution profile was registered at 280 nm; fractions containing rOmpF (0.5-0.7 ml) were collected.
In all experiments, rOmpF concentration was determined from absorbance at 280 nm using molar extinction coefficient of 1.27.
SDS electrophoresis in polyacrylamide gel (SDS-PAGE) was performed by the method of Laemmli [8]. Protein samples were mixed with sample buffer and loaded on a gel without prior boiling. The gels were stained with Coomassie Brilliant Blue R-250 solution in 10% acetic acid and 45% ethanol. Protein standards (11, 17, 24, 33, 40, 55, 72, 100, and 130 kDa) were used as molecular mass markers (Fermentas, Lithuania).
N-terminal sequencing. The N-terminal sequence of rOmpF (20 a.a.) was determined in two repeated experiments by the Edman method with a Procise 492c LC amino acid sequencer (Applied Biosystems, USA) equipped with a Series 200 UV/VIS detector (Perkin Elmer, USA). Amino acid phenylthiohydantoin derivatives were analyzed as recommended by the manufacturer.
Circular dichroism and fluorescence spectroscopy. CD spectra were recorded on a Chirascan plus CD spectrometer (Applied Photophysics Ltd, UK) in the peptide bond and aromatic spectrum regions in 0.1- and 1-cm quartz cuvettes, respectively. For the CD spectrum of the peptide region (190-240 nm), the ellipticity [θ] of an average residue was calculated assuming the molecular mass of the residue to be 110 Da using the formula:
[θ] = [θ]observed S × 110/(Cd) (deg·m2/dmol),
where S is the spectrometer sensitivity; C is the protein concentration, mg/ml; d is the cuvette width, cm. Molar ellipticity [θ]M for the aromatic part of the CD spectrum (240-320 nm) was calculated assuming molecular mass of the protein to be 37.1 kDa. The rOmpF samples for recording CD spectra in water and PBS were prepared by gel filtration as described above. rOmpF samples in buffer D and buffer D containing 35% PEG 20 kDa were obtained by diluting protein solution in buffer A (1.8 mg/ml) 10 times with the corresponding buffer.
Fluorescence spectra were registered with a Hitachi 850 spectrophotometer (Hitachi, Japan) at 25°C in a 1-cm quartz cuvette (λex = 280 and 296 nm) with a monochromator slit width of 5 nm.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). Mass spectra were obtained by MALDI-TOF MS with an Ultraflex III TOF/TOF mass spectrometer (Bruker Daltonics, Germany) equipped with a high-mass detector HMD-1 (CovalX AG, Switzerland) in linear regime with registration of positively charged ions. Saturated solution of 3,5-dimethoxy-4-hydroxycinnamic (sinapinic) acid (10 mg/ml) in acetonitrile/0.1% TFA (1 : 1 v/v) was used as the matrix. Before mass spectrometry, buffer A in the samples of homogenous rOmpF in buffer A was replaced by deionized water by gel filtration on the HiTrap Desalting column. The samples were mixed with the matrix and loaded on the target by the dried droplet method.
Aggregation index (AI). To calculate changes in the AI of protein solutions with time, optical absorbance of these solutions at 280 and 340 nm was determined with a µ-Quant spectrophotometer (Bio-TEK Instruments, USA) in a 96-well quartz microplate every 3 min for 2 h. The AI was calculated using the formula:
AI = 100 × A340/(A280 – A340) [9].
The samples (total volume, 300 µl) were prepared by mixing 30 µl of rOmpF (0.55 mg/ml) in buffer A, 70 µl of 50 mM buffer B, and 200 µl of 100 mM buffer B buffer or buffer C.
Dynamic light scattering (DLS). The size of the IBs and porin particles in aqueous solutions was determined by the dynamic light scattering method with a ZetaSizer Nano ZS spectrometer (Malvern, UK) equipped with a He-Ne laser (λ = 633 nm; 4 mW) at 173° angle. The hydrodynamic radius (RH) of the protein particles was calculated using software provided with the spectrometer. Before the DLS measurements, samples of denatured rOmpF in buffer A (0.55 mg/ml) were transferred to aqueous medium (water or PBS) by gel filtration or by 10× dilution with the corresponding buffer (buffers B or E) as described above. All measurements were performed in a 10 × 10-mm cuvette. The time for accumulation of the correlation function (2.5-10 min) was selected automatically by the spectrometer software.
