REVIEW: On the Cause and Mechanism of Phenoptosis
R. F. Walker
ProSoma LLC, 2206 Beach Trail, #6, Indian Rocks Beach, FL 33785, USA; E-mail: drrwalker@gmail.com
Received June 26, 2017
Based upon evolvability theory, phenotypes like aging that offer no apparent individual benefit may evolve nonetheless. Pursuant to that concept, the evolution of a hypothetical, genome-based aging program called phenoptosis was proposed. However, while aging may facilitate evolvability, it need not result from a program specifically selected for that purpose. Instead, it is possible that the potential for aging is conserved within the genome as a part of a beneficial program that orchestrates and integrates developmental transformation of the soma from conception to maturation. Because somatic remodeling is inherently unstable, its continued non-programmatic expression beyond young adulthood (developmental inertia) erodes internal order, initiating and exacerbating aging. Thus, aging may result paradoxically from post-maturational expression of the same programmatic function for somatic transformation that previously provided individual benefit. It did so by ensuring acquisition of reproductive competence, post-reproductive development of parents to nurture offspring and thereby, to guarantee species survival. In an attempt to identify genes capable of controlling developmental inertia, we sequenced DNA from a series of subjects displaying extreme neoteny, i.e. retention of youthful characteristics during adulthood. We hoped to identify mutations associated with delayed development and to compare each subject’s biological and chronological ages. De novo mutations of coding-genes were found in all the subjects, but they could not be definitively identified as a cause of developmental delay. Nonetheless, genetic and epigenetic studies of neotenic subject’s DNA are on-going. We are attempting to determine if phenoptosis specifically evolved to cause aging, or rather if it exists as a cryptic component of the developmental program that expresses its lethal potential serendipitously and only after individual benefit is realized.
KEY WORDS: developmental inertia, temporal order, neoteny, homeodynamics, somatic transformation, DNA sequencingDOI: 10.1134/S0006297917120069
August Weismann is remembered for his theory that aging emerged as a programmed trait serving to eliminate old individuals, increase population turnover, reduce competition for resources, and thereby promote evolution [1]. However, at the time he lived, Darwin’s view that population evolution resulted from the genetic diversity originating within and benefiting individuals, prevailed. Presumably, such variants then spread as the result of differential survival and reproduction. Accordingly, Weismann abandoned his theory because it was incapable of explaining how aging benefited individuals. But, Darwin also felt that “our ignorance of the laws of variation is profound” [2], a concern that was relieved to some extent by Mendel and others working to better understand mechanisms of genetic variation and heredity.
Subsequently, attempts to explain the relationship between genetic variations and selectable phenotype variations led to the development of an alternative theory of evolution that was formally introduced in 1996 [3]. It dealt with observed discrepancies in traditional evolutionary mechanics, but not specifically with the problem of aging. Rather, it focused on the surprising observation that through mutation, recombination and development, organisms can produce offspring that are more fit than themselves [4]. The alternative theory of evolvability considers how genetic change leading to phenotype modification is conditioned by multiple levels of organization within the phenotype itself. Through bias of certain cellular, developmental and physiological processes of the organism, random mutation sometimes makes favorable non-lethal phenotypic variations available for evolution. Evolvability has at its core a genotype-phenotype map which relates genotypes to the range of phenotypes they are capable of producing [5]. This then provides a means to explain how genetic traits that do not necessarily convey individual benefit, can nonetheless evolve in groups and thereby how populations can adapt through natural selection.
Perhaps recognizing that evolvability could deal with the problematic issue of how aging and death might be beneficial, Vladimir Skulachev revived Weismann’s concept of programmed aging one year after Wagner’s publication [3, 6]. He suggested that programmed aging would gradually increase challenges to survival and reproduction, thereby enhancing selection of beneficial characteristics and assisting in the evolutionary process. Furthermore, he argued that “the balance between concepts of programmed and non-programmed aging seems to be really shifted to the programmed” [7] by likening natural death of organisms from intrinsic causes to “apoptosis”; a morphologically distinct form of programmed cell death [8]. Since apoptosis was first described, other forms of programmed cell death have also been recognized [9]. However, feeling that organismal aging was analogous to cellular apoptosis, he coined the term “phenoptosis” to describe death of individuals resulting, hypothetically, from the expression of lethal, genetically programmed mechanisms favored by natural selection [10]. Since its introduction to the nomenclature of aging, four types of phenoptosis have been proposed. These are classified in terms of their meanings, implications and comparisons with classical concepts of senescence. A classification relevant to the current discussion is slow phenoptosis which is defined as “obligatory and slow when it is characterized by an age-related progressively increasing probability of death that is a progressively decreasing fitness” [11, 12].
In support of programmed aging, Theodore Goldsmith proposed that an evolved design characteristic could benefit group survival or the evolution process and thereby could offset disadvantage to the individual [13]. He also argued that by increasing the generation rate, and thereby the evolution rate, determinate lifespan could prevent older, less intelligent, but more experienced individuals from dominating the gene pool. Thus, selection of beneficial inheritable characteristics, such as higher intellect or greater immunity of younger individuals, would not be overridden by experience of the elders. In this way, aging would improve the process of evolution [14].
Although the main proponents of programmed aging are Skulachev and Goldsmith, others have also supported this concept, claiming that aging accelerates accumulation of novel adaptive genes in local populations [15] and that the rate of aging itself can be an adaptation [16].
HOW CONVINCING IS THE EVIDENCE THAT AGING IS PROGRAMMED?
Since evolvability is an accepted alternative to traditional evolutionary theory, it is not my purpose to discuss the subject, nor the obvious existence of aging and its benefits for adaptive evolution. Instead, I propose that there exists a genome-based program for phenoptosis that is more “evolutionarily” logical than the one which purportedly evolved to kill per se. Accordingly, I use the word “phenoptosis” not specifically as intended by Skulachev [10], i.e. as a program designed to cause aging, but rather as a part of an evolved program in which the potential to cause aging exists because it is invisible to natural selection.
As a prelude to that argument, I suggest that it is a leap of faith to assume aging is “programmed” based upon evolvability theory and adaptive evolution. While determinate life span may indeed promote evolution, why need it be the result of programmed aging? In essence, such conclusion circumvents the basic question of how can aging and death be beneficial to the individual? Must evolvability considerations always negate that evolutionary obligation? Goldsmith argued that, indeed, it might in saying that an evolved design characteristic could offset disadvantage to the individual [14]. However, to cite the evolvability benefits of phenoptosis as a partial “proof” that an aging program exists is reminiscent of Darwin’s response when asked to reconcile aging with individual benefit and evolutionary fitness. His reply, as published in a book by Goldsmith, was “My theory says there must be some hidden compensating (individual, theory conforming) benefit so therefore there must be one”. Goldsmith criticized Darwin’s argument as being circular because “The theory is being used to predict the observation as opposed to observations resulting in a theory. This same “explanation” could be used to “explain” any instance of an apparent individually adverse organism design characteristic” [17]. Likewise, when programmed aging theory explains aging as a mechanism to prevent overpopulation, speed evolution and/or benefit young animals by eliminating old (“less valuable”) animals that become old precisely because of aging itself, it employs circular reasoning [18]. While aging may facilitate evolvability, that benefit does not answer the question of how it (aging) is initiated, nor describes the mechanism by which it proceeds. Instead, the theory (phenoptosis) is used to explain the observation (determinate life span) rather than the observation being used to provide the most logical basis for a plausible theory.
As previously noted, Skulachev proposed that phenoptosis is analogous to apoptosis, a recognized program of cellular death that “purifies a tissue from unwanted cells” [7]. Presumably, a program for organismal death encoded in the genome causes “aging to clear the population of ancestors and free space for progeny carrying new useful traits” [19].
The flaw in this argument is that when properly regulated, apoptotic cell death is balanced with mitosis as a vital mechanism to regulate development and cell numbers and to prevent accumulation of perilous tumor cells. In other words, apoptosis is essential for survival of an organism during development and only becomes detrimental during aging. So, by that logic, unregulated apoptosis is a consequence of aging, not a cause. One could argue that it was selected and conserved because of its survival benefits during development, which is more logical than for its actions in terminating life of cells, tissues and organs as organisms grow old. These degenerative effects of apoptosis are more likely “accidents” of its expression in metabolically and physiologically deranged internal environments.
The programmed aging theory isolates development from aging as two separate and independent programs affecting organisms. If programs for development and aging were separate entities, yet both residing in the genome of every individual, then why are they always expressed sequentially? It would seem possible that through chance, the programs might be expressed coincidentally or intermingled, such that aging might begin in a five-year-old and elements of development in a 50-year-old. The latter case of course was portrayed by Brad Pitt in the fictional 2008 film “The Curious Case of Benjamin Button” [20]. The absolute requirement for sequential expression suggests that the programs are not independent entities.
Furthermore, if phenoptosis evolved solely and specifically to purify communities of unwanted individuals, it then has no apparent link with development and survival as does apoptosis. On the other hand, and like the regulated effects of apoptosis, it is possible that the degenerative effects associated with slow phenoptosis are aberrant manifestations of processes that were beneficial to survival earlier in life. These suggestions are consistent with the idea that aging is a deviant expression of the developmental program and also with antagonistic pleomorphic theory whereby beneficial factors during development become detrimental later in life [21]. Thus, association between development and aging better cements phenoptosis to apoptosis as analogous phenomena affecting organisms and cells, respectively. As such, and as demonstrated for apoptosis, they would both have offered some degree of individual benefit and thus evolved through natural selection.
