REVIEW: The Ribosome as an Allosterically Regulated Molecular Machine
T. M. Makarova1,2 and A. A. Bogdanov1,2*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; E-mail: bogdanov@belozersky.msu.ru2Lomonosov Moscow State University, Chemistry Department, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received August 16, 2017
The ribosome as a complex molecular machine undergoes significant conformational rearrangements during the synthesis of polypeptide chains of proteins. In this review, information obtained using various experimental methods on the internal consistency of such rearrangements is discussed. It is demonstrated that allosteric regulation involves all the main stages of the operation of the ribosome and connects functional elements remote by tens and even hundreds of angstroms. Data obtained using Förster resonance energy transfer (FRET) show that translocation is controlled in general by internal mechanisms of the ribosome, and not by the position of the ligands. Chemical probing data revealed the relationship of such remote sites as the decoding, peptidyl transferase, and GTPase centers of the ribosome. Nevertheless, despite the large amount of experimental data accumulated to date, many details and mechanisms of these phenomena are still not understood. Analysis of these data demonstrates that the development of new approaches is necessary for deciphering the mechanisms of allosteric regulation of the operation of the ribosome.
KEY WORDS: ribosome, allostery, allosteric regulation, tRNA, rRNADOI: 10.1134/S0006297917130016
Abbreviations: DC, decoding center of the ribosome; FRET, Förster resonance energy transfer; GAC, GTPase-associated center of the ribosome; PTC, peptidyltransferase center of the ribosome; RT, ribosomal tunnel; smFRET, Förster resonance energy transfer of single molecules; SRL, sarcin-ricin loop, or ribosomal helix 95.
The ribosome is a molecular machine that synthesizes all cellular
proteins by decoding information contained in the nucleotide sequence
of messenger RNA (mRNA). The ribosome is a huge macromolecular complex:
in bacteria it contains three molecules of ribosomal RNA (rRNA) with
general length of more than 4000 nucleotide bases, more than 50
ribosomal proteins and, in addition, at every moment of its work,
auxiliary protein factors. The ribosome synthesizes protein chains from
activated amino acids delivered to its peptidyltransferase center (PTC)
by transfer RNA (tRNA). For this goal, the ribosome must scan an mRNA,
select the aminoacyl-tRNA (aa-tRNA) with the anticodon that is
complementary to the codon in the mRNA, orient the substrates of
peptide synthesis in the PTC in the correct manner in order to catalyze
the transpeptidation reaction, and after that carry out the concerted
advance (translocation) of tRNAs and the mRNA by one codon for
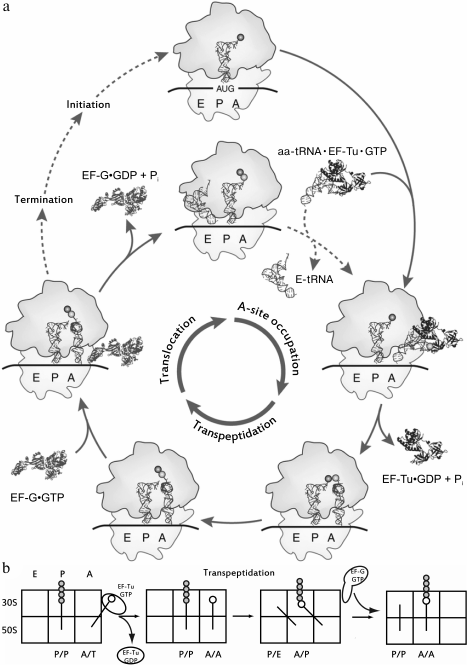
releasing the site for a new aa-tRNA (Fig. 1a). At every stage of this multistage process, the
conformation of the ribosome and its ligands, tRNA and protein
translational factors, undergo certain changes (for reviews see [1, 2]). Allosteric features are
revealed in ribosomes of all living organisms, which is not surprising
if we consider its exclusively high conservativity during biological
evolution. However, now enough documented data about allostery in the
translation apparatus are found in the literature only concerning
bacterial and yeast ribosomes. So, in this review only facts obtained
for these organisms are analyzed.
Fig. 1. a) The cycle of the ribosome. Three main stages of an elongation are labeled in the inner circle; the elongation factors involved in the cycle are labeled as part of the complexes, both in the interaction with the ribosome, and dissociated after the completion of the stage. The initiation of the synthesis of a new peptide chain, just like the termination of translation on the stop codon, is in an external cycle. Adapted from [92]. b) Sequential advancement of tRNA along the sites of the ribosome during elongation. The sites occupied by tRNA are labeled from below, the subunits of the ribosome are labeled to the left of the figure; the newly synthesized polypeptide chain is indicated as gray circles, the new amino acid is open one.
The terms “allostery” and “allosteric” initially appeared when it was established that the activity of proteins could be stimulated or inhibited by distant factors, binding sites of which in the protein molecule are remote from its functional center and do not overlap with it [3]. These terms originate from Greek word “allos” – “another”. Further, it was realized that allosteric phenomena are fundamental features of living matter, its susceptibility and changeability, and its ability to perceive, transmit, and remember information consisting in conformational states of macromolecules. Activity of many proteins and enzymes (for example [4]), different stages of gene expression [5, 6], ligand–receptor interactions, ion channel activity [7], and many other processes are known to be regulated in allosteric manner. This phenomenon was found not only in proteins, but also in DNA–protein complexes [8, 9] and RNAs from small aptamers [10] up to, as already mentioned, the largest ribonucleoprotein complex in a cell – the ribosome.
Allosteric regulation exists first due to flexible and mobile structure of biological macromolecules, and second due to conformational coherency of its residues: cooperative dynamics provides several permitted states near global energetic minima between which temporal switching is possible. In the simplest model, there are two such the states, and they are designated as R, the relaxed one, and T, the tensed one, which are in equilibrium when the molecule is vacant, but introduced into the system allosteric effector can bias this equilibrium due to preferential affinity to one certain state. Allosteric effectors are usually low molecular weight ligands, but since about the 1990s, point mutations, modifications, change of general conditions, such as pH or ionic strength of the media, are also considered as allosteric effectors. If upon changing the protein state geometry of an active or binding to another ligand center switches through the threshold separating active and inactive states, then allosteric regulation of its activity is observed [11].