Identification of hydrophobic surfaces in rOmpF in aqueous media. To identify hydrophobic surfaces in rOmpF in aqueous media of varying composition, we used binding of the hydrophobic fluorescent probe 8-ANS to the porin molecule. The fluorescence of the protein solution was determined at λem = 530 nm (λex = 360 nm) with an FL-600 spectrophotometer (Bio-TEK Instruments) in PolySorp 96-well polystyrene plates (Nunc). Porin samples were in water, buffer D, and buffer D containing 35% PEG 20000; rOmpF was in buffer A (0.18 mg/ml, 4.9 mM). The samples were prepared as described above (see “Circular dichroism and fluorescence spectroscopy”) and incubated for 24 h at 25°C. Then, 8-ANS was added to the protein solutions to a final concentration of 85 μM, and the solutions were incubated at 25°C for 60 min. Relative fluorescence was calculated using the formula:
FR = (F – F0)/F0 [10],
where FR is relative protein fluorescence in the aqueous solution; F is fluorescence intensity of protein-bound 8-ANS in the aqueous solution; and F0 is fluorescence intensity of 8-ANS in the same aqueous solution in the absence of protein.
RESULTS AND DISCUSSION
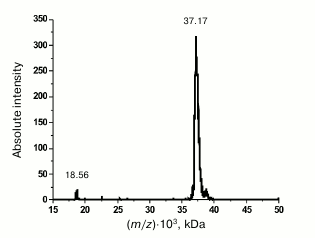
Purification and characterization of rOmpF. Recombinant porin OmpF (rOmpF) from Y. pseudotuberculosis was synthesized in E. coli cells as inclusion bodies (IBs) [7]. Inclusion bodies are insoluble aggregates of the expressed target protein that also contain admixtures of lipids, nucleic acids, and some cell proteins [11]. Existing procedures for OmpF purification, including treatment of IBs with enzymes, detergents, or chaotropic agents, are insufficient to completely remove these contaminants. According to gel filtration and DLS data, even after dissolving the IBs in such strong chaotropic agent as 8 M urea, rOmpF still remained in aggregates associated with various contaminants and exhibited multimodal particle distribution (Fig. 1, a and b, thin solid line). Average hydrodynamic radius (RH) of the particles in this sample was 50.5 nm according to the number size distribution or 158 nm according to the volume size distribution. Most of the contaminants were removed by the anion-exchange chromatography; however, rOmpF still remained in aggregates that could be only broken with a high concentration of strong anionic detergent (1% SDS) in the presence of 1 M NaCl and 8 M urea (Fig. 1, a and b, thick solid line). According to SDS-PAGE, mass spectrometry, and N-terminal sequencing, the purified rOmpF was a homogenous protein with a molecular mass of 37.1 ± 0.3 kDa (as determined by MALDI-TOF MS). The mass spectrum of the protein had signals from single- and double-charged protein ions (m/z = 37.1 and 18.6 kDa, respectively) (Fig. 2).
Fig. 1. Gel filtration (a) and number size distributions (b) of IBs (solid thin line) and unfolded rOmpF (solid thick line) in buffer A.
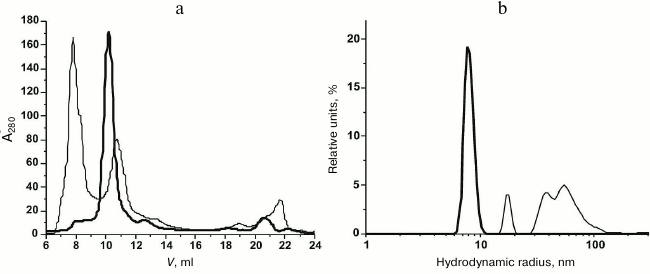
Fig. 2. MALDI-TOF MS spectrum of rOmpF.
The N-terminal sequence of rOmpF (20 a.a.) was determined by solid-phase Edman sequencing to be AEIYNKDGNKLDLYGKVDAR, and this coincides with the N-terminal sequence of Y. pseudotuberculosis OmpF from the UniProtKB database (accession number Q5EMM5). According to the DLS data, rOmpF in buffer A containing 8 M urea exhibited monomodal particle-size distribution with average RH of 8.2 ± 0.3 nm (Fig. 1b, broad solid line). The CD spectrum of porin in 8 M urea in the far-UV region had a negative minimum at 200 nm; the ellipticity of porin in this solution was close to zero at 222 nm, which is typical for fully denatured proteins [12]. Therefore, rOmpF from Y. pseudotuberculosis was obtained in monomeric form and was completely denatured in the buffer containing 8 M urea and 10 mM NaH2PO4/NaOH (pH 8.0).
Dynamic light scattering and gel filtration. To describe qualitatively and quantitatively the processes related to changes in the size of porin molecules (folding, denaturation, and aggregation), we used DLS and gel filtration.