Cellular damage resulting from accumulation of toxic metabolites has been long embraced as a possible aging mechanism [22-24]. Pursuant to this concept, Skulachev posits that phenoptosis results from expression of an aging program which is encoded in the genome as a chain of ultimately lethal biochemical events. Presumably, this degenerative process is initiated within the mitochondria through the action of reactive oxygen species (ROS). These cause oxidative damage to the mitochondrial inner compartment leading to their destruction and resulting in apoptosis, which in turn, decreases cellular content of organs and tissues [25, 26]. This idea is based in part upon Harman’s theory of a mitochondrial program that increases ROS thereby causing slow destruction of the body over the course of a lifetime [27]. Others have also identified accumulation of irreparable damages from spontaneous, molecular level side reactions including free radical/glycation, induced carbonyl stress and accumulation of age pigments as the “essential and profound nature of higher animals' aging mechanisms” [28]. Also, genes [29] and proteins [30] are thought to contribute to the aging process. Indeed, any number of such events having potential to cause organismal death are known. However, no evidence has been provided to show that any of these initiate aging, nor to describe specific sequences by which they accomplish it. Also, while pharmacological alteration of their activities can change the rates of aging, none can stop its ultimate progression [31]. These observations indicate that it is more likely that the lethal factors are consequences rather than primary causes of aging. Thus, while one or many of them may contribute to death, why should it be assumed that they constitute a program encoded in the genome specifically to kill? If any one or several of them were the cause(s) of aging rather than contributors to the decay of an organism, then induction or inhibition of their effects in developing and aging animals, respectively, should initiate or completely stop the process in those animals. Indeed, while erosion of cellular and genetic integrity could characterize generalized deterioration of all organs and systems during senescence [32, 33], the questions of why it occurs, how it is activated, coordinated or defined by any uniform pattern or sequence of a lethal program that coincides with the progression of senescence remain unanswered. Also, while chemical and metabolic events that are maladaptive can occur during aging, they, like apoptosis, play normal supportive roles in function of a young organism.
If these doubts of programmed aging are valid for iteroparous animals, are they also valid for semelparous animals that experience “fast phenoptosis”? As an example, to support his theory, Skulachev cites the fact that during their upstream reproductive migration, salmon display numerous typical traits of aging including amyloid plaques in the brain [34, 35]. Their death soon after spawning is taken as an evidence that it is programmed [31]. While salmon are cited as a prime example of animals that experience “fast phenoptosis”, other species, such as the Australian marsupial mouse (Antechinus), Labord’s chameleon and others, are similarly affected. After exposure to stressors, the hypothalamic-pituitary-adrenal (HPA) axis in these species and many other semelparous animals is hyperactivated. This causes the adrenal glands to increase secretion of steroids that rapidly impact the entire body, including the brain [36]. Subsequently, death results from global deterioration of the body following excessive exposure to intolerable and fatal concentrations of circulating glucocorticoids [37]. However, glucocorticoids are not elevated in these animals specifically to kill them. Rather the gluconeogenic hormones are adaptive as a source of energy, for memory, smell and other functions needed for success in courting, aggression, defense and reproductive migration. It is true that chronic glucocorticoid excess erodes somatic structure by converting amino acids from muscle to sugars. However, this effect provides energy necessary for the fish to successfully make the treacherous upstream journey to its breeding ground, where it can reproduce and ensure another generation of its species. This is an example of a programmed event that evolved to benefit survival. Death is secondary to the physical exhaustion and degeneration required to complete the task of reproduction. Perhaps this is semantics, but it is important to distinguish those events that are an essential part of life and species survival from the idea that their sole purpose is to kill an organism/animal. That the lethal events in salmon are specifically linked to the reproductive effort can be seen in the fact that “removal of the adrenals immediately after spawning will allow them to live for a year afterward” [38]. Thus, even though adrenal steroids degrade their bodies, salmon gain survival advantage during migration and reproduction as a terminal part of their developmental program. This is true of all semelparous animals, whether the mechanism by which they die is glucocorticoid excess or any other process that evolved as a beneficial trait for the individual and its species but subsequently results in death. So yes, the lethal glucocorticoid excess in salmon and other semelparous animals is programmed, but not for the purpose of fast phenoptosis. Rather, it has evolved as a neuroendocrine mechanism necessary for continuation of life, not as an intentional act of nature to kill an organism because it is no longer of use to the population.
Finally, if an aging program evolved due to evolvability, the process would have had to take an incredibly long time, since single traits take tens of thousands of generations to affect adaptive phenotypes [39]. For complete programs to evolve such time requirements would be expected to expand logarithmically. If this premise is true, then immortal organisms must have existed well before the first aging program ever evolved. If so, then what of the immortals? Is there any evidence of their existence? There does not seem to be, suggesting that aging was present upon emergence of the first complex organism.
Based upon the preceding arguments, I feel that there is not very strong evidence to support the theory that aging is specifically programmed to terminate life. Rather, even as proponents of the theory acknowledge, preexisting mechanisms intrinsically associated with development, maturation and survival go rogue, adopting destructive tendencies once young adulthood is reached. In the case of semelparous and iteroparous animals, there are two different and distinct causes of aging and death. Since the focus of much work on aging has been on iteroparous animals and especially on slow phenoptosis to the extent that Skulachev and Goldsmith have suggested that medical benefits may result from understanding and intervening in the program for aging, the remainder of this presentation will be devoted to that group.
THE LIFE/DEATH PARADOX OF ORGANISMAL DEVELOPMENT
The epic of Gilgamesh tells of an ancient Sumerian king who set out to discover the secret of immortality [40]. While on this quest, he met an immortal angelic being created by God who told him that his search for human immortality was futile. The immortal explained, “Unlike that of the divine, human creation itself contains the seed of death”, making it inescapable. Thus, this ancient text reveals the life/death paradox: aging, the seed of death, resides within the very process by which we inherit life. Is there any truth to this allegory? Indeed, there is!
Despite the untenable premise that senescence has individual benefit, respect for the genius of Charles Darwin, along with the beauty, simplicity, and brilliance of natural selection as an explanation for the myriad observations of life, the search for an evolutionary theory of aging has been sustained since Weismann’s was first presented [1]. The relatively recent “evolution of evolvability” theory has restored interest in programmed aging by providing a means to explain how determinate life span might be beneficial to populations if not to individuals and thus favored by natural selection [13]. However, because limitation of life span by aging might be beneficial for evolvability, it does not require that a program for aging evolved. Instead, it is possible that while aging does not have individual beneficial, its potential is conserved in the genome nonetheless, because the process by which it occurs is essential for life. Paradoxically, aging and death could result from expression of the same specific programmatic function that provides individual benefit by ensuring reproductive success, post-reproductive development and survival of the species. Such a mechanism would not have to face the unlikely prospect of evolving as a separate aging program to accommodate evolvability after traits directly beneficial to survival had been selected. What if such were a cryptic component of a survival mechanism that revealed its lethal potential only after its benefit was realized. We proposed that the developmental program fits those criteria [41, 42] and others agree that the mechanism of aging is linked to development [43-45].
In 1932, Bidder suggested that “continued action of a regulator after growth ceases” may cause aging. He wrote further that “the regulator does efficiently all that concerns the welfare of the species” and “this negative growth is the unimportant by-product of a regulating mechanism evolved by selection that is necessary to the survival of any race” [46]. Using similar verbiage, Blagosklonny and Hall wrote more recently, “manipulations that decrease growth also decrease aging and prolong life span” and “aging and growth may be linked in a way that growth produces aging” [47]. Comfort [32] and later Williams [21] proposed that there is a “fixed developmental sequence which if completely arrested would eliminate senescence”. Others have argued that development and ageing are linked and directed by the same gene set [48].
Organisms are born, grow, reproduce, age and eventually die. Through all these phases of life, they continually experience change involving both internal activities and environmental influences. Previously, we proposed that a potentially lethal aspect of development is unremitting physical and functional transformation of the soma from conception to young adulthood [41]. Such uninterrupted transition in form and function of the body, from simple to complex must reach an end point. Also, its very nature is unstable and cannot continue indefinitely without regulatory oversight. Most importantly, the transition is not from one fixed and stable state to another. Rather growth and maturation are dynamically seamless and while they are occurring, the interactive systems of the body are transitioning and adapting to provide essential life functions. Thus, these two very critical levels of development, including the continuum of growth/maturation and dynamic performance of internal functions, must operate under strictly controlled parameters or else they can degrade into chaos and disorder. During youth, the genome-based, developmental program prevents such disintegration by maintaining a dynamic yet stable state despite the moment-to-moment exigencies and unpredictable changes in external conditions. Such stability is a universally accepted view of healthy organismal function and is basic to continuance of life. The traditional conceptual model to describe this process that has long dominated biology, physiology and medicine is homeostasis. However, the homeostasis model is somewhat inadequate because its defining principle is “stability through constancy”, which does not take into account that the internal milieu of complex biological systems is not permanently fixed, nor at equilibrium. Rather it exits under conditions of dynamic regulation and interaction among various levels of organization that has been more appropriately called homeodynamics [49, 50].
An even more accurate term, allostasis, has been used to describe the internal conditions of living organisms [51]. The allostasis model considers “reciprocal trade-offs between various cells, tissues and organs, accommodative sensing and prediction with respect to the severity of potential stressors, and the final cost of making a response and readjustment to bring about the necessary change” [52].
Unsuccessful maintenance of homeodynamics resulting in unrepaired molecular damage, reduced energy supplies and progressively less efficient or unstable structural and functional components of the body among other things, degrade its integrity and increase its allostatic load. It has been proposed that aging, senescence and death are the final manifestations of unsuccessful homeodynamics or failure of allostasis [52]. Thus, the need for homeodynamic constancy as organisms change both in response to their own internal/external developmental modifications and to environmental influences is essential for maintenance of life. Paradoxically, while essential for life, the requirement for dynamic change represents a potential threat to its indeterminate existence. The reason for this limitation is that there comes a point in time when the developmental program, a component of which specifically evolved to orchestrate and maintain internal order, must end. Upon reaching the age of maturity when programmed developmental information is exhausted, continued expression of non-programmed somatic transformation/remodeling becomes maladaptive. Such non-programmed change, residual from the developmental program, degrades homeodynamic maintenance, causing progressive loss of coordinated complex dynamics in multiple system functions, such as cardiovascular control, temporally coordinated hormone release, electroencephalographic potentials, etc. These changes slowly impair the body’s ability to adapt to physiologic stress, a universal characteristic of aging. Thus, a single process with the potential to initiate progressive systemic dysfunction, somatic degeneration, leading to disease and ultimately death, would result from unremitting somatic remodeling causing gradual and progressive loss of internal order [18, 41, 42, 44, 53, 54].