The first mathematical models describing allosteric effects were created for exploration of cooperative effects in multisubunit proteins, the classic example being the Bohr effect, or cooperative binding of a proton by four hemoglobin subunits. The earliest, so called concerted model, or MWC model according to the author’s surnames abbreviation (Monod–Wyman–Changeux) [12] considers a multisubunit protein as an indivisible allosteric switch, so that all its subunits adopt active or inactive state simultaneously and in concerted manner, which defined the model. The so-called sequential model, published shortly after the previous one, designated by abbreviation KNF also due to the author’s surnames (Koshland–Nemethy–Filmer) [13], allows the possibility for different subunits of the protein to be in different states, but in this model conformational change in one of the subunits shifts the equilibrium for the others. The KNF model in distinction from the MWC model enables description of not only positive but also negative cooperativity, found, for example, in ATPases of the multisubunit bacterial chaperon GroEL [14].
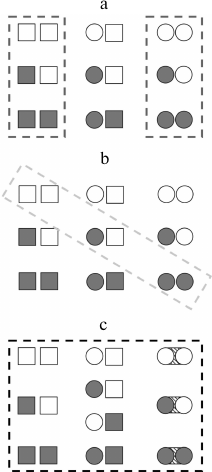
Rather recently the thermodynamic ensemble allosteric model (EAM) was elaborated [15], following the approaches of the 1960s, but already allowing explanation of more complicated phenomena up to sign changes of allosteric effects of a ligand depending on certain conditions. This phenomenon is particularly observed in the case of certain transcriptional factors, which can be agonists or antagonists in shifting conditions [16, 17]. With this model, the protein is subdivided into conditional “domains” (like subunits in predecessor models) connecting two ligands A and B, which have their T and R states, and the change in free energy in the transition between the states is composed of differences between energies of states of the domains themselves and change in energies of the interdomain interactions. In general, this new thermodynamic model can take into account many more states than the models of the 1960s (Fig. 2).
Fig. 2. Schematic representation of individual subunits states considered in different thermodynamic models for a multisubunit protein. Here the square denotes the T (tensed) state, the circle – R (relaxed) state; closed figure designates a domain with bound ligand, open figure represents a ligand-free domain. a) MWC model (concerted); b) KNF model (sequential); c) EAM model (energy ensembles). Adapted from [15].
These models assume only a “quaternary” level of changes in the protein through a switching between the states of the subunits, leaving the “tertiary” level, the level of certain intermolecular interactions and conformations of individual residues, a black box that only provides a particular state of the subunit. Meanwhile, the “tertiary” level itself independently is able to provide information exchange in single-subunit proteins, and, presumably, makes allostery a universal property of all proteins except fibrillar ones [18]. At this level, for some systems, specific pathways for signal propagation and networks of interactions were identified, using both computational approaches [19] and experimental ones, detecting conformational changes by means of NMR relaxation methods [20]. In this case, the overall picture of interactions of remote functional regions of proteins often implies a set of intertwined paths organized in global communication networks (GCN), as well as the interaction between the “quaternary” and “tertiary” levels, thus encompassing the comprehensive whole of the molecule [21].
Allosteric states might differ not only in equilibrium positions of atoms, but also in a degree of motion in vibrational and rotational modes, in other words, conformational entropy [22]. This possibility was predicted theoretically back in 1984 and was called “allostery without conformational changes” [23]. The presence of the entropic component makes conventional structural methods insufficient for investigation of allostery. The simplest estimate of principle modes ranges, based on B-factor values extracted from structural data for an irredundant sample of 91 proteins bound with ligands and vacant ones, revealed a significant difference in about half of the investigated structures. Thus, in these proteins the entropic factor is important for allosteric switching [24].
In summarizing the above, it is noteworthy that biological macromolecules can occupy several resolved states near a global minimum, and these states differ between each other in conformational and entropic characteristics. The switching of the macromolecule between these states occurs in a holistic manner, so that it becomes possible to regulate a functionally important site by impact on remote regions stabilizing a certain state by a small effector ligand at the scales of the biomolecule, as well as a mutation or modification of its monomeric units.
ALLOSTERIC EFFECTS OBSERVED BY INTERACTION OF THE RIBOSOME WITH
tRNAs
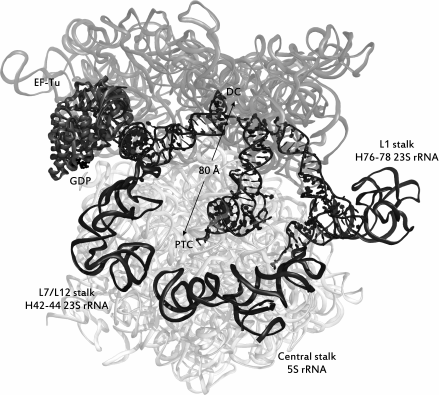
Binding of tRNA to the A-site of the ribosome. As seen from Fig. 1, tRNA, acylated by an amino acid residue, sequentially occupies three binding sites in the ribosome, namely the A (acyl)-, P (peptidyl)-, and E (exit)-sites. The ribosome consists of two subunits – large and small; each subunit of the ribosome has three specific areas corresponding to each of the mentioned sites. Upon translation, tRNAs can simultaneously occupy not only areas of the same site on the small and large subunits, but also be in one of the so-called hybrid states, i.e. be bound with regions belonging to different sites (Fig. 1b). Aminoacyl-tRNA is delivered to the ribosome as part of a ternary complex with the elongation factor EF-Tu (in bacteria) bound to GTP (aa-tRNA•EF-Tu•GTP complex); it occupies the A-site region of the small subunit, and it establishes contacts with the L7/L12-stalk on the large subunit in complex with EF-Tu•GTP that is designated as the hybrid A/T-state of aa-tRNA. The correct geometry of the codon–anticodon complex upon the interaction of the anticodon of this tRNA with the corresponding codon in the mRNA stimulates EF-Tu GTPase activity and GTP hydrolysis, after which the ternary complex dissociates and the aminoacylated CCA-terminus of the tRNA occupies the A-site region of the large subunit. This stage of the translation cycle is called an accommodation of aa-tRNA. In this case, the ribosomal P-site is already occupied by tRNA, acylated with a newly synthesized peptide (nascent peptide), or, in the case of a translation initiation, with an N-formylmethionine residue. The mutual arrangement of E-, P-, and A/T-tRNA, as well as of EF-Tu in a complex with the ribosome, is shown in Fig. 3.