According to the DLS data, the transfer of rOmpF from 8 M urea (monomodal distribution; RH, 8.2 nm) into water by gel filtration resulted in drastic changes in the distribution and size of the protein particles. During the first two hours, we observed the multimodal distribution with dominating content of particles with RH of 3.6 and 13.2 nm (96.9% of total particle volume) (Fig. 3a). Note that the small particles were comparable in size to molecules of hydrophilic proteins of similar molecular mass. For example, Skp3 chaperones from Y. pseudotuberculosis and E. coli with molecular masses of 48.3 and 47 kDa have RH values of 4.2 and 3.5 nm, respectively [13, 14]. After incubation for 24 h, the rOmpF solution contained mostly 13.4-nm particles (86.8% by volume), whereas small (<10 nm) particles disappeared due probably to aggregation into larger ones. The content of very large particles (>90 nm) increased from 3.1% (2-h incubation) to 13.2% (Fig. 3a). After 48 h, the contribution of large particles (88 and 445 nm) to the total volume increased to 26%, but relatively small particles (15.2 nm) still dominated (74%) in the solution (Fig. 3a). Within the time interval from ~5 min from the beginning of the experiment to 48 h of incubation, we observed formation of structures with more compact monomolecular structure than that of the fully unfolded protein. These structures formed small stable water-soluble associates (oligomers) and very large aggregates, the concentration of which increased relatively slowly.
Fig. 3. Particle-size volume distribution of rOmpF in (a) water and (b) PBS. The porin was incubated in the solution for 2 (solid thick line), 24 (solid thin line), or 48 h (dashed line).
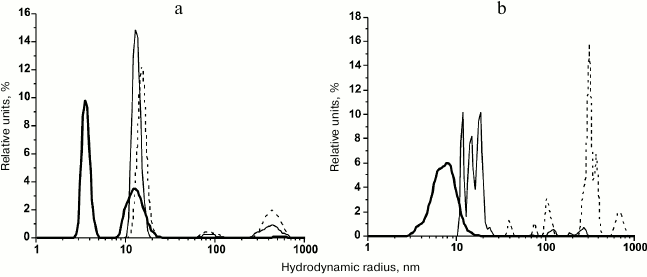
When incubated for 1 or 1.5 h in PBS, unfolded rOmpF demonstrated rather dynamic behavior (compared to the same protein in water) (Fig. 3b). The polymodal distribution of particles (3-16 nm) with maxima at 3.4, 6.5, and 7.8 nm became monomodal after 2 h of incubation (average RH, 5.6 nm; number and volume content, 100 and 99.7%, respectively) and stayed the same for at least another 30 min. Incubation of the porin for 24 h in PBS (Fig. 3b) or water resulted in the disappearance of particles <10 nm in size and formation of larger particles with RH of 10-25 nm. Unlike the particles in water, they exhibited polymodal distribution with maxima at 11.8, 14.8, and 18.7 nm (volume distribution) and were characterized by nearly the same relative content – 26.3, 29.2, and 38.2%, respectively (93.7%). The fraction of very large particles (>70 nm) increased from 0.3% (2.5 h) to 6.3%. After 48-h incubation, the porin solutions in PBS significantly differed from its water solution – in the presence of salt, the rOmpF formed particles of 39.1, 74.6, 103.1, and >300 nm (3.3, 2.4, 10.7, and 83.8% by volume, respectively). Fractions of relatively small particles of 39 and 74 nm comprised 22 and 19.5% by number, respectively.
When completely unfolded rOmpF (8 M urea) was subjected to gel filtration on a Superdex 200 column, the profile of protein elution with PBS contained two poorly resolved peaks with maxima at 12 and 14 ml (Fig. 4, peaks 1 and 2, respectively). Note that denatured protein was eluted from the same column with 10.3 ml of the elution buffer (Fig. 1a, thick solid line). Increase in the elution volume caused by rOmpF transfer into the buffer with no urea indicated more compact structure of the protein (peak 2) and, probably, the onset of aggregation (peak 1). According to the DLS data, after 2 h of incubation, peak 2 contained a population of particles of 2-9 nm and exhibited multimodal particle-size distribution with prevalence of 5.4-nm particles (90 and 89.6% of total number and volume of all particles, respectively). Interestingly, particles of similar size (~5 nm), i.e. more compact than the unfolded protein, dominated in the unfractionated solution that had been incubated for 2 h. This suggested that they were the most stable folding intermediates among all the compact particles observed. However, when isolated from the solution, these particles formed more compact intermediates. Indeed, further incubation of peak 2 shifted the distribution curve toward smaller particles: a peak of 2.5-nm particles (51.2%) appeared in addition to the 5.4-nm peak (48.3%). Incubation for 3 h resulted in the appearance of three peaks at 3.1, 3.5, and 6.2 nm (48.1, 41.2, and 10.7% of total amount of particles, respectively). After 72 h, the sample demonstrated monomodal size distribution of particles with RH of 376 nm.