In iteroparous animals, programmed somatic change continues beyond the age of reproduction and to the point of maturation so as to provide parental care for dependent offsprings. However, if somatic transformation that is essential for development does not cease, then at some point such change becomes maladaptive, eroding internal order and promoting chaos. Thus, the dynamics of development expressed beyond maturation is inconsistent with indeterminate life span, and physiological evidence of declining homeostasis is detectable at or shortly after adulthood is reached. In fact, the beginning of senescence can be observed in humans as early in life as the mid 20’s [55]. Muscle loss is especially evident in high performance athletes, such as 5 gold medal winner Michael Phelps, former competitive swimmer and the most decorated Olympian of all time, who at 25 years of age faced the inevitability of retirement before 30.
Evidence of age-related erosion of internal order and progressive chaos in humans and animals has been quantified with a statistical measure of “nonlinear complexity” called approximate entropy (ApEn) [56]. Using ApEn, aging is shown to be associated with progressively decreased orderliness and reduced complexity of physiological systems [57]. Particularly relevant was the finding that serial disorder within the growth hormone/insulin-like growth factor (GH/IGF) system increases during aging, as assessed by ApEN statistics. This means that functional integration within the GH/IGF-1 neuroendocrine axis, which is associated both with development and aging, becomes progressively dissociated over time [58].
Thus, there exists a potentially degenerative process for aging within the developmental program that is inextricably linked with individual survival. However, its maladaptive effects are invisible to natural selection because they begin well after puberty, upon reaching maturation. In iteroparous animals that means the developmental program containing the cause of aging continues to run after reproductive competence is reached. In other words, unlike programmed aging which would have had to be anticipated by the genome after reproductive potential is reached when natural selection is no longer operable, a cryptic degenerative aspect of the developmental program, not visible to natural selection, would not require programming and thus, would be more evolutionarily logical.
From an anthropomorphic perspective, the fact that somatic restructuring continues after maturation could be considered a “design flaw” in the developmental program. This of course is nonsense, since evolution makes no special accommodations for human wishes of immortality. The absence of genomic “stop commands” for those developmental genes whose expression drives maladaptive somatic change and erodes homeodynamics after maturation when species survival has been guaranteed, is totally irrelevant to nature.
DEVELOPMENTAL INERTIA: THE LINK BETWEEN DEVELOPMENT AND
AGING
Why does aging begin when development is complete? As previously stated, the answer lies in two important functions of the developmental program. The first is that every sexually reproducing animal begins life as a single cell which after fertilization transforms into a complex organism consisting of a multitude of interactive systems. For the transition to occur, the body must undergo continuous modification of structure and function orchestrated by the genome. I call this force imposed upon the soma “developmental inertia”, proposing an analogy with Newton’s First Law as being “matter continues in motion unless acted upon by an external force” [59]. Assuming the absence of a “stop command” for genes controlling developmental inertia, it will continue throughout life. After maturation, its effects are maladaptive, since remodeling/restructuring of the body has finite limits beyond which disorganization and chaos must ensue.
The second function of the developmental program is to coordinate and integrate change resulting from developmental inertia throughout the period of development. Because unregulated change is inherently unstable, there must also be an informational component of the developmental program to properly control the body’s constantly changing form and function. Upon reaching maturity and completion of the developmental program, the information in genes controlling such change is exhausted. Unlike developing animals, adults require homeodynamic stasis, not remodeling, in order to sustain optimal health and vitality. These differences in survival requirements underscore the fact that at some point in the lifetime of every sexually reproducing multicellular animal, the dynamics of development becomes inconsistent with indeterminate life span.
A major error in the programmed aging theory is the premise that programmed aging evolved specifically to kill, and thus is independent of the developmental program. A more evolutionarily compatible program for phenoptosis is one that evolved because of its benefit to survival and reproduction, but secondarily causes aging and death [18, 42]. The developmental program meets all those criteria.
It is important to understand that the developmental program for regulating coordinated somatic remodeling in iteroparous animals is primarily optimized through evolution to allow successful achievement of reproductive competence. Developmental somatic transformation beyond the age of reproductive competence is programmed to ensure individual survival only so long as parental care is provided for dependent offspring. This period of time lasts until young adulthood, when information for coordinating further structural and functional change is depleted. However, the soma continues to change thereafter due to non-programmed post-maturational “developmental inertia”. Since natural selection becomes inoperable beyond the age of reproductive competence, a mechanism to stop expression of developmental inertia could not have evolved. As a result, continued expression of the previously adaptive gene(s) for initiation and progression of non-programmed change is maladaptive. This functionally “biphasic” application of antagonistic pleiotropy meets evolutionary criteria for selection and avoids the problem(s) inherent in the hypothesis of explaining how an aging program and/or “death genes” were selected and stringently conserved [21, 60]. In fact, evolutionary theory would support the concept that unchecked and unregulated post-maturational developmental inertia initiates the process of aging.
Post-maturational expression of the developmental program has been called a “quasi programmed hyperfunction” [61]. In that regard, Blagosklonny posits that “when developmental growth is finished, growth-signaling pathways may continue to run on inertia”, and that aging is driven thereafter by overactive mechanistic target of rapamycin (mTOR) signaling which causes animals to develop faster and also senesce faster [44]. I agree that the developmental program has beneficial and detrimental effects before and after maturation, respectively. Such antagonistic pleiotropy of the developmental program would favor its selection and conservation by natural selection [62]. However, I disagree that the cause of aging is mTOR hyperactivity, even though Blagosklonny claims that onset and progression of aging and its related diseases can be delayed by deactivating mTOR signaling with caloric restriction, genetic manipulations and drugs. Indeed, inhibition of mTOR with rapamycin extends maximal and median life span in mice even when initiated late in life [63]. But that effect does not prove that mTOR causes aging nor maintains its progression. The reason is that none of the mTOR interventions that slow the rate of aging and/or onset of intrinsic disease, stop the process of senescence. Intuitively this means that some other mechanism, e.g. the effects of developmental inertia on the temporal order as described below, causes aging to continue when mTOR is deactivated. However, since the rate of aging is slowed by deactivation of mTOR, then this signaling pathway must be a consequence of aging that exacerbates the degenerative process like all other molecular and cellular events associated with aging.
Thus, while agreeing in principle with the “quasi program” concept that development and aging are linked through unregulated expression of the developmental program in adults, it is not mTOR hyperexpression that initiates the aging process nor sustains it. Instead, the cause of aging is unremitting, non-programmed remodeling of the whole body after it achieves physical and functional perfection. Such unregulated change erodes homeodynamics and internal order, slowly at first but progressively increases chaos and organizational decay. If the “inertial” component of the developmental program could be stopped at the point of maturation, then form and functional relationships of body would cease to change. Such would be essential for biological immortality. Thus, because somatic remodeling continues after maturation, i.e. when development is complete and programmatic information is exhausted, then non-programmed developmental inertia can be considered the “causal link” between development and aging. It is responsible for progressive somatic chaos which has been described by others as a characteristic of aging [57].
SUPPORTING EVIDENCE: POST-MATURATIONAL DEVELOPMENTAL
INERTIA
Specific changes in physical appearance that occur across the lifespan provide visual evidence of the effects of post-maturational developmental inertia. For example, general trends in timing and patterns of facial change during aging have been documented [64]. These age-related changes, which are not degenerative, are nonetheless so obvious that even an untrained eye does not confuse a twenty-year-old adult with one who is forty, even though forty is not considered elderly. Of course, many later changes in appearance are degenerative, due to decreased muscle tone, diminished collagen and elastin in skin, wrinkling and sagging. However, facial remodeling due to continued expression of developmental genes occurs well before the late effects of aging are manifest [64]. For example, altered bone shape in the craniofacial region results from age-related increases in head circumference, length, cheekbone-to-cheekbone width and facial height. Also, increases in anterior facial height and certain changes in the dentoalveolar region progressively alter appearance of lower facial areas with advancing age [65].
Evidence that post-maturation developmental inertia affects lifespan is derived from studies of microRNAs (miRNAs) and transcription factors in the nematode C. elegans. Because gene expression is regulated at the level of transcription, which in turn is determined by factors that bind specific sequences in promoter and enhancer regions, developmental inertial changes in transcription factor binding or post-transcriptional regulation by miRNAs could account for the age-related alterations in gene action [66]. In fact, non-programmed activity of miRNA-regulated networks due to post-maturational developmental inertia may modulate aging rates, since their differential expression alters life span in C. elegans [67]. Temporal shifts in expression of genes affecting brain differentiation during development were also found to be associated with age-related functional decline in monkeys and humans [68]. The age-related changes in miRNAs and gene expression represent extensions (distortions) of the developmental patterns suggesting that the regulatory processes that are beneficial during development become detrimental thereafter. The authors found that few changes in gene expression were unique to aging further arguing that senescence follows non-programmed expression of developmental inertia, not the activation of “aging genes”.