Fig. 3. The ribosome immediately after hydrolysis of GTP into GDP during recognition of aa-tRNA. rRNA is shown in the form of transparent ribbons, while ribosome ligands are represented as follows: tRNAs – in the form of helices with leaves, elongation factor – in the form of opaque ribbons with GDP displayed in black. Dark gray opaque ribbons display the rRNA of the stalks of a large ribosomal subunit. rRNA of the small subunit (16S rRNA) is depicted in a darker shade of gray than the rRNA of the large subunit. tRNAs occupy the sites (from left to right): A/T, P/P, E/E. The distance in angstroms between the decoding center (DC) and the peptidyltransferase center (PTC) is shown. The source of the structure is PDB id: 5AFI.
Binding of the tRNA to the ribosome in the A-site was found to be disturbed by distant mutations, and therefore it depends on the conformation of the ribosomal elements surrounding this site.
Thus, the substitution of adenine A2531, playing an important role in the tertiary contact of helices H91 and H95 in 23S rRNA to pyrimidine bases, considerably reduced the affinity of the ternary complex (in which the role of aa-tRNA was carried out by Phe-tRNAPhe) to the ribosome. Meanwhile, the affinity of a peptidyl-tRNA to the P-site did not altered. The distance from the point of the mentioned mutation to the A-site of the large subunit of the ribosome is not less than 50 Å [25].
In addition, a mutation that distorted the secondary structure of the H89 helix by replacing two nucleotide residues of UU2492-3 by a single C residue could disturb the binding of the A-tRNA [26].
It is noteworthy that in both the cases the ribosome retained its functionality, and several translocation stages and affinity for P-tRNA were not damaged by these mutations.
Is there allosteric connection between the A and E sites of the ribosome? The fact that the binding of an aa-tRNA to the A-site can be selectively inhibited by remote point mutations implicitly suggests its ability to be found in two states – an “open” state that can accept a tRNA, and a “closed” state that prevents this binding. Therein, is there any functional significance for the switchover between the states? Might this switch be a sensor or, on the contrary, a regulator that coordinates the binding of the aa-tRNA in the A-site and other stages of protein synthesis in the ribosome?
It would seem that a sufficiently definite answer to this question was made many years ago by Nierhaus et al., who discovered the phenomenon of negative cooperativity of tRNA binding in the A- and E-sites [27-29]. According to those authors, the affinity of deacylated tRNA to the E-site, which is largely determined by the codon–anticodon interactions with mRNA, declined dramatically when the aa-tRNA (for instance, Thr-tRNAThr) occupied its site. Vice versa, if conditions were created conducive to the binding of deacylated tRNA to the E-site of the ribosome, whose A-site was already occupied by aa-tRNA, the complex of the latter with the ribosome was destroyed [28].
Subsequently, Nierhaus et al. obtained data suggesting that the allosteric interaction of tRNA in the A- and E-sites via mRNA is expected to support the reading frame [30] and reduce the frequency of decoding errors [31, 32]. Indeed, on one hand, tRNA in the E-site prevented a random shift of the mRNA reading frame [33]; on the other hand, it facilitated its shift if it was programmed [34, 35]. Thus, it is expected for these interactions to be directly related to the control of the movement of mRNA during translocation.
The hypothesis of Nierhaus on the mutually exclusive binding of A- and E-tRNA to the ribosome has long been the subject of bitter controversy. Thus, it was shown in the laboratory of Rodnina and Wintermeyer that in a cell-free system with a high concentration of Mg2+ and polyamines, the ternary complex involving Phe-tRNAPhe did not accelerate the dissociation of tRNAfMet from the E-site, firmly connected with it due to the introduction of polyamines into the system [36].
Strictly speaking, the discussion about Nierhaus’ hypothesis is not considered to be complete (for a review, see [2]), especially since in the literature from time to time there appear data seemingly reconciling two opposing points of view. Thus, studying the binding and dissociation of tRNA in real time on individual ribosome molecules by smFRET, which allows tracing several initial elongation cycles of translation with different mRNA sequences, it was found that the competition between the A- and E-sites of the ribosome in fact depends on of the length of the synthesized peptide. Leaving aside the details, based on the results of that work, it is to be concluded that the mechanism of Nierhaus acted at the earliest stages of polypeptide synthesis, and the Rodnina–Wintermeyer mechanism at subsequent ones [37]. It is also noteworthy that the dilemma discussed above can be solved by analyzing a system with an undissociated into subunits ribosome in the presence of mRNA and entire tRNA molecules, since there is no prohibition of simultaneous binding of CCA-fragments of tRNA to the A- and E-sites of the large subunit, while the occupation of the E-site of the large subunit did not distort in any way the geometry of binding of the tRNA terminus fragment in the A-site of the 50S subunit of Haloarcula marismortui [38]. In favor of the requirement to continue investigations to resolve this problem, the allosteric relationship of A- and E-sites was established by site-directed mutagenesis: a mutation in the A-site of the yeast ribosome C2820U (C2452 E. coli) increased the availability for chemical modification of two adenines (A2778 and A2779) in helix H88 of 25S rRNA located near the E-site [39].