Fig. 4. Gel filtration of denatured rOmpF in PBS (solid thick line) and buffer D (solid thin line). Peaks 1 and 2 on both curves are designated.
Therefore, in PBS porin aggregated faster and formed considerably larger particles than in water, probably because salt shielded charges on the protein particles and lessened electrostatic repulsion between them. A similar effect of sodium chloride on α-chymotrypsinogen A aggregation was described [15]. The behavior of porin in acidic buffer (pH 5.0, close to the protein isoelectric point (pI) 4.94) also confirmed this suggestion.
Using optical spectroscopy, we determined the aggregation index (AI) of the protein solution [9] at acidic pH 5.0 in the presence of 0.8 M urea. As follows from the changes in the protein AI (Fig. 5, curve 1) resulting from the dilution of the concentrated solutions of denatured porin (8 M urea) with the acidic buffer (pH 5.0), the rate of protein aggregation within the first 20 min after dilution was the highest (AI = 88%) with following slow increase in optical density at 340 nm.
Fig. 5. Aggregation index (AI) of denatured rOmpF and dependence of average RH on the time of porin incubation in 0.1 M buffer B containing 0.8 M urea. Curves: 1) AI of the rOmpF solution; 2) average RH according to the volume size distribution; 3) average RH according to the number size distribution.
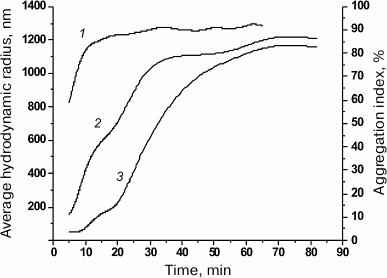
According to the DLS data, already within the first few minutes (5-9 min) of incubation in the acidic buffer in the presence of 0.8 M urea, fully denatured porin (RH, 8.2 nm) formed particles with RH of 44-53 nm (95.6-92.6% of total number of particles) as well as larger particles of 169 nm (7 min) and 240 nm (9 min). To follow the aggregation dynamics, we plotted the average RH of particles (obtained from data on their number and volume size distribution) against the time of porin incubation in the acidic buffer (Fig. 5). As seen from the graphs, changes in the particle size reflected two stages of the aggregation process: nucleation (formation of multiple relatively small particles via association of free molecules that served as aggregation centers) and aggregate growth (particle enlargement). The growth rate of large particles was high (according to the volume size distribution data) and mostly followed the increase in AI (Fig. 5, curves 1 and 2). Note that already at the first time-point (5 min), large particles with RH of 109 and 243 nm comprised up to 59% of the total particle volume (AI, 59%). The nucleation stage occurred almost immediately; no changes in the average RH were observed within the first 9 min (according to the number size distribution data) (Fig. 5, curve 3). The aggregation rate in this case was probably slowed due to the presence of 0.8 M urea in the solution. Further growth of protein aggregates can proceed by two mechanisms: attachment of individual protein molecules and association of small aggregates. It is possible that the rapid rise of the curves within 20 to 40 min of incubation (Fig. 5, curves 2 and 3) might be due to the second mechanism. The increase in aggregate size was accompanied by multimodal particle-size distribution at each time (data not shown). By minute 43, both size distribution curves had converged and then changed in a similar way (Fig. 5, curves 2 and 3); the particle distribution of at any time became monomodal, and the particle growth significantly slowed, which corresponded to the latest stage of the aggregation process, the so-called sedimentation stage.