Interventions that alter the rate of somatic change and duration of life but do not completely stop aging, provide further evidence that post-maturational development inertia causes senescence. If developmental inertia initiates and drives progressive aging after maturation, slowing or accelerating the rate of somatic change it causes should have comparable effects on life span. Indeed, that seems to be true, since changes in production of hormones within the growth hormone neuroendocrine axis that are known to accelerate somatic structural and functional change, alters the rate of living. For example, life span of C. elegans is increased when expression of daf-2, a specific gene that encodes a protein resembling the insulin/IGF-1 receptor, is decreased [69, 70]. Similarly, in other species that are genetically altered to reduce production of the somatotropic/metabolic hormone insulin-like growth factor-1 (IGF-1), aging is delayed, resistance to oxidative stress increased and life span is prolonged, whereas it is shortened if IGF-1 exposure is increased [71, 72]. The well-recognized effects of caloric restriction (CR) on life extension support these effects and the proposal that non-programmed developmental inertia is the driving force behind aging. CR which limits food intake to about 70% of ad libitum consumption, alters a multitude of processes, including energy expenditure, oxidative damage, neuroendocrine function, gene expression, etc. and is perhaps the most widely reported method for extending life span in such diverse species as fruit flies, spiders, fish, and rodents (see [73]). It slows but does not stop the progression of developmental change nor senescence, so that animals stay physically and functionally younger for longer periods of time but still age and die. As previously discussed, CR also extends life by deactivating mTOR and other contributing factors to senescence thereby postponing onset of most major intrinsic diseases, such as cancer, kidney and heart disease, cataracts, etc. Presumably these beneficial effects result in part from prolonging youthful immunosurveillance [74] by attenuating the rate of post-maturational developmental inertia. The pluripotent nature of CR’s ability to extend life indicates that its mechanism of action cannot be attributed to any one of the multitude of processes it alters. Instead, they must all result from a higher level of control capable of influencing each specific effect en masse. The global effect of CR is not due solely to the developmental delay, because restriction extends longevity even when it is initiated after adulthood, although proportionally less the later in life it begins. These data suggest that no matter when initiated, CR slows a common and general process of developmental inertia that is in progress from conception until death. As a result, the extent to which longevity can be extended depends upon how far non-programmed post-maturational change has eroded internal order. It has been suggested that this global-type effect with differential age-dependent outcomes results from slowing of the entire genetic program thereby indirectly affecting aging [43]. Perhaps instead and more specifically, expression of genes causing developmental inertia is slowed by reduced calories/metabolism thereby slowing progression of age-associated internal disorder whenever it begins.
LOSS OF TEMPORAL ORDER: THE MECHANISM OF AGING CAUSED BY
NON-PROGRAMMED DEVELOPMENTAL INERTIA
If non-programmed post-maturational expression of developmental inertia is the primary cause of aging, then what is the mechanism by which its maladaptive effects are expressed?
During development, levels of organization increase in complexity as cells become tissues, then organs, then systems and ultimately unit organisms. The necessity for appropriate structure of molecules that constitute and ensure integrity of cells and tissues is obvious. If the structure of a specific protein were not appropriate, that imperfection might adversely affect function of the cell for which it was originally intended. Such effects are typical of metabolic and cellular defects that are consequences of aging and can cause dysfunction and disease. However, such defects in individual components of the body do not necessarily have global impact, but instead, might only disrupt discrete functions. Considering the whole organism, it is important to note that to natural selection, the aggregate of systems no matter how numerous or scattered, but having the same genotype, comprises a single individual. Thus, the systems require processes or means to coordinate their independent functions with the needs of the unit organism so as to guarantee its survival. A complex of biological rhythms evolved to provide functional coordination among the interdependent elements to serve that purpose. These specific temporal dynamic processes link functions of the body with environmental oscillations. The existence of such rhythms has been demonstrated in diverse multicomponent functions ranging from enzyme activities to neuroendocrine secretory patterns [75, 76]. Their ubiquitous presence indicates that temporal organization evolved early as an essential part of metazoan somatic integrity. During youth, the rhythms in such systems are regular and predictable. Furthermore, since orderly sequence of functions is required to ensure survival, then time structure itself must change during development to support body’s changing needs. This requirement for distinct rhythms that are essential for health and vitality is indicated by their absence in cases of developmental retardation with reduced functional capacity in fetal physiological systems [77]. Thus, an integral part of the developmental program selected during evolution would have been to sustain temporal order during dynamic transformation from embryo to adult.
Consistent with the strategic role of temporal order, structural and functional deterioration of the whole soma, which is the hallmark of senescence, does not seem to be comparably reflected at lower levels of organization, especially during the early stages of aging. This inconsistency suggests that a common mechanism of aging is one initiated at the organismal level, rather than in any of its specific cells or tissues. In other words, aging could result from a disruptive process that negatively and progressively impacts the body’s ability to sustain coordinated function among its multitude of parts. Since the body is inextricably involved with and vitally dependent upon environmental input, distortion of intrinsic time structure is inherently pathogenic.
With advancing age, slight deviations in timing among interdependent functions due to post-maturational developmental inertia are additive and perhaps synergistic. These maladaptive shifts in phase relationships result in loss of coordination that progressively becomes detrimental not only to interdependent processes, but also to functions dependent upon them. If temporal order is required to maintain health and vitality, then progressive disturbance of time structure during senescence must increase physical and functional decline. It also increases the risk for “diseases of aging” through negative effects on function in the autonomic nervous, sensorimotor, neuroendocrine and other systems [75]. The degree of complexity of such a communication/response network and the importance of its proper performance to maintain health and vitality for the entire body cannot be overstressed. Comparing a machine with an organism can be useful in demonstrating why strict temporal order is of an utmost importance for maintaining optimal function, good health and longevity. Using an automobile engine for example, it is obvious that if all its parts are properly manufactured and brand new, they have the potential to operate perfectly. However, in addition to the quality of “things” composing it, proper function of the engine requires a “process” for ensuring precise sequence timing of events involving the interdependent parts. Timing is important to ensure integration of the four-cycle engine including: 1) mixing and delivering air and fuel to the combustion chamber; 2) moving a piston upward to compress the mixture; 3) combusting the fuel mixture with a spark exactly when compression is maximal, thereupon pushing the piston downward; 4) opening a valve to exhaust the burnt fuel gases.
If the spark does not occur at the proper time, symptoms of incorrect ignition timing will result, including hard starting, backfiring, “pinging” or “spark knock”, poor fuel economy, and sluggish acceleration [78]. As an analogy for living organisms, these acute operational engine deficiencies could represent early stages of aging, when suboptimal physical and function begins and progresses thereafter. It is important to note that none of these initial deficiencies in engine performance are due to defects in the parts involved but rather in the timing of their operation. Initially it is not a matter of “things” that cause poor performance, but rather defects in the “process”. However, after an extended period of poor ignition timing, the cylinders will become excessively heated and the engine can suffer damage. The initial overheating could be analogous to stress and inflammation that in organisms advance with aging and precede the onset of disease. Subsequently, serious engine problems, such as cylinder cracking, coolant leaking, bent valves, broken pistons, damaged cylinder heads and engine blocks, can result from excess heat initiated by improper ignition timing. At this point, process has negatively affected things, such that the parts which initially were perfect became defective themselves. These mechanical problems demonstrate that disruption of proper timing among interdependent functions could subserve the genesis of disease. Failure of the engine, like death of an organism, will follow age-related disease initiated by temporal disorder. This simple example is used to differentiate between the quality of individual parts and the quality of the whole in order to stress the fact that senescence, or the process of physical decay that we associate with aging, need not result initially from malfunction or disease of individual cells, tissues, and organs. It can and does begin in the organism with slow but progressive erosion of temporal organization driven by non-programmed post-maturational developmental inertia. This process damages the functional relationships between various parts of the body and eventually imposes a fatal allostatic load upon them [52].
Thus, even when gene products are properly synthesized, they may still be ineffective if their presence is not perfectly timed with that of appropriate substrates and cofactors. It is also logical that when temporal order is altered, so functions must be altered. As deterioration of sensing and communicating systems that are linked with interdependent hierarchies of oscillating functions that collectively represent a living unit organism progresses, an organism’s integrity and vigor would collapse, and death would be inevitable. In other words, it is possible that a global insult originating in the organism itself could impair the homeodynamic processes that coordinate and integrate somatic structure and function. If such insult worsened with unremitting expression of non-programmed post-maturational developmental inertia, it would progressively erode internal order causing metabolic, cellular and systemic inefficiency. If that assumption is true, then the numerous and negative cellular and molecular events associated with global deterioration during aging are consequences of senescence and a higher-order, primary and general pathophysiological mechanism of temporal disorder underlies them all.
So, to sustain youthful health and vitality, there must exist within the body a perfect temporal equilibrium within and among various integrated functional components, including the reproductive, circulatory, respiratory, excretory, digestive, immune, integumentary, nervous, and endocrine systems. It is important to understand that none of these parts work independently of each other, and that each is supported and influenced by the performance of the others. This functional interdependence is ensured by coordination of regulatory signals provided in strict accordance with qualitative, quantitative, and temporal requirements. It is only after temporal disorder begins, that aberrant cellular and metabolic events take place. Thus, aging first and foremost is an organismal function that is then exacerbated by cellular malfunction. This is the reverse of what has been the popularly held belief. Aging starts first within the whole organism as progressive loss of temporal order which then initiates cellular discoordination and degeneration.
Clinical examples of temporal distortion in the brain during old age includes a non-linear age-related increase in the subjective rate of time passage [79], while a decline in future perspective [80], and decay of the sleep–wake cycle [81]. Taken along with recent findings of clock genes [82], these penultimate changes support the view that temporal disorganization is a hallmark of the aging process.
SUPPORTING EVIDENCE: LOSS OF TEMPORAL ORDER AS THE
PATHOPHYSIOLOGICAL PROCESS UNDERLYING THE SEQUELAE OF AGING
The pathogenic potential of temporal disorganization has been confirmed through research on animals and studies of human shift-work experience. One of the earliest studies linking internal disorder with age-associated intrinsic disease reported that parabiosis of time-displaced with normally-timed cockroaches produced intestinal cancer [83]. Since tumors in insects are rare, it was concluded that the two out-of-phase pacemakers operating simultaneously disrupted temporal order and subsequently homeodynamics sufficiently to cause cancer in the creatures.
Loss of temporal order in transcription factors that are vital in youth have been shown to promote aging when temporally distorted later in life. In the nematode C. elegans, the elt-3/elt-5/elt-6 GATA transcription circuit that is responsible for controlling skin and intestine development becomes “unbalanced in exerting influence over gene combinations” as the worms age [84, 85]. Kim noted that “it looked as though worm aging was not a storm of chemical damage. Instead, key regulatory pathways optimized for youth have drifted off track in older animals”. Subsequently, the age effect on this circuit was termed developmental drift because the sequence of events normally occurring during youth “drifts” during aging [86]. Furthermore, one of the authors expressed the idea that such developmental drift occurred in tissue-specific transcription factors. This implies that a more general phenomenon affecting temporal signaling among elements of the transcription circuits is at play. More recently, temporal changes in expression of genes affecting brain differentiation during development were also found to be associated with age-related functional decline in monkeys and humans [68].