To conclude this section, we note that it is possible to obtain T. thermophilus ribosomes in a crystal state as a complex containing tRNAs in all three classical sites simultaneously. However, in many cases (PDB id: 5J4C, 5J4B, 5J8B, 4WPO, 4V51, 5VP2, 4V5C, 4V5D, 4V8N, 4WT8) this is achieved due to the contact of E-tRNA with mRNA, hardly like “correct” codon–anticodon interactions.
Allosteric regulation of the ribosomal decoding center activity. The selection of tRNA with an anticodon corresponding to the codon in mRNA occurs in two main stages. First, an aa-tRNA in a ternary complex (with EF-Tu and GTP) occupies its site on the ribosome, i.e. binds to a hybrid A/T-site. The first two pairs of the codon–anticodon complex are stabilized by interaction with nucleotide residues A1492 and A1493 of the h44 16S rRNA and the hydrogen bond with the G530 residue of helix h18. The formation of the “cognate” codon–anticodon complex serves as a signal for the conformational rearrangement of a vast region of the small subunit of the ribosome, which in turn stimulates the GTPase activity of EF-Tu, hydrolyses GTP into GDP, and reduces the interactions of the ternary complex with the ribosome. In the second stage, the so-called aa-tRNA accommodation occurs, during which, in the case of the “cognate” codon–anticodon complex formation, the aa-tRNA vacates EF-Tu, and its CCA-terminus occupies the A-site site in the PTC. During this, the aa-tRNA remains bound to the mRNA. If there was an error at the decoding stage, the aa-tRNA dissociates from the ribosome.
It is not surprising that at this very important stage of genetic information translation, accompanied by various conformational rearrangements of the ribosome and its ligands, the allosteric effects manifest themselves quite obviously and are particularly variable.
The decoding center of the small subunit of the ribosome, where the codon–anticodon complexes are recognized and discriminated, is located about 80 Å from the GTPase center of EF-Tu. The participants in the transmission of the signal about the formation of the “cognate” codon–anticodon complex to this center are well known: first, it is a tRNA molecule. Ramakrishnan and coworkers demonstrated that after binding of the ternary complex to the ribosome, i.e. in the hybrid A/T-state, the tertiary structure of tRNA differs significantly from the structure in the free complex [40]. Accordingly, mutations in the tRNA molecule that alter the macromolecular structure of the tRNA in the A/T-state, and which are sufficiently remote from the decoding center, allosterically influence the decoding accuracy [41]. The best-studied mutation for this case is a so-called “Hirsch suppressor”, the G24A mutation in the D-loop of tRNATrp, which recognizes the stop codon UGA as Trp [42].
Second, this is a rather large RNA–protein domain of the 30S subunit (sometimes called the “arm” of this subunit), on the surface of which the decoding process itself takes place. In the formation of the “cognate” codon–anticodon complex, the “shoulder” of the small subunit assumes a more compact form (transits from an “open” to a “closed” state), which acts as a “trigger” for transmitting the allosteric signal about this event to the GTPase center of EF-Tu. When this occurs, a connection of one of the 16S rRNA segments of this domain with EF-Tu is established [43]. The following events are not yet studied at the level of individual nucleotide and amino acid residues. However, it is well known that an important role in the conformational transformations of the “arm” of the 30S subunit is played by the mutually contacting uS4 and uS5 proteins. For many years, Dalberg, Gregory, and O’Connor studied the effect of mutations in these proteins on the course of the decoding process and the effectiveness of the action of the antibiotics that inhibit this process [44-46]. They established that a disruption of contact between uS4 and uS5 by deletion of certain uS5 protein residues resulted in a decrease in the accuracy of mRNA decoding and a change in the sensitivity to an antibiotic of the uS12 protein, an important participant in the regulation of this process. These effects are surely transmitted in an allosteric manner, while the uS4-uS5 tandem does not interact immediately with the uS12 protein or with the decoding center.
It is significant that the tracking of the GTPase activity of EF-Tu appeared to be a very convenient approach for searching for sources of allosteric signals in the ribosome. Thus, mutations in components of an intersubunit “bridge” B8 consisting of h8 and h14 of the 16S rRNA, on one hand, and the uL14 and uL19 proteins of the large ribosome subunit, on the other, negatively affected the GTPase activity of the EF-Tu [47, 48]. The distance from the mutated residues to the GTPase center was several tens of angstroms.
Decoding accuracy can be influenced not only by mutations in RNA and proteins of the small subunit, but also those in RNA molecules of the large subunit, both in a molecule of 5S rRNA [49] and 23S rRNA, for example, in its helices H92 or H89, which are remote from the decoding center [50].
ALLOSTERIC REGULATION OF THE PEPTIDYLTRANSFERASE CENTER OF THE
RIBOSOME
The peptidyltransferase center (PTC) of the ribosome, catalyzing the transpeptidation reaction, is composed predominantly of the nucleotide residues of rRNA of the large subunit. Therefore, ribosomes are considered as a class of ribozymes. These nucleotide residues, like in any other enzyme, provide optimal relative arrangement of the reaction substrates, exactly the substrates of the transpeptidation reaction: the amino group of the amino acid bound to the A-tRNA (A-substrate) and the carbonyl group of the C-terminal amino acid residue of the synthesized peptide (P-substrate), bound to tRNA, located in the P-site of the ribosome. Since the P-substrate, which is subjected to a nucleophilic attack by a free NH2-group of the A-substrate, has already been activated in the previous stages of translation, the main contribution to the catalytic function of the PTC is created by the entropy factor.