Renaturation of proteins synthesized in vivo as IBs is often carried out in 0.8 M urea: IBs are dissolved in 8 M urea, and the solution is then diluted 10-fold. To study rOmpF renaturation under these conditions, rOmpF was transferred into buffer D (pH 8.0) containing 0.8 M urea. According to the DLS data, the rOmpF solution had a broad multimodal number distribution of particles (RH, 8-60 nm) already within the first 20 min of the experiment. Prolongation of the incubation time was accompanied by an increase in relative content of small particles (while preserving the above-mentioned particle size range). In solutions incubated for 20 min, 1.5 h, or 2.5 h, the dominating particles were of 41 and 47 nm (63%), 28 and 37 nm (83%), or 15 and 27 nm (65%), respectively (Fig. 6). The particle-size distribution was established after 2.5 h of incubation (average RH, 21 nm) and did not change significantly for another 24 and 48 h: average RH values of 25.8 and 26.6 nm, respectively, with dominating particles of 26 and 37 nm (60%) and 39 nm (42%). Apparently, low urea concentrations stabilized the formed particle population and prevented generation of very large aggregates (RH > 80 nm). These results correlated well with earlier data that low urea concentrations stabilize proteins in the molten globule state [16]. Large particles (100-600 nm) were present at all times of incubation and considerably differed in their size from the dominating population of smaller particles. Indeed, the fraction of these aggregates did not exceed 0.7% of the total number of particles; however, they represented 25-35% of the total particle volume. Note that the content of small particles (3-8 nm) that usually dominate in porin solutions in water and PBS was low within the first 2.5 h of incubation. We suggest that these particles were also formed in the presence of urea, but could not be detected due to rapid aggregation with the formation of larger and more stable particles, as confirmed by gel filtration data (see below).
Fig. 6. Number size distribution of rOmpF particles in buffer D. The porin solution was incubated for 20 min (solid thick line), 1.5 h (solid thin line), or 2.5 h (dashed line).
Porin was loaded on a Superdex 200 column in 8 M urea and eluted with buffer D. The elution profile featured two major peaks – 1 and 2 with elution volumes of 11 and 14 ml, respectively; peak 2 coincided with the peak in the gel filtration profile of rOmpF in PBS (Fig. 4). The AI for the porin solution in 0.8 M urea was typical of slightly aggregated solutions (AI < 8%). It increased insignificantly within 2 h of incubation and then stayed the same during incubation of the porin for another 24 h (data not shown).
Our experiments clearly showed that rOmpF solutions in 0.8 M urea, PBS or water formed sufficiently stable particles (folding intermediates) that were more compact than the unfolded protein. However, in PBS these particles associated with the formation of a dynamic population of oligomers, while in the presence of urea they formed larger and more stable associates.
Structure of intermediates. Formation of intermediates during porin folding in our experiments was confirmed by optical spectroscopy. Twenty-four hours after transferring into water, unfolded porin had a far-UV CD spectrum typical for proteins with residual secondary structure [12], i.e. with a strong negative band at 200 nm ([θ]200 = –10,000 deg·cm2·dmol–1) and a shoulder at 210-230 nm ([θ]220 = –4000 deg·cm2·dmol–1) (Fig. 7a, curve 1). Similar spectra are characteristic of proteins in an intermediate conformation state between molten globule and random coil, the so-called molten globule precursor (pre-molten globule) [17]. An increase in the time of porin incubation in water solutions did not affect the shape of the CD spectrum, except it caused a slight decrease in the absolute value of ellipticity of the 200-nm band (from –10,400 to –9045 deg·cm2·dmol–1).
Fig. 7. CD spectra of denatured rOmpF. a) Far-UV CD spectra in (1) water (24 h), (2) PBS (30 min), (3) PBS (24 h), (4) buffer D (30 min), (5) buffer D (24 h), and (6) buffer D containing 35% PEG 20 kDa (24 h). b) Near-UV spectrum in buffer D containing 35% PEG 20 kDa (24 h).
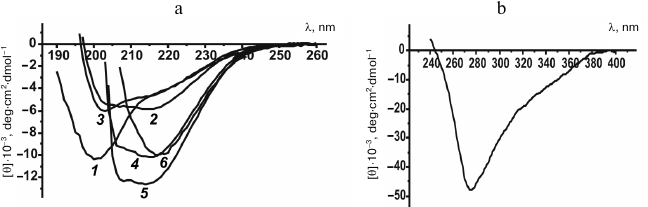
The CD spectrum of unfolded porin after incubation for 30 min in PBS had a broad negative band with the major minimum at 216 nm and a less pronounced minimum with a smaller amplitude at ~207 nm; [θ]197 = 0 is typical for α-β proteins (Fig. 7a, curve 2). Prolongation of porin incubation in PBS to 24 h changed the shape of the CD spectrum: the ellipticity of the 216-nm band decreased in absolute value, and the 207-nm minimum shifted to 203 nm (Fig. 7a, curve 3), which might be explained by a decrease in the content of β-fold regions and increase in content of random coil conformation in the protein.