Blagosklonny wrote in his theoretical paper on the mechanism of reproductive aging: “And here is another puzzle: why women undergo menopause” [87]. It is not a puzzle. The cause of female cyclic reproductive failure has been known for years.
Originally it was presumed that with advancing age, the ovary becomes depleted of its finite stock of follicles. However, during the 1970s, animal studies on the functional relationship between somatic integrity and organ function cast doubts on that conclusion. In those studies, it was observed that aging laboratory rats failed to ovulate, even though some antral follicles remained present in their ovaries. This suggested that ovarian depletion of gametes was not the prime cause of age-related loss of reproductive cycles. Also, it appeared that ovarian aging itself was not the primary cause of female reproductive dysfunction, because when ovaries from young rats (32 days old) were transplanted into ovariectomized old rats (26-30 months old), estrous cycles were not restored [88, 89]. However, when the reciprocal procedure was performed, i.e. when ovaries from old rats were transplanted into young rats, then ovarian functional capacity was restored, and the animals displayed cyclic estrus behavior [89]. These findings indicated that the dysfunctional ovaries from the old rats were rejuvenated by the young body. Thus, the primary effect of ovarian failure was not inherent in the organ, but rather in the environment of the old body that influenced its function.
Upon expanding these studies, we found that a remarkably early indicator of impending ovarian acyclicity and reproductive senescence is progressive delay in timing, as well as attenuation of the pre-ovulatory luteinizing hormone (LH) surge [76]. The temporal changes resulted in lengthening of the follicular phase without ovulation. These changes were similar to those induced in young women by rotating shift-work that disrupted time structure [90]. In our studies, the LH surge occurred in a majority of young animals at approximately the same time of day (Fig. 1). However, in middle-aged animals the LH surge was delayed, attenuated and shifted out of phase with follicular ripening (Fig. 2). The changes in ovulatory signal timing correlated with progressively irregular cycles as the animals grew older. Eventually cycling stopped when pituitary signals were sufficiently out of phase to cause even occasional ovulation, even though the ovaries still contained antral follicles. Our findings were corroborated by others demonstrating that progressive failure of timing in neuroendocrine signaling beginning soon after young adulthood eventually leads to the termination of the reproductive cycles. Similar blunting and loss of precise timing in sequential physiological events in other functional systems has been reported, representing a seemingly primary and universal effect of aging in animals [91].
Fig. 1. Precise timing of the pre-ovulatory surge of luteinizing hormone (LH) with the light/dark cycle in young rats. Blood samples were collected 3 h before (a), at (b), and 3 h after (c) onset of darkness and assayed for LH content. Peak serum levels of LH (the pre-ovulatory “LH surge”) occurred in most young females at the light dark transition (reprinted from [76]).
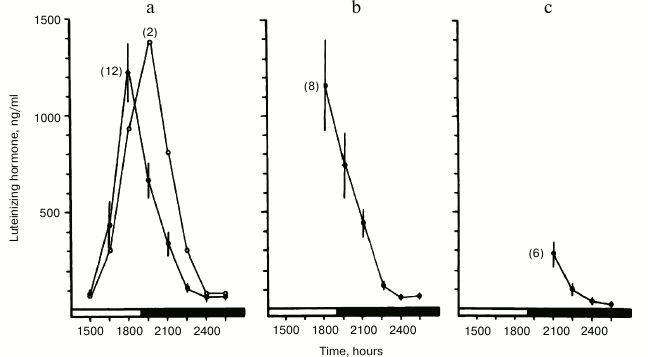
Fig. 2. Decay of temporal order in reproductive cycles of middle-aged rats. Blood samples were collected and assayed for LH content as described in Fig. 1. Impending age-related failure of reproductive cycles was foretold by progressive delay in the onset and attenuation of the pre-ovulatory “LH surge” (reprinted from [76]).
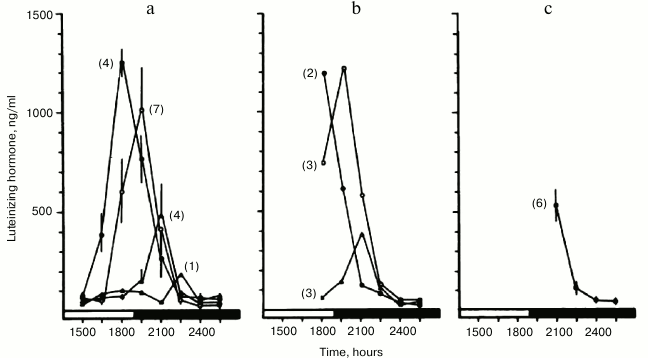
Other studies have shown that women also exhibit a temporally-linked deficit in neuroendocrine function at menopause, and that these changes can contribute to adverse health outcomes associated with aging [92-94].
Shift work has been linked with irregular menstrual cycles and accelerated bone resorption, which coincidentally are characteristics of female senescence [90]. In fact, disturbed timing of ovulatory signals preceding menopause/estropause in female mammals provides an excellent example of how loss of temporal order progressively erodes functional capacity and may thus be the primary physiological disturbance initiating senescence. Contrary to William’s opinion that “it is improper to regard menopause as part of the aging syndrome” [21], progressive failure of reproductive cycles is perhaps the best example of how senescence occurs, because unlike other systems, ovarian functions cease in the absence of disease. Thus, the menopause represents a “pure” aging effect resulting from progressive temporal disorder and erosion of homeostasis that precedes and increases the risk for developing intrinsic disease.
In humans, shift work or chronic jet lag that disrupts phase relations of fluctuations in behavioral, hormonal and metabolic variables, damage temporal order. In the long term, such forced disorder in time structure has been linked with increased risk for cardiovascular disease, peptic ulcer, sleep disturbance, breast cancer, and pregnancy complications [95, 96]. Disturbed temporal order has also been identified as a strong promoter of obesity and metabolic syndrome [97]. Thus, if the functional potential of biological systems depends upon well timed signals that preside over a vast complex of interdependent processes, it is likely that progressive loss of temporal order during aging (due to developmental inertia) is the mechanism of senescence that inevitably causes declining vigor, intrinsic disease and eventually death.
Therefore, aging did not evolve as a trait per se, but instead arose incidentally as a consequence of “living too long”. In other words, aging is the result of a “design flaw” in the developmental program that allows expression of genes causing non-programmed developmental inertia to continue at a time when stability, not change, is essential for biological immortality. However, it should be noted that the term “design flaw” is not intended to mean that there really is a flaw in the developmental program. It is only flawed from the human perspective which covets longevity and biological immortality. The failure to stop developmental inertia upon completion of the developmental program is irrelevant to all other species in which aging rarely occurs, if ever. If appropriate technology currently existed, this theory could be tested in young adult animals by identifying and silencing the putative genes responsible for developmental inertia.
THE THEORY
Emergence of the earliest sexually reproducing multi-cellular eukaryote was undoubtedly dependent upon the evolution of a developmental program. It was needed to direct sequences of cellular and molecular events throughout the period of dynamic somatic transformation from conception to adulthood. As somatic complexity increased during the course of metazoan evolution, genes to coordinate physical and functional changes throughout the unit organism and to oversee development of novel cells, tissues and organs were undoubtedly selected for inclusion in the program. This progression of genome complexity suggests that a functional hierarchy of developmental genes evolved, in which the most ancient ones provide general or global oversight of the soma. If so, such genes might serve at least two essential functions. One (or a specific set) could be to orchestrate a “developmental inertia” for integrating somatic change during the dynamic developmental transformation from conceptus to adulthood. Once evolved, the developmental program would have been stringently conserved in every subsequent species because of its absolute survival benefit. Haeckel’s recapitulation theory (ontogeny recapitulates phylogeny) reflects this concept of developmental gene conservation [98].
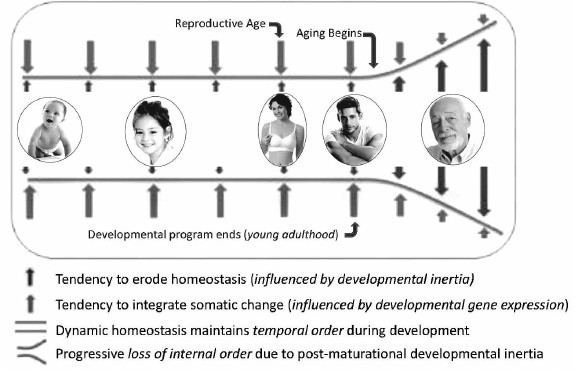
In contrast to development, aging did not evolve. Instead, it always existed within the developmental program as a maladaptive potential of developmental inertia. Because predation and other external factors, not extreme consequences of aging, are the common causes of death in natural populations, the aging potential of developmental inertia was rarely if ever fully realized. It was only after the advent of human socialization and animal domestication that individuals survived beyond ages when their developmental programs become depleted of information for directing coordinated developmental inertia. Thereafter, continued expression of non-programmed developmental inertia causes inappropriate unregulated somatic change that is inconsistent with maintenance of health and vitality. However, this non-programmed expression of the cause of aging could not be eliminated from the developmental program, because it is expressed after reproductive competence and adulthood were reached, when natural selection is inoperable. Thus, the lethal potential of unregulated somatic change resulting from post-maturational non-programmed developmental inertia erodes temporal order, causing progressive chaos among functionally interdependent systems. Paradoxically, aging results from expression of the very same developmental genes and processes that are essential for survival before that time. The only difference is that developmental inertia is essential for life before maturation and yet is the source of aging and death thereafter. As a result, unremitting and undirected somatic change resulting from residual expression of those specific developmental genes responsible for physical and functional remodeling, i.e. developmental inertia, continues after the organism becomes optimally functional, reproductively competent and capable of protecting its offspring. At this point in life, indeterminate life span requires somatic stasis, not persistent change which erodes temporal order, homeodynamics, weakens body defenses, causes frailty, increases risk for intrinsic diseases and makes death inevitable (Fig. 3).
Fig. 3. Diagrammatic representation of the development/aging continuum. Dynamic transformation of structure and function begins at conception and continues throughout life. Somatic change is sustained and coordinated throughout the unit organism by information from developmental genes until young adulthood. After reproduction and young adulthood is reached, information that coordinates change and sustains homeodynamics becomes exhausted. Thereafter, developmental inertia continues to force somatic change without benefit of programmatic oversight, ultimately eroding temporal order causing physical and functional decline of senescence (reprinted from [42]).