In the PTC of the ribosome, at least two layers of nucleotide residues forming it are commonly distinguished: nucleotides interacting immediately with the A- and P-substrates (the first layer, in E. coli ribosomes it is a set of residues A2451, U2585, and A2602), and an another one contacting the nucleotides of the first layer (the second, more spacious layer; in E. coli ribosomes, these are residues A2447, C2063, G2061, A2450, C2452, U2506, and also nucleotide residues G2553, G2251, and G2252 forming complementary pairs with the CCA-termini of the tRNAs in the A- and P-sites, respectively, ensuring their optimal conformation in the active center) [51-53]. It is clear that the interactions of the nucleotide residues from the first and second layers should not be considered as allosteric. However, 10-20 Å from the nucleotides of the first layer, the nucleotide and amino acid residues of rRNA and proteins of the large subunit, whose mutations affect the activity of both the PTC and inhibitors of the transpeptidation reaction are defined as allosteric (see, for example, [54]). The description of a detailed mechanism for the transmission of allosteric signals in these cases seems to be a realistic task, but to the best of our knowledge, it has not been completed.
It is well established that the activity of the PTC is regulated by allosteric signals received from the adjacent region of a ribosomal tunnel (RT). This important functional element of the ribosome serves to transport the polypeptide chains synthesized in the PTC to the surface of the large subunit of the ribosome, where the first stages of protein processing are carried out. In addition, binding sites of many inhibitors of protein synthesis are located in the RT, including several antibiotics used in clinical and veterinary medicine. In rather rare but very important cases, the growing peptide interacting with the elements of the RT walls (itself or in the presence of an antibiotic) completely inhibits the PTC and thereby stops the synthesis of the protein on the ribosome. This phenomenon has been considered in several reviews [55-58]. It is also important to note that the nucleotide residues of 23S rRNA located on the walls of the RT that participate in the transmission of the signal aimed to stop the synthesis of the polypeptides in the PTC were identified by Mankin et al. by means of directed mutagenesis. They were found to be nucleotides A2058, A2503, A2062, U1782, and U2609 [59]. Nucleotide residue A2058 is located about 20 Å from the nucleotides of the first layer of the PTC. It participates in the formation of the binding site of the antibiotic erythromycin with the RT. Via binding to this site, erythromycin transmits an allosteric signal to the PTC about the alteration in the position of the U2585 residue in relation to the other residues of the first and second layers of this center [60]. The stoppage of protein synthesis in this case is quite understandable, since it was previously shown that the distortion of the native conformation of the first layer of the PTC (including U2585) inevitably entails its inactivation [61].
Recently, using molecular dynamics simulations of a ribosome fragment containing the entire PTC and the whole RT, an opportunity for coordinated transmission of the allosteric signal through the formation of continuous stacking interactions for the base sequence A2058-A2059-m2A2503-G2061-A2062-C2063-U2585 was found by Makarov et al. [62]. For the complete formation of such a cascade, base A2058 was to separate from the stack of bases of helix H73, which was associated with stacking interaction with base A2057. At the end of the cascade, nucleotide residue A2062 occupied an unconventional position between G2061 and C2063, and the U2585 residue from the first layer of the PTC was in stacking interaction with C2063.
It is still unclear whether the PTC can exchange allosteric signals with other functional centers of the ribosome. Regarding this, as in many other cases, useful information was obtained by combining the method of directed mutagenesis of ribosome components with an assay of changes in the availability of rRNA nucleotide residues to various chemical agents (so-called “chemical probing”) induced by the certain mutation. Thus, mutation UU2492-3C in the part of the helix H89 adjacent to the PTC, which was already discussed above, affected its functional activity and, expectedly, induced a conformational rearrangement of the ensemble of nucleotide residues forming it. However, the distortion of the geometry of the H89 helix simultaneously noticeably affected the accessibility to modifying agents of the bases in the so-called sarcin-ricin loop of 23S rRNA (SRL) or H95 at the binding site of elongation factors far distant from the PTC [26].
Another example was found in the paper of Dinman and coworkers, who systematically studied allosteric phenomena in yeast ribosomes by means of both mutagenesis and “chemical probing”. They investigated the effects of mutations in the structural element of the ribosome that connects the small and large subunits in the 80S ribosome named “bridge B1b/c”. The eL11 protein of the central stalk of the 60S subunit of the ribosome takes part in its formation. It is located rather far from the PTC. Nevertheless, mutations in eL11 affect the PTC and the whole chain of 25S rRNA helices, along which, according to the authors, the allosteric signal is transmitted [63].
The peptidyltransferase reaction is also sensitive to breakdown of the ribosome structure: for example, the absence of 5S rRNA connecting the II and V domains of 23S rRNA ultimately disturbs its fulfilment, but the effect of the deletion of this rRNA can be partially compensated by the antibiotic connecting these domains [64].
ALLOSTERIC EFFECTS ACCOMPANYING THE TRANSLOCATION PROCESS
Deacylated upon the transpeptidation reaction, tRNA is gradually moved from the P-site to the E-site, and peptidyl-tRNA, respectively, from the A-site to the P-site through a series of intermediate conformational changes both in the tRNA itself and in the ribosome [65]. This process is called translocation.
Interaction of the L1 stalk with tRNAs. In the process of translocation, the large elements of the ribosome are shifted in relation to each other. Thus, upon the transition of tRNA to the hybrid P/E-state, the L1 stalk of the 50S subunit (named after the uL1 protein in its structure) is bent inward to the interface of the subunits, shifting by ~20 Å. This state is considered the “closed” conformation of the L1 stalk, in contrast to the “open” conformation, when tRNAs in the PTC are in the canonical A/A- and P/P-states. Accordingly, the equilibrium between these states is defined as a pre-translational. The most significant event in this stage of translation is the rotation of the small subunit by ~8 Å counterclockwise relative to the large subunit and lateral displacement of the “head” of the small subunit by ~20 Å relative to its “body”. The mRNA moves to one codon forward in the same direction [66]. The coherence of the motion of the L1 stalk and tRNAs was experimentally established by the smFRET method for several basic translocation steps: the transition of tRNA from the canonical P/P- to the hybrid P/E-state [67], the binding of the tRNA by the stalk [68], and the rotation of the subunits relative to each other [69, 70]. Thus, two global states can be distinguished, 1 and 2 (GS1 and GS2), between which the ribosome oscillates. In the first global state the tRNA molecules are in the canonical A/A- and P/P-sites, the L1 stalk is in the “open” conformation, and the subunits are not displaced relative to each other. The second global state is the next stage of translocation, namely, the shift of the CCA-termini of tRNAs in one position toward the exit of the tRNAs from the ribosome, the formation of hybrid states, the rotation of the L1 stalk to the “closed” position described above, which is required for retention of the tRNA in the P/E-state, the transition of the ribosome itself to the unlocked state, accompanied by “disconnection” of a set of intersubunit “bridges” [71], which is indispensable for relative intersubunit rotation (the GS2 state is depicted in Fig. 4).