Far-UV CD spectra of porin solution in buffer D containing 0.8 M urea after 0.5, 1.0, 1.5, 2.0, 2.5, and 24 h of incubation of the denatured protein had similar shapes (minima at 206 and 215-217 nm), relatively high negative ellipticity (from –10,000 to –12,000 deg·cm2·dmol–1), and [θ]203 = 0 (Fig. 7a, curves 4 and 5). The CD spectra of protein solutions incubated for 0.5-2.5 h almost coincided in their shape and amplitude (data not shown), while the porin incubated for 24 h had a relative increase in the absolute value of the ellipticity of the 208-nm band with a background increase in the CD signal in the peptide bond region of the spectrum. The spectral data indicated the presence of a developed secondary structure in the studied porin. The shape of the spectra indicated a high content of β-structure (minimum at 215-217 nm) and suggested an increase in the relative content of α-helical structure with prolongation of porin incubation in the solution. It should be noted that porin solutions incubated for different time periods (especially within the time interval from 20 min to 2.5 h) differed in size of protein associates but had virtually identical CD spectra, which suggests structural similarity between the multimeric forms of porin.
Total fluorescence spectra of denatured porin in buffer D had a maximum at 334.7 nm and similar fluorescence intensity (Fig. 8a, curve 2) irrespectively of the incubation time. Moreover, they significantly differed from the spectrum of completely unfolded protein that displayed two maxima at 307 nm (tyrosine residues) and 348 nm (tryptophan residues) (Fig. 8a, curve 1). Under the same conditions, the tryptophan fluorescence spectra of the porin solutions were shifted to shorter wavelength (maximum at 338.7 nm) compared to the spectrum of the unfolded protein, but they had similar intensity (Fig. 8b, curve 2). This indicates that the environment of tryptophan residues in partially folded porin is more hydrophobic that in the fully unfolded protein. However, total and tryptophan fluorescence spectra of the wild-type Y. pseudotuberculosis OmpF have maxima at 322 ± 2 and 332 ± 1 nm, respectively [18]. Therefore, the observed porin folding intermediates had sufficiently compact tertiary structure, although less compact than that of the native protein.
Fig. 8. Intrinsic fluorescence spectra of denatured rOmpF in (1) buffer A, (2) buffer D, and (3) buffer D containing 35% PEG 20 kDa: a) total fluorescence; b) tryptophan fluorescence.
One of the challenges in experiments on protein folding is identifying and deciphering structures of intermediate folding states (folding intermediates). To observe formation of an intermediate known as the molten globule, researchers use the fluorescent probe 8-ANS whose affinity for this intermediate is considerably higher than for the native or fully unfolded protein [19]. We found that relative intensity of 8-ANS fluorescence in the solution of denatured rOmpF in buffer D was 14 times higher than for the solution of the same protein in water or 8 M urea, which indicated the presence of solvent-accessible hydrophobic clusters (i.e. the absence of compact tertiary structure) in the intermediates formed by rOmpF in buffer D and the absence of such clusters in the intermediates formed in water or 8 M urea.
Therefore, the rOmpF folding intermediate in the buffer with 0.8 M urea was more compact than fully unfolded protein (according to gel filtration), had the secondary structure close to the native structure and tertiary structure less fixed than in a native protein and represented the molten globule.
Summarizing our results, we conclude that in PBS and 0.8 M urea unfolded porin formed intermediates with significantly organized spatial structure. In intermediates formed in PBS, the content of the ordered structure was lower than in the intermediates in 0.8 M urea, and the structure of such intermediates was less stable. Gel filtration and DLS data suggest that porin solutions can simultaneously contain intermediates that differ in levels of spatial organization. Comparative analysis of the size and structure of porin folding intermediates in water and buffer containing 0.8 M urea supports the earlier observation that self-association of these intermediates promotes formation of additional structural elements and even new intramolecular structural levels in the protein molecule [20]. Moreover, these data suggest that a porin solution can simultaneously contain several conformational intermediates with different structures.