NEOTENY AND AGING: TESTING THE THEORY
It has been suggested that highly social mammals are long-lived due to neoteny, a term which Skulachev proposed represents “prolongation of youth”. Thereby it may be a mechanism underlying human longevity. If so, he mused that specific drugs targeting genes causing neoteny might be a promising approach to retard aging and prolong healthspan [99]. However, others have reported that neoteny is not a ubiquitous feature of the human phenotype [100]. Instead, humans are peramorphic, displaying both neotenic, as well as non-neotenic traits [101]. So, in the context of our research, it was not clear whether neoteny slows developmental inertia and thus, represents a mechanism to identify genes that control “global aging” or if it is only a retardation of development in selected parts of the body, such as the head and extremities, that provides evolutionary benefit [101, 102].
Our theory of phenoptosis predicts that expression of genes responsible for sustaining non-programmed developmental inertia beyond maturation thereby resulting in progressive loss of temporal order, is the cause of aging in iteroparous animals. If true, then silencing those genes or in some other way suppressing their activity in young adulthood should prevent them from initiating senescence. While such technology is not yet perfected, research is proceeding and may become possible in the future. However, the basic problem with performing such intervention is that the identity of putative gene(s) effecting developmental inertia is unknown. It is possible that mutation might provide a marker with which they can be identified and described. However, spontaneous mutation of the genes during gametogenesis would be lethal because it would prevent them from guiding ontogenesis of a functionally cohesive soma. On the other hand, rare cases in which such genetic mutation does not completely destroy function may occasionally occur and thereby provide a marker. If so, one would expect that mutational damage would cause severe disruption of the developmental program characterized by growth retardation and possibly by disorganized development of structural, functional and integrative characteristics of physiological systems. Remarkably, a girl fitting that description was born in 1993 [41]. She was suffering extreme developmental delay associated with structural and functional defects of her nervous, gastrointestinal, respiratory, cardiac, and musculoskeletal systems. Most interesting was the striking neoteny she displayed, causing her to appear as a toddler rather than a teenager; her actual chronological stage of life. Because of dysmorphic features and her unusually juvenile appearance associated with multiple congenital anomalies, her pediatrician assumed that there was a genetic basis for her clinical condition. However, based upon karyotyping and comparative genomic hybridization analysis, her chromosomes appeared to be that of a normal female. Also, during the course of her life, multiple examinations failed to support a diagnosis of any known genetic syndrome, yet she persisted in a condition resembling that expected to result from damage to genes controlling developmental inertia. In fact, at twenty-one years of age she still appeared physically as an infant. Thus, based upon her poorly integrated and significantly slow rate of somatic remodeling, we proposed that mutation of the gene(s) affecting her development/aging might be responsible and possibly identified by DNA sequencing. However, this initial subject died from complications of tracheomalacia before we could sequence her genome.
Nonetheless, due to public interest our research was publicized by the media in several documentaries and news reports. After such press coverage and public exposure to our work, we were contacted by many families seeking information about their children with similar developmental delay. Most of those cases were explicable as being caused by endocrine or metabolic deficiencies and clinically were quite different from the proband. However, of the families responding, seven presented with neotenous children having clinically similar congenital anomalies. The physical appearances of all the new subjects were that of infant/toddler children despite chronological ages reaching ages of 25 years or more (Fig. 4). Their retarded growth rate of height standard deviation score (SDS) was more than 3 SDS below the mean for chronological age and sex. They all failed hormonal and/or dietary interventions intended to overcome their growth deficits and they retained juvenile characteristics such as baby teeth, failure to reach menarche, absence of language, failure to walk independently, etc., throughout the course of study. Thus, after meeting the IRB approved inclusion criteria for study, we undertook investigation of two relevant questions including: 1) did there exist discrete mutations in one or more genes common to all subjects that might be responsible for influencing the development/aging continuum; 2) were their biological ages younger than their chronological ages?
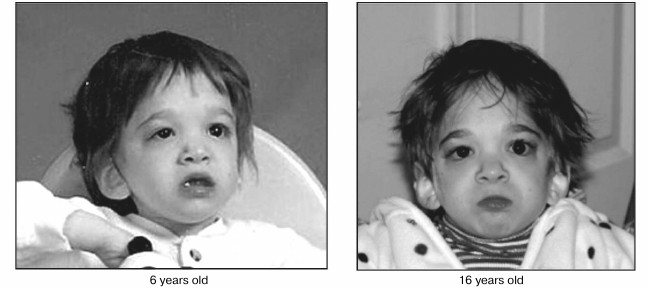
Fig. 4. Changes in facial appearance over the course of a decade in a girl suffering putative mutation of genes orchestrating the development/aging continuum. Photographs taken at 6 and 16 years of age showing minimal changes in appearance during the time of transition from childhood to adolescence when physical effects of maturation are normally striking (reprinted from [42]).
Several de novo mutations were found in five different genes. There were no large de novo or inherited structural variations shared between our patients, nor were there any small inherited variants we could identify that contributed to their condition. Thus, the first objective of our efforts to find genes whose expression would control putative “developmental inertia” was not achieved. However, our clinical and genetic findings allowed us to name the novel clinical syndrome; neotenic complex syndrome (NCS), based upon its most obvious characteristic, i.e. neoteny and the genes involved in its clinical pathology [103]. Based upon the genes associated with NCS, we also concluded that the neotenic features of these patients are caused by changes in development and should be differentiated from slowed aging and extended healthy life span that are mostly caused by reducing damage to cell components and improving tissue maintenance. Further support for this conclusion derived from aging biomarker data obtained using an “epigenetic clock” process based upon DNA methylation levels [104]. No statistically significant differences in chronological and epigenetic ages were detected in any of the newly discovered cases demonstrating that while our subjects maintained the façade of persistent toddler-like features even into young adulthood, their blood DNA was not younger than their chronological ages [53]. The only caveat to this conclusion is that the epigenetic clock was only tested in blood, so that it may be possible that the rate of development/aging in other tissues may be delayed. Future studies will assess whether other tissue types from these subjects (or their bodies as a whole) evade epigenetic aging. Thus, while we cannot exclude that tissue-specific ageing is causal of NCS, our current findings suggest that the observed delay in whole body development and possibly aging results from other yet undiscovered factors.
In closing, it is important to note that our inability to find a common mutation in coding genes does not automatically negate the hypothesis that post-maturational developmental inertia and loss of temporal order are the cause and consequences, respectively of aging, nor that the former is DNA-based. Getting back to my prior discussion of “things’ versus “processes”, it is important to recognize that coding genes produce “things”, i.e. proteins, and make up only a minority of the entire genome sequence, i.e. roughly 2% in humans. The remainder was once dismissed as “junk”, mostly because its function remained elusive. However, study of “junk” or “dark matter” DNA might gradually shed light on the cause and mechanism of phenoptosis. Non-coding DNA sequences may be far from useless in effecting development and aging, since they contain so-called regulatory regions or enhancers that determine when and where each gene is expressed. In other words, they regulate “processes” and may in fact be the loci of those bits of DNA that regulate “developmental inertia”. In this regard, it may be relevant to note that cis-regulatory non-coding DNA elements that control transcription may also be involved in the evolution and control of development. If so, they may also promote the process that eventually becomes the cause of aging [105, 106]. In the early online edition of Nature on October 12, 2011, researchers reported the sequencing of 20 new mammalian genomes and that they compared with 9 others that were previously described, including humans. They found that at least 5% of the genome appears to be constrained by evolution and that 3.6 million specific elements under constraint make up over 4% of the human genome. These elements include hundreds of new families of RNA, thousands of previously undetected segments of protein-coding DNA, and 2.7 million elements thought to play a role in controlling gene expression [107, 108]. Thus, it is possible that within these newly discovered areas of “dark DNA” the answers may reside. It is also possible that unique epigenetic modification of gene expression rather than alteration of the genetic code itself may be effective in stopping post-maturational developmental inertia. Continued research is ongoing to determine if our hypothesis on the cause and mechanism of phenoptosis is valid or not.
REFERENCES
1.Weismann, A. (1889) in Essays Upon Heredity and
Kindred Biological Problems, Clarendon Press, Oxford, p. 304.
2.Darwin, C. (1859) Laws of Variation, in On the
Origin of Species by Means of Natural Selection or the Preservation of
Favored Races in the Struggle for Life, Chap. 5, Murray Publishers,
London.
3.Wagner, G. P., and Altenberg, L. (1996)
Perspective: complex adaptations and the evolution of evolvability,
Evolution, 50, 967-976.
4.Altenberg, L. (1994) The evolution of evolvability
in genetic programming, in Advances in Genetic Programming
(Kinnear, J. K. E., ed.) MIT Press, Cambridge, MA, pp. 47-74.
5.Alberch, P. (1991) From genes to phenotype:
dynamical systems and evolvability, Genetica, 84,
511.
6.Skulachev, V. (1997) Aging is a specific biological
function rather than the result of a disorder in complex living
systems: biochemical evidence in support of Weismann’s
hypothesis, Biochemistry (Moscow), 62, 1191-1195.
7.Skulachev, V. (2011) Aging as a particular case of
phenoptosis, the programmed death of an organism (A response to
Kirkwood and Melov “On the programmed/non-programmed nature of
ageing within the life history”), Aging, 3, No.
11.
8.Kerr, J. F., Wyllie, A. H., and Currie, A. R.
(1972) Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics, Br. J. Cancer, 26,
23-57.
9.Sperandio, S., De Belle, I., and Bredesen, D. E.
(2000) An alternative, non-apoptotic form of programmed cell death,
Proc. Natl. Acad. Sci. USA, 97, 1476-1481.
10.Skulachev, V. P. (1999) Phenoptosis: programmed
death of an organism, Biochemistry (Moscow),
64, 1418-1426.
11.Libertini, G. (2012) Classification of
phenoptotic phenomena, Biochemistry (Moscow), 77,
707-715.