Fig. 4. Ribosome in a pre-translocational state. tRNAs are situated in hybrid sites (from left to right): A/P, P/E. The distance in angstroms from the E-site of the large subunit to the GTPase center of EF-G is shown. The image style of the elements of the complex is the same as in Fig. 3. The source of the structure is PDB id: 4V7D.
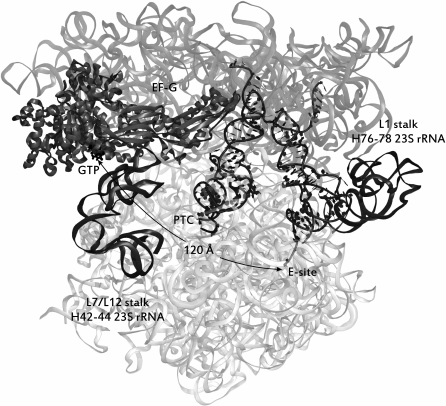
The efficiency of the translocation process depends critically on the presence of elongation factor G (EF-G) in the translating ribosome in a complex with GTP (see Fig. 1). It binds to the GTPase center of the large subunit. In this case, we encounter a record distance along which allosteric signals are transmitted. It has been found that the bacterial factor EF-G regulates the conformational state of the L1 stalk, which is distant from it by more than 170 Å: even in the absence of acylated tRNA in the A- (or A/P-) site, the formation of the EF-G complex with the ribosome displaces the equilibrium between the “open” and “closed” states of the L1 stalk towards the latter [72]. The mechanism of this allosteric connection is still to be established.
Apart from EF-G, aminoglycoside antibiotics can also interfere in the pre-translational equilibrium of the ribosome. These drugs bind near the decoding center and the A-site of the small subunit of the ribosome. It is known that in addition to malfunction the very decoding center, they inhibit the translocation process [73, 74]. By the same FRET method that detects intersubunit rotation, it was found that the aminoglycoside kanamycin froze the ribosome in a closed state, and viomycin froze the ribosome in an open state. The antibiotic neomycin, which binds to the H69 helix, located rather adjacent to the decoding center and forming an important intersubunit B2 bridge, also fixed the closed conformation of the ribosome at low concentrations [75].
There are other examples of how the position of the L1 stalk relative to the surface of the subunits can be altered in allosteric way due to disruption of intersubunit contacts. Thus, mutations that disturb the contact between proteins uS13 and uL5, composing certain ribosomal “bridge”, and thereby inducing rotational displacement of the small subunit, simultaneously stabilized the “closed” position of the L1 stalk. It is important that this connection can also be observed in vacant ribosomes in the absence of tRNA. Again, the allosteric link between the L1 stalk and the decoding center of the ribosome was established in relation with antibiotics. Thus, the aminoglycoside viomycin, binding near this center, stabilizes both the “closed” state of the L1 stalk and the rotational displacement of the ribosomal subunits relative to each other both in the acting and in the vacant ribosome [76].
In addition to the dynamic equilibrium inside the so-called pre-translational state (PRE), the ribosome can spontaneously fluctuate into the terminal state of translocation – the post-translocational state designated as POST in the literature, where tRNAs completely occupy the canonical P/P- and E/E-sites, and the ribosome subunits return to an unshifted position. In the absence of EF-G, the rate of this transition is insufficient to provide the normal functioning of the ribosome: the PRE state is more energetically favorable than the POST state (if EF-G is removed from the system, the ribosome spontaneously returns to the PRE state). Several aminoglycosides also affect this balance: for example, neomycin increases the rate of transition from PRE to POST by 3-fold and streptomycin by 14-fold, whereas tetracycline reduces it 4-fold [77]. Sparsomycin binding in the PTC also strongly shifts this equilibrium towards the POST state, resulting in a spontaneous factor-less translocation [78]. Similarly, spontaneous translocation is triggered by several antibiotics with affinity for the A-site of the large subunit [79].
Like the transition between the global states of the pre-translocational complex, the conversion to the post-translocational state evidently affects the main active centers of the ribosome: the PTC, the decoding center, and the A-site of the large subunit.
Interconnection of EF-G and the pre-translocational state of the ribosome. It appeared that the choice of the pre-translocational state of the ribosome influenced the catalytic activity of EF-G: until the ribosome is not in the GS2 state with the hybrid sites of tRNAs, the hydrolysis rate of GTP required for release of EF-G from the ribosome remained extremely low. It was previously known that the binding of factor G to a vacant ribosome faintly catalyzed the hydrolysis of GTP: to stimulate it, a deacylated tRNA in the P-site, capable of transferring its CCA-terminus to the E-site, was required [80], although the factor can occupy the ribosome in any of the pre-translational states and initiate translocation starting from any of them [81]. Nucleotide residue mutation C2394G, which consists of substitution of the base located opposite the 3′-terminal the A76 tRNA residue, when it is located in the E-site, decreased the affinity of the deacylated tRNA for this site. As a result of this mutation, the affinity of the tRNA for the E-site of the ribosome was ultimately reduced not only in the spontaneous binding of tRNAfMet in the presence of mRNA and fMetPhe-tRNAPhe in the P-site, but the mutation also inhibited the post-translocational retention of the deacylated tRNA in the E-site and the formation of the hybrid P/E-state of the pre-translocational complex. Ribosomes with such a mutation, where the formation of GS2 was hindered, significantly faintly catalyzed the hydrolysis of the GTP of EF-G and, in addition, the affinity of the factor to these ribosomes was reduced [82]. Besides stabilization of the GS2 state of the ribosome due to the catalytic function of the elongation factor, it controls the concordance of the translocation: the GTPase center of the EF-G is sensitive to the state of the ribosome before translocation, being an allosteric sensor. It is difficult to determine from experimental data which of its binding sites on the ribosome is responsible for receiving the allosteric signal, or if this role is played by their combination activating the hydrolysis of GTP in mutual coordination, because, as discussed above, pre-translocational rearrangements in the ribosome itself also occur in concert.