Effect of macromolecular restrictions (crowding) on protein folding. In nature, protein folding occurs inside the cell, where the environment considerably differs from that in dilute buffer solutions used in this study. The intracellular space is filled with macromolecules (intracellular concentration, 80-400 mg/ml) and therefore has a limited amount of free water molecules and almost no free space [21, 22]. This environment significantly affects all aspects of protein behavior, including folding. To estimate the effect of macromolecular crowding on porin folding, we studied conformational behavior of unfolded porin in 35% PEG 20 kDa that is traditionally used as a molecular crowding agent [22]. Structural conversion of rOmpF was followed by CD spectroscopy and intrinsic fluorescence spectroscopy 0.5, 1.0, 2.5, and 24 h after 10× dilution of the unfolded porin in 8 M urea with 35% PEG in buffer D. The rOmpF intermediate formed under these conditions had a CD spectrum with a pronounced minimum at 217-218 nm in the peptide bond region that did not change within the time interval from 0.5 to 2.5 h. Twenty-four hours after dilution of the unfolded porin, its spectrum narrowed, and the ellipticity at 217 nm increased in absolute value by 12% (Fig. 7a, curve 6). The shape of this spectrum resembled that of the native OmpF dissolved in a detergent solution [18]. The near-UV CD spectrum of this folding intermediate had a characteristic negative band at 274 nm with molar ellipticity of –48,000 deg·cm2·dmol–1, which indicated an asymmetric environment of aromatic residues in the protein molecule (Fig. 7b). To characterize the tertiary structure of the porin folding intermediate formed in the presence of PEG, we recorded the spectra of its intrinsic fluorescence. The total fluorescence spectrum of this protein had a maximum at 345 nm that coincided with the long-wavelength maximum in the spectrum of fully unfolded porin in 8 M urea, while the maximum for the tryptophan fluorescence (359 nm) was shifted to longer wavelength compared to the spectrum of the fully unfolded protein (Fig. 8, a and b, curve 3). The results of fluorescence spectroscopy suggested that this intermediate lacked any organized tertiary structure. However, this suggestion did not correlate with the near-UV CD spectra of the protein, and our conclusions on the tertiary structure of porin based on the spectral data might be incorrect, because PEG is able to bind the protein and affect its intrinsic fluorescence [23]. This controversy requires further studies.
Recombinant porin rOmpF from Y. pseudotuberculosis was purified from IBs as fully unfolded protein (random coil) in 8 M urea. When transferred to aqueous solutions containing no chaotropic agents or low concentrations of a chaotropic agent (0.8 M urea), the rOmpF folded with formation of particles that were smaller than the unfolded protein. These particles differed in size, and probably shape, and represented partially folded porin molecules. The folding process at this stage resembled the first stage of the folding of the soluble protein. As observed for aqueous porin solutions, rOmpF folding into more compact structures was not necessarily accompanied by the formation of considerable amounts of secondary structure.
Compact monomeric structures could form soluble associates (multimers), whose size varied depending on the solvent – it was minimal in water and maximal in 0.8 M urea. Analysis of the particle size changes in the population of multimers with time suggested that these multimers were prone to association/dissociation. It is possible that an important role in protein self-association belongs to hydrophobic interactions. Indeed, monomeric folding intermediates of porin in water that lacked hydrophobic surface regions (did not bind 8-ANS) associated poorly, unlike the same particles with exposed hydrophobic clusters (efficiently bound 8-ANS) in 0.8 M urea.
According to the optical spectroscopy data, multimer formation not only favored preservation of the structure in partially folded protein molecules, but promoted further structuration of these molecules. Oligomeric folding intermediates displayed a high content of secondary structure similar to that in the native protein, as well as pronounced tertiary structure.
Aggregation of protein molecules in solutions occurred in parallel to their association and resulted in the accumulation of very large aggregates. (RH, 100-400 nm). This process was very slow and depended on the medium composition, i.e. it was promoted by the presence of salts and, especially, by acidic pH 5.0, which is close to isoelectric point of porin.
This study showed that the behavior of fully unfolded porin molecules is greatly influenced by the composition of the medium. Medium constituents affect size and spatial organization of monomeric intermediates, structure of their associates, and rate of porin self-organization.
Our results contribute to understanding of folding mechanisms and allow future development of methods for efficient renaturation of recombinant proteins that form inclusion bodies when synthesized in vivo.
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (project No. 16-08-00679) and by the Far East Program (project No. 15-I-5-004).
REFERENCES
1.Uversky, V. N., and Fink, A. L. (1998) Structural
effect of association on protein molecules in partially folded
intermediates, Biochemistry (Moscow), 63, 456-462.
2.Fink, A. L. (1998) Protein aggregation: folding
aggregates, inclusion bodies and amyloid, Fold. Des., 3,
R9-R23.
3.Henning, U., Sonntag, I., and Hindennach, I. (1978)
Mutants (OmpA) affecting a major outer membrane protein of
Escherichia coli K12, Eur. J. Biochem., 92,
491-498.
4.Eisele, J.-L., and Rosenbusch, J. P. (1990) In
vitro folding and oligomerization of a membrane protein. Transition
of bacterial porin from random coil to native conformation, J. Biol.
Chem., 265, 10217-10220.
5.Finkelstein, A. V., and Ptitsyn, O. B. (2005) in
Protein Physics [in Russian], KDU, Moscow, pp. 259-273.
6.Capaldi, R. A., and Vandercooi, G. (1972) The low
polarity of many membrane proteins, Proc. Natl. Acad. Sci. USA,
69, 930-932.