12.Skulachev, M. V., and Skulachev, V. P. (2014) New
data on programmed aging: slow phenoptosis, Biochemistry
(Moscow), 79, 977-993.
13.Goldsmith, T. C. (2004) Aging as an evolved
characteristic-Weismann’s theory reconsidered, Med.
Hypotheses, 62, 304-308.
14.Goldsmith, T. C. (2008) Aging, evolvability, and
the individual benefit requirement; medical implications of aging
theory controversies, J. Theor. Biol., 252, 764-768.
15.Yang, J.-N. (2013) Viscous populations evolve
altruistic programmed ageing in ability conflict in a changing
environment, Evol. Ecol. Res., 15, 527-543.
16.Lenart, P., and Bienertova-Vasku, J. (2016)
Keeping up with the Red Queen: the pace of aging as an adaptation,
Biogerontology, doi: 10.1007/s10522-016-9674-4.
17.Goldsmith, T. C. (2014) The Evolution of
Aging, 3rd Edn., Amazon, New York, p. 32.
18.Blagosklonny, M. V. (2013) Aging is not
programmed. Genetic pseudo-program is a shadow of developmental growth,
Cell Cycle, 12, 3736-3742.
19.Skulachev, V. P. (2002) Programmed death
phenomena: from organelle to organism, Ann. N.Y. Acad. Sci.,
959, 214-237.
20.Fitzgerald, F. S. (2008) The Curious Case of
Benjamin Button and Other Jazz Age Stories, Penguin Classics,
London.
21.Williams, G. C. (1957) Pleiotropy, natural
selection, and the evolution of senescence, Evolution,
11, 398-411.
22.Harman, D. (1956) Aging: a theory based on free
radical and radiation chemistry, J. Gerontol., 11,
298-300.
23.Bjorksten, J., and Tenhu, H. (1990) The
crosslinking theory of aging: added evidence, Exp. Gerontol.,
25, 91-95.
24.Orgel, L. E. (1963) The maintenance of the
accuracy of protein synthesis and its relevance to ageing, Proc.
Natl. Acad. Sci. USA, 49, 517-521.
25.Korshunov, S. S., Skulachev, V. P., and Starkov,
A. A. (1997) High protonic potential actuates a mechanism of production
of reactive oxygen species in mitochondria, FEBS Lett.,
416, 15-18.
26.Skulachev, V. P. (1999) Phenoptosis: programmed
death of an organism, Biochemistry (Moscow), 64,
1418-1426.
27.Harman, D. (1972) The biologic clock: the
mitochondria? J. Am. Ger. Soc., 20, 145-147.
28.Yin, D., and Chen, K. (2005) The essential
mechanisms of aging: irreparable damage accumulation of biochemical
side reactions, Exp. Gerontol., 40, 455-465.
29.Liu, X., Jiang, N., Bigras, E., Shoubridge, E.,
and Hekimi, S. (2005) Evolutionary conservation of the clk-1-dependent
mechanism of longevity: loss of mclk1 increases cellular fitness and
lifespan in mice, Genes Dev., 19, 2424-2434.
30.Holley, C. L., Olson, M. R., Colon-Ramos, D. A.,
and Kornbluth, S. (2002) Reaper eliminates IAP proteins through
stimulated IAP degradation and generalized translational inhibition,
Nat. Cell Biol., 4, 439-444.
31.Skulachev, V. P. (2012) What is
“phenoptosis” and how to fight it? Biochemistry
(Moscow), 77, 689-706.
32.Comfort, A. (1956) in The Biology of
Senescence, Rinehart and Co., Inc., Lanham, MD, p. 257.
33.Lansing, A. I. (1952) in Cowdry’s
Problems of Ageing, 3rd. Edn., Williams & Wilkins, Baltimore,
p. 1061.
34.Dong, H., Yuede, C. M., Yoo, H. S., Martin, M.
V., Deal, C., Mace, A. G., and Csernansky, J. G. (2008) Corticosterone
and related receptor expression are associated with increased
beta-amyloid plaques in isolated Tg2576 mice, Neuroscience,
155, 154-163.
35.Jones, R. E., and Norris, D. O. (2002) Cortisol
and pacific salmon: a new look at the role of stress hormones in
olfaction and home-stream migration, Integr. Comp. Biol.,
42, 574-581.
36.McEwen, B. S. (2000) The neurobiology of stress:
from serendipity to clinical relevance, Brain Res., 886,
172-189.
37.Dickhoff, W. W. (1989) Salmonids and annual
fishes: death after sex, in Development, Maturation, and Senescence
of Neuroendocrine Systems: A Comparative Approach (Schreibman, M.
P., and Scanes, C. G., eds.) Academic Press, New York, pp. 253-268.
38.Sapolsky, R. (2004) in Why Zebras Don't Get
Ulcers, 3rd Edn., Holt Paperbacks, New York, pp. 245-247.
39.Diaz Arenas, C., and Cooper, T. F. (2013)
Mechanisms and selection of evolvability: experimental evidence,
FEMS Microbiol. Rev., 37, 572-582.
40.Mitchell, S. (2004) Gilgamesh: A New English
Version, Free Press, New York, NY.
41.Walker, R. F., Pakula, L., Sutcliffe, M. J.,
Kruk, P. A., Graakjaer, J., and Shay, J. W. (2009) A case study of
disorganized development and its possible relevance to genetic
determinants of aging, Mech. Aging Dev., 130,
350-356.
42.Walker, R. F. (2011) Developmental theory of
aging revisited. Focus on causal and mechanistic links between
development and aging, Rejuvenation Res., 14,
429-436.
43.De Magalhaes, J. P., and Church, G. M. (2005)
Genomes optimize reproduction: aging as a consequence of the
developmental program, Physiology, 20, 252-259.
44.Blagosklonny, M. V. (2006) Aging and immortality:
quasi-programmed senescence and it’s pharmacologic inhibition,
Cell Cycle, 5, 2087-2102.
45.Zwaan, B. J. (2003) Linking development and
aging, Sci. Aging Knowledge Environ., 47, p. 32.
46.Bidder, G. P. (1932) Senescence, Brit. Med.
J., 2, 583-585.
47.Blagosklonny, M. V., and Hall, M. N. (2009)
Growth and aging: a common molecular mechanism, Aging, 1,
357-362.
48.Lints, F. A. (1978) Genetics and ageing, in
Interdisciplinary Topics in Gerontology, Karger, Basel.
49.Yates, F. E. (1994) Order and complexity in
dynamical systems: homeodynamics as a generalized mechanics for
biology, Math. Comput. Model., 19, 49-74.
50.Lloyd, D., Aon, M., and Cortassa, S. (2001) Why
homeodynamics, not homeostasis, Sci. World, 1,
133-145.
51.Sterling, P., and Eyer, J. (2004) in
Allostasis, Homeostasis, and the Costs of Adaptation (Schulkin,
J., ed.) Cambridge University Press, Cambridge, UK.
52.Suresh, I., and Rattan, S. (2006) Homeostasis,
homeodynamics and aging, in Encyclopedia of Gerontology, 2nd
Edn. (Birren, J., ed.) Elsevier Inc., UK.
53.Walker, R. F., Liu, J. S., Peters, B. A., Ritz,
B. R., Wu, T., Ophoff, R., and Horvath, S. (2015) Epigenetic age
analysis of children who seem to evade aging, Aging, 7,
1-6.
54.Walker, R. F. (2013) Why We Age: Insight into
the Cause of Growing Old, Dove Medical Press.
55.Lexell, J., Taylor, C. C., and Sjostrom, M.
(1988) What is the cause of the ageing atrophy? Total number, size and
proportion of different fiber types studied in whole vastus lateralis
muscle from 15‐ to 83‐year‐old men, J. Neurol.
Sci., 84, 275-294.
56.Pincus, S. M. (1991) Approximate entropy as a
measure of system complexity, Proc. Natl. Acad. Sci. USA,
88, 2297-2301.
57.Lipsitz, L. A., and Goldberger, A. L. (1992) Loss
of “complexity” and aging potential applications of
fractals and chaos theory to senescence, JAMA, 267,
1806-1809.
58.Veldhuis, J. D. (1995) Differential impact of
age, sex steroid hormones and obesity on basal versus pulsatile growth
hormone secretion in men as assessed in an ultrasensitive
chemiluminescence assay, J. Clin. Endocrinol. Metab., 80,
3209-3222.
59.Galili, I., and Tseitlin, M. (2003)
Newton’s first law: text, translations, interpretations and
physics education, Sci. Educat., 12, 45-73.
60.Iafrate, A. J. (2004) Detection of large-scale
variation in the human genome, Nat. Genet., 36,
949-951.
61.Blagosklonny, M. V. (2012) Answering the ultimate
question “what is the proximal cause of aging”?
Aging, 4, 861-877.
62.Blagosklonny, M. V. (2010) Revisiting the
antagonistic pleiotropy theory of aging: TOR-driven program and
quasi-program, Cell Cycle, 9, 3151-3156.
63.Laplante, M., and Sabatini, D. M. (2012) mTOR
signaling in growth control and disease, Cell,
149, 274-293.
64.Zimbler, M. S., Kokosa, M., and Thomas, J. R.
(2001) Anatomy and pathophysiology of facial aging, Facial Plastic
Surg. Clin. North Am., 9, 179-187.
65.Behrents, R. G. (1985) An atlas of growth in the
aging craniofacial skeleton, in Craniofacial Growth Series, Ann
Arbor, Michigan.
66.Helenius, M., Hanninen, M., Lehtinen, S. K., and
Salminen, A. (1996) Changes associated with aging and replicative
senescence in the regulation of transcription factor nuclear
factor-κB, Biochem. J., 318, 603-608.
67.Boehm, M., and Slack, F. (2005) A developmental
timing microRNA and its target regulate life span in C. elegans,
Science, 310, 1954-1957.
68.Somel, M., Guo, S., Fu, N., Yan, Z., Hu, H. Y.,
Xu, Y., Yuan, Y., Ning, Z., Hu, Y., Menzel, C., Hu, H., Lachmann, M.,
Zeng, R., Chen, W., and Khaitovich, P. (2010) MicroRNA, mRNA and
protein expression link development and aging in human and macaque
brain, Genome Res., 20, 1207-1218.