In another report from the same group, it was shown that deletion of a fragment of helix H38 of 23S rRNA forming the intersubunit bridge B1a by interaction with the protein S13 of the small subunit only partially reduced the efficiency of the GTP hydrolysis activation by deacylated tRNA located in the P/E-site. It can therefore be expected that signal transmission occurs in this case along several pathways, which duplicate and complement each other [83].
The L7/L12 stalk, consisting of three helices of 23S rRNA (H42-44) and proteins uL10 and uL11, is doubtless involved in the formation of the correct state of the ribosome, which activates the hydrolysis of the GTP by the GTPase EF-G. Its elongation by one pair at the very bottom of helix H42 via insertion of an additional Watson–Crick base pair C1030/G1124 reduced the GTPase activity of the EF-G. It is noteworthy that upon this mutation alterations in the accessibility to the modifying agents occurred in many rRNA bases rather remote from L7/L12 as a consequence of significant rearrangements in the structure of the ribosome [84].
Additional evidence for the existence of allosteric coherency in the ribosome. We mention here that to date there have been accumulated many examples of the propagation of the effects of mutations in rRNA over long distances along the ribosome that are detected by means of chemical probing.
The A960C mutation affects several bases of the PTC, exposing the P-tRNA-binding bases Gm2251 and G2252 and shielding the nucleotide residues of A2518 and U2249 at the same time [85].
The mutations A2531U and A2531C, disturbing the tertiary interaction of the vertices of helices H95 and H91, altered the modification profile of several GAC bases (GTPase-associated center, helices H42-44, forming the stalk required for GTP-dependent translation factor activity), including residue U1033 forming a tertiary contact with helix H97, as well as two bases of the PTC – U2585 and A2572 [25].
The nucleotide residue A2518 is also deprotected for modification by dimethyl sulfate in the ribosome when the integrity of SRL (helix H95) is disrupted, in addition to a number of nucleotides of helices H89, H90-92, H39 (including A960), H80, H72, certain residues of the PTC, and of the stem of H42, participating in the organization of the GAC [86].
In summary, data about changes in the degree of modification of the E. coli bases presented above suggest a total linkage between the SRL, a three-piece plug of helices H90-92, the nucleotide residue A2518, which is situated in a stack of four nucleotide residues together with A1127-A1129 tethering helices H90 and H89, the GAC, as well as PTC and H39 leading towards the E-site from the PTC.
In the yeast ribosome, eL3 protein mutations increasing the affinity of A- and P-tRNA to the ribosome induce certain changes in the helices distant by more than 100 Å: they increase the availability of a significant number of the H85 residues and entail the hyperprotection of a number of bases of helices H89 (A2901, A2920), H91-92 (A2948, A2958, A2966, G2978 (G2554 in the numeration accepted for E. coli ribosomes)), H93 (A3015), and U3009 (U2584 according to the numeration for E. coli ribosomes), i.e. located near the PTC [87].
Profound dynamic coherency of the ribosomal residues enables Nature to create antibiotic-resistant strains of bacteria employing mutations remote from their binding sites to the ribosome. In the cases when small distances are implicated (see, for example, [54]), it is possible to trace a succession of disruptions of previous bonds and the formation of new ones in the sequence of rRNA or protein residues from the mutation point to the affinity region to the antibiotic. The successive changes revealed in this way could serve as a model for understanding allostery at larger distances.
The single deletion ΔSer145 in uL3 protein of Staphylococcus aureus provides resistance to linezolid, the binding site of which is more than 20 Å apart from the mutation point. Reduction of one of the protein loops resulted in displacement of helix of H90 of the 23S rRNA from the PTC by about 2 Å, which induced rearrangements in the linezolid-binding site. The largest conformational changes in the comparison of structures obtained by cryoelectron microscopy were observed in residues 2504-2506, G2576, G2581, and U2584 of the 23S rRNA, and mutations along these nucleotides can provide resistance to this antibiotic [88].
Another antibiotic, anisomycin, normally binding with its aromatic fragment in the A-gap between A2451 and C2452 according to the numbering of E. coli, lost its affinity for the H. marismortui ribosomes as a result of mutations G2581A and G2576U located at a distance from the binding site of at least 12 and 7 Å, respectively. The first mutation resulted in the formation of a new hydrogen bond of A2581 with G2576 (the site of the second mutation) upon disruption of the bond with U2506, which developed into bending of the whole block of bases 2504-2507. U2504 is further connected to A2453, and this displacement eventually narrows the so-called A-gap. The second of the mentioned mutations, G2576U, also destroys one of the bonds of residues 2504-2507 (namely G2576 with G2505), resulting in even greater bending of this block, similarly to that in the previous case [89].
The data discussed in this review clearly demonstrate that the ribosome is a flexible and adaptive molecular machine. At the heart of its action are well-coordinated mechanisms whose details are still to be deciphered.
Modern experimental structural methods do not yet reveal micromovements at different stages of the work of such a complicated macromolecular complex as the ribosome, although clear progress has been observed for simpler biological structures [90].
In general, the aforementioned empirical evidences indicated two features of the ribosome as an allosteric system:
1. Exchange of allosteric signals is observed at various stages of elongation for different sites of the macromolecular complex; for the realization of a whole set of allosteric transmissions, the ribosome is to either be divided into several allosteric “modules”, or to occupy a set of near-equilibrium conformations, and in the process of elongation to switch within this set. A combination of these variants is also possible when some of the conditional “modules” are interdependent and form their discrete set of near-equilibrium states, where only certain combinations of states of individual “modules” are permitted.