7.Khomenko, V. A., Portnyagina, O. Yu., Novikova, O.
D., Isaeva, M. P., Kim, N. Yu., Likhatskaya, G. N., Vostrikova, O. P.,
and Solovyeva, T. F. (2008) Isolation and characterization of
recombinant OmpF-like porin from the outer membrane of Yersinia
pseudotuberculosis, Bioorg. Khim., 34,
177-184.
8.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
9.Dunn, J. S., Nayar, R., Campos, J., Hybertson, B.
M., Zhou, Y., Manning, M. C., Repine, J. E., and Stringer, K. A. (2005)
Feasibility of tissue plasminogen activator formulated for pulmonary
delivery, Pharm. Res., 22, 1700-1707.
10.Tapan, K. C., Kali, P. D., and Nirmal, K. S.
(1993) Surface hydrophobicity of a low molecular weight basic trypsin,
J. Biochem., 113, 729-733.
11.Wulfson, A. N., Tikhonov, R. V., and Pechenov, S.
E. (2001) General approach to renaturation of recombinant proteins
produced as inclusion bodies, Dokl. Akad. Nauk, 380,
400-403.
12.Uversky, V. N. (2002) Natively unfolded proteins:
a point where biology waits for physics, Protein Sci.,
11, 739-756.
13.Sidorin, E. V., Sidorova, O. V., Tishchenko, N.
M., Khomenko, V. A., Novikova, O. D., and Solovyeva, T. F. (2015)
Chaperone-like activity of immunoglobulin-binding protein from
Yersinia pseudotuberculosis, Biol. Membr., 32,
217-220.
14.Qu, J., Mayer, C., Behrens, S., Holst, O., and
Kleinschmidt, J. H. (2007) The trimeric periplasmic chaperone Skp of
Escherichia coli forms 1 : 1 complexes with outer
membrane proteins via hydrophobic and electrostatic interactions, J.
Mol. Biol., 374, 91-105.
15.Li, Y., Ogunnaike, B. A., and Roberts, C. J.
(2010) Multi-variate approach to global protein aggregation behavior
and kinetics: effects of pH, NaCl and temperature for
α-chymotrypsinogen A, J. Pharm. Sci., 99,
645-662.
16.Edwin, F., Sharma, Y. V., and Jagannadham, M. V.
(2002) Stabilization of molten globule state of papain by urea,
Biochem. Biophys. Res. Commun., 290, 1441-1446.
17.Uversky, V. N., Gillespie, J. R., Millett, I. S.,
Khodyakova, A. V., Vasiliev, A. M., Chernovskaya, T. V., Vasilenko, R.
N., Kozlovskaya, G. D., Dolgikh, D. A., Fink, A. L., Doniach, S., and
Abramov, V. M. (1999) Natively unfolded human Prothymosin R adopts
partially folded collapsed conformation at acidic pH,
Biochemistry, 38, 15009-15016.
18.Kim, N. Yu., Novikova, O. D., Khomenko, V. A.,
Likhatskaya, G. N., Vostrikova, O. P., Emelyanenko, V. I., Kuznetsova,
S. M., and Solovyeva, T. F. (2007) Effect of pH on the structure and
functional activity of porin from the outer membrane of Yersinia
pseudotuberculosis. 1. Functionally important conformational
transitions of yersenin, Biol. Membr., 24, 150-158.
19.Semisotnov, G. V., Rodionova, N. A., Razgulyaev,
O. I., Uversky, V. N., Gripas’, A. F., and Gilmanshin, R. I.
(1991) Study of the “molten globule” intermediate state in
protein folding by a hydrophobic fluorescent probe, Biopolymers,
31, 119-128.
20.Kuznetsova, I. M., Biktashev, A. G., Khaitlina,
S. Yu., Vassilenko, K. S., Turoverov, K. K., and Uversky, V. N. (1999)
Effect of self-association on the structural organization of partially
folded proteins: inactivated actin, Biophys. J., 77,
2788-2800.
21.Kuznetsova, I. M., Turoverov, K. K., and Uversky,
V. N. (2014) What macromolecular crowding can do to a protein, Int.
J. Mol. Sci., 15, 23090-23140.
22.Chebotareva, N. A., Kurganov, B. I., and
Livanova, N. B. (2004) Biochemical effects of molecular crowding,
Biochemistry (Moscow), 69, 1522-1536.
23.Lai, J., Yan, H., Liu, Y., and Huang, Y. (2015)
Effects of PEG molecular weight on its interaction with albumin,
Chin. J. Polym. Sci., 33, 1239-1251.