69.Kenyon, C. (2002) Regulation of life-span by
germ-line cells in Caenorhabditis elegans, Science,
295, 499-502.
70.Lin, K., Hsin, H., Libina, N., and Kenyon, C.
(2001) Regulation of the Caenorhabditis elegans longevity
protein DAF-16 by insulin/IGF-1 and germline signaling, Nat.
Genet., 28, 139-145.
71.Bartke, A., Brown-Borg, H., Mattison, J., Kinney,
B., Hauck, S., and Wright, C. (2001) Prolonged longevity of
hypopituitary dwarf mice, Exp. Gerontol., 36, 21-28.
72.Holzenberger, M., Dupont, J., Ducos, B., Leneuve,
P., Geloen, A., Even, P. C., Cervera, P., and Le Bouc, Y. (2003) IGF-1
receptor regulates life span and resistance to oxidative stress in
mice, Nature, 421, 182-187.
73.Heilbronn, L. K., and Ravussin, E. (2003) Calorie
restriction and aging: review of the literature and implications for
studies in humans, Am. J. Clin. Nutr., 78, 361-369.
74.Weindruch, R., Walford, R. L., Fligiel, S., and
Guthrie, D. (1986) The retardation of aging in mice by dietary
restriction: longevity, cancer, immunity and lifetime energy intake,
J. Nutr., 116, 641-654.
75.Samis, H. V. (1968) Aging: the loss of temporal
organization, Perspect. Biol. Med., 12, 95-102.
76.Cooper, R. L., Conn, P. M., and Walker, R. F.
(1980) Characterization of the LH surge in middle-aged female rats,
Biol. Reprod., 23, 611-615.
77.Kintraia, P. I., Zarnadze, M. G., Kintraia, N.
P., and Kashakashvili, I. G. (2005) Development of daily rhythmicity in
heart rate and locomotor activity in the human fetus, J. Circadian
Rhythms, 3, 5-17.
78.Popular Mechanics Complete Car Care Manual
Updated & Expanded (2005) Hearst Communications, Inc., CA.
79.Gallant, R., Fidler, T., and Dawson, K. (1991)
Subjective time estimation and age, Percept. Motor Skills,
72, 1275-1280.
80.Dawson, K. A. (1992) Opposing changes in past and
future time orientations with age, Percept. Motor Skills,
75, 1242-1251.
81.Dawson, D. A. (2004) Temporal organization of the
brain: neurocognitive mechanisms and clinical implications, Brain
Cogn., 54, 75-94.
82.Piggins, H. D. (2002) Human clock genes, Ann.
Med., 34, 394-400.
83.Harker, J. (1956) Experimental production of
midgut tumors in Periplaneta americana (L.), J. Exp.
Biol., 35, 251-259.
84.Budovskaya, Y. V., Kendall, W., Southworth, L.
K., Min, J., Tedesco, P., Johnson, T. E., and Kim, S. K. (2008) An
elt-3/elt-5/elt-6 transcription circuit guides aging in C.
elegans, Cell, 134, 1-13.
85.Pincus, A., and Slack, F. J. (2008)
Transcriptional (dys)regulation and aging in Caenorhabditis
elegans, Gen. Biol., 9, 233.
86.Kim, S. (2008) What is the developmental drift
theory?, in Sage Crossroads. 53. Evolution of Aging.
87.Blagosklonny, M. V. (2010) Why men age faster but
reproduce longer than women: mTOR and evolutionary perspectives,
Aging, 2, 265-273.
88.Aschheim, P. (1964) Results provided by
heterochronic grafts of the ovaries in the study of the
hypothalamo-hypophyso-ovarian regulation of senile rats,
Gerontologia, 10, 65-75.
89.Peng, M. T., and Huang, H. H. (1972) Aging of
hypothalamic-pituitary-ovarian function in the rat, Fertil.
Steril., 23, 535-542.
90.Lohstroh, P. N., Chen, J., Ba, B., Ryan, L. M.,
Xu, X., Overstreet, J. W., and Lasley, B. L. (2003) Bone resorption is
affected by follicular phase length in female rotating shift workers,
Environ. Health Perspect., 111, 618-622.
91.Gibson, E. M., Williams, W. P., 3rd., and
Kriegsfeld, L. J. (2009) Aging in the circadian system: considerations
for health, disease prevention and longevity, Exp. Gerontol.,
44, 51-56.
92.Eskin, B. A., Trivedi, R. A., Weideman, C. A.,
and Walker, R. F. (1988) Feedback disturbances revealed by analysis of
serum reproductive hormones in women during aging, Am. J. Gyn.
Health, 2, 9-15.
93.Weiss, G., Skurnick, J. H., Goldsmith, L. T.,
Santoro, N. F., and Park, S. J. (2004) Menopause and
hypothalamic-pituitary sensitivity to estrogen, JAMA,
292, 2991-2996.
94.Santoro, N., Banwell, T., Tortoriello, D.,
Lieman, H., Adel, T., and Skurnick, J. (1998) Effects of aging and
gonadal failure on the hypothalamic-pituitary axis in women, Am. J.
Obstet. Gynecol., 178, 732-741.
95.Nakamura, K., Shimai, S., Kikuchi, S., Tominaga,
K., Takahashi, H., Tanaka, M., Nakano, S., Motohashi, Y., Nakadaira,
H., and Yamamoto, M. (1997) Shift work and risk factors for coronary
heart disease in Japanese blue-collar workers: serum lipids and
anthropometric characteristics, Occup. Med. (London), 47,
142-146.
96.Tuchsen, F., Christensen, K. B., Lund, T., and
Feveile, H. (2008) A 15 year prospective study of shift work and
disability pension, Occup. Environ. Med., 65,
283-285.
97.Escobar, C., Salgado-Delgado, R.,
Gonzalez-Guerra, E., Osorio, A. T., Angeles-Castellanos, M., and Buijs,
R. M. (2011) Circadian disruption leads to loss of homeostasis and
disease, Sleep Disord., doi: 10.1155/2011/964510.
98.Haeckel, E. (1899) Riddle of the universe at the
close of the nineteenth century, https://archive.org/details/riddleofuniverse00haecrich.
99.Skulachev, V. P., Holtze, S., and Vyssokikh, M.
Y. (2017) Neoteny, prolongation of youth from naked mole rats to
“Naked Apes” (humans), Physiol. Rev., 97,
699-720.
100.Shea, B. T. (1989) Heterochrony in human
evolution: the case for neoteny reconsidered, Am. J. Phys.
Anthropol., 32, 69-101.
101.Somela, M., Franz, H., and Yana, Z. (2009)
Transcriptional neoteny in the human brain, PNAS, 106,
5743-5748.
102.Andrew Fire, A., Xu, S., Montgomery, M. K.,
Kostas, S. A., Driver, S. E., and Mello, C. C. (1998) Potent and
specific genetic interference by double-stranded RNA in
Caenorhabditis elegans, Nature, 391, 806-811.
103.Walker, R. F., Ciotlos, S., Mao, Q., Chin, R.,
Drmanac, S., Barua, N., Agarwal, M. R., Zhang, R. Y., Li, Z., Wu, M.,
Sun, K., Lee, K., Nguyen, S., Liu, J. S., Carnevali, P., Drmanac, R.,
and Peters, B. A. (2017) Clinical and genetic analysis of a rare
syndrome associated with neoteny, Genet. Med., DOI:
GIM.2017.140:1-8.
104.Horvath, S. (2013) DNA methylation age of human
tissues and cell types, Genome Biol., 14,
R115.
105.Carroll, S. B. (2008) Evo-devo and an expanding
evolutionary synthesis: a genetic theory of morphological evolution,
Cell, 134, 25-36.
106.Arnold, C. D., Gerlach, D., Stelzer, C., Boryn,
L. M., Rath, M., and Stark, A. (2013) Genome-wide quantitative enhancer
activity maps revealing complex cis-regulation of transcription,
Science, 339, 1074-1077.
107.Wein, J. (2011) Genome comparison casts light
on dark areas of DNA, NIH Research Matters, October 24,
2011.
108.Lindblad-Toh, K., Garber, M., Zuk, O., Lin, M.
F., Parker, B. J., Washietl, S., Kheradpour, P., Ernst, J., Jordan, G.,
Mauceli, E., Ward, L. D., Lowe, C. B., Holloway, A. K., Clamp, M.,
Gnerre, S., Alfoldi, J., Beal, K., Chang, J., Clawson, H., Cuff, J., Di
Palma, F., Fitzgerald, S., Flicek, P., Guttman, M., Hubisz, M. J.,
Jaffe, D. B., Jungreis, I., Kent, W. J., Kostka, D., Lara, M., Martins,
A. L., Massingham, T., Moltke, I., Raney, B. J., Rasmussen, M. D.,
Robinson, J., Stark, A., Vilella, A. J., Wen, J., Xie, X., Zody, M. C.;
Broad Institute Sequencing Platform and Whole Genome Assembly Team,
Baldwin, J., Bloom, T., Chin, C. W., Heiman, D., Nicol, R., Nusbaum,
C., Young, S., Wilkinson, J., Worley, K. C., Kovar, C. L., Muzny, D.
M., Gibbs, R. A.; Baylor College of Medicine Human Genome Sequencing
Center Sequencing Team, Cree, A., Dihn, H. H., Fowler, G., Jhangiani,
S., Joshi, V., Lee, S., Lewis, L. R., Nazareth, L. V., Okwuonu, G.,
Santibanez, J., Warren, W. C., Mardis, E. R., Weinstock, G. M., Wilson,
R. K.; Genome Institute at Washington University, Delehaunty, K.,
Dooling, D., Fronik, C., Fulton, L., Fulton, B., Graves, T., Minx, P.,
Sodergren, E., Birney, E., Margulies, E. H., Herrero, J., Green, E. D.,
Haussler, D., Siepel, A., Goldman, N., Pollard, K. S., Pedersen, J. S.,
Lander, E. S., and Kellis, M. (2011) A high-resolution map of human
evolutionary constraint using 29 mammals, Nature, 478,
476-482.