2. Allosteric information propagates along the ribosome for distances that are large by molecular scale. Thus, the E-site of the large ribosome subunit, and the EF-G factor binding sites, GTPase activity of which requires a certain pre-translocational state, are separated by more than 100 Å, which is comparable to the linear dimensions of the large ribosomal subunit. The coordinated mechanism of translocation can be controlled by small effectors – antibiotics – both from the decoding and from the peptidyltransferase centers, which in itself indicates high coherency of the translocation process. Deep coordination of translocation is also confirmed by numerous smFRET experiments, which revealed large-scale displacements of macro-elements or large ligands (tRNAs) of the ribosome. Regulation of the GTPase activity of another elongation factor, EF-Tu, also implicates at least a significant region of the small subunit of the ribosome extending to 70-80 Å and propagates up to the intersubunit bridge region, and this is also affected by rRNA mutations of the large ribosomal subunit. As for a negative allosteric connection between the A- and E-sites, if it indeed exists, the large subunit alone is not sufficient for its realization; therefore, the coordinated operation of the entire translational mechanism is required. Changes in the degree of nucleotide modifications are also able to occur at tens of angstroms from a point mutation. Therefore, if independent allosteric modules exist in the ribosome, their dimensions are comparable with the dimensions of the complex itself. Besides, the stable propagation of the signals for such distances requires extended rigid RNA elements.
The main sensors of allosteric switching that are available for identifications from the whole array of experimental data are:
As for the mechanism of propagation of allosteric signals, the most obvious candidate for signal transmission over long distances is stacking interactions between heterocyclic rRNA bases. On one hand, they stabilize long and strong helices as rigid elements, potential signal conductors. On the other hand, nucleotide bases that can form or leave stacking interaction with extended rRNA elements, for example, A2062 and U2585 in the PTC, were revealed by molecular dynamics simulations, and it is not surprising that mutations at these bases are critical for the ability of the PTC to release the peptide from tRNA in the P-site [51]. However, hypothetical “switches” are also able to rely on other interactions – hydrogen bonds, salt bridges, etc.
How far are the allosteric pathways extended and how much are they branched and interconnected? If we consider the relationship of the allosteric centers listed above, then, for example, the antagonism of A- and E-sites is enhanced by the elongation of the peptide chain by more than three amino acid residues, indicating an allosteric connection between the A-site and the upper part of the RT [37], in which, in turn, there is a potential “switch” that regulates the activation of the PTC.
Obviously, the large dimensions of a minimal allosteric mechanism do not enable in the foreseeable future to investigate the details of switching between the states experimentally, for example, using NMR or other spectral methods. However, the performance of modern supercomputers already enables calculating the full-atom model of the ribosome by the molecular dynamics method in nano- and even microsecond time intervals (the record calculation was 1.2 ms). Of course, such times do not cover completely any of the stages of the elongation cycle. To reproduce the operation of a ribosome, various methods of non-equilibrium dynamics are often employed, such as aligning the coordinates of the selected system into correspondence with the low-resolution electron density of the ribosome in the functional state of interest. The electron density required for this technique is obtained by means of cryoelectron microscopy of ribosomes with analysis of individual images (for a review, see [91]). In addition, even equilibrium molecular dynamics could be an instrument for comparative analysis of the states of ribosome complexes with ligands (antibiotics, tRNA, elongation factors) and vacant ribosomes, wild-type ribosomes and those containing mutations. It is likely that it will help us to distinguish the conformations and the degrees of motion of individual atoms and residues, and at least partially answer the questions raised above. Despite all the difficulties, the solution of such problems is extremely important, since it would enable not only deeper understanding of the mechanism of action of the ribosome, but also the development of fundamentally new approaches to rational design of new allosteric ribosomal antibiotics.
Acknowledgments
This work was supported by the Russian Science Foundation (projects Nos. 15-04-00006, section devoted to allosteric effects accompanying the translocation process; and 14-24-00061-P, section devoted to allosteric effects observed upon interaction of the ribosome with tRNAs) and by the Russian Foundation for Basic Research (project No. 16-04-00709-a, section devoted to a regulation of the peptidyltransferase center of the ribosome).
REFERENCES
1.Steitz, T. M. (2008) A structural understanding of
the dynamic ribosome machine, Nat. Rev. Mol. Cell Biol.,
9, 242-253.
2.Voorhees, R. M., and Ramakrishnan, V. (2013)
Structural basis of the translational elongation cycle, Annu. Rev.
Biochem., 82, 203-236.
3.Changeux, J. P. (2013) 50 years of allosteric
interactions: the twists and turns of the models, Nat. Rev. Mol.
Cell. Biol., 14, 819-829.
4.Cornish-Bowden, A. (2014) Understanding allosteric
and cooperative interactions in enzymes, FEBS J., 281,
621-632.
5.Kleckner, I. R., Gollnick, P., and Foster, M. P.
(2012) Mechanisms of allosteric gene regulation by NMR quantification
of µs-ms protein dynamics, J. Mol. Biol., 415,
372-381.
6.Stower, H. (2013) Gene regulation: allosteric
effects, Nat. Rev. Genet., 14, 238.
7.Cecchini, M., and Changeux, J. P. (2015) The
nicotinic acetylcholine receptor and its prokaryotic homologues:
structure, conformational transitions and allosteric modulation,
Neuropharmacology, 96, 137-149.
8.Kim, S., Brostromer, E., Xing, D., Jin, J., Chong,
S., Ge, H., Wang, S., Gu, Ch., Yang, L., Gao, Y. Q., Su, X., Sun, Y.,
and Xie, X. S. (2013) Probing allostery through DNA, Science,
339, 816-819.
9.Chen, I. (2013) Allostery through DNA, Nat.
Struct. Mol. Biol., 20, 410.
10.Soukup, G. A. (2004) Aptamers meet allostery,
Chem. Biol., 11, 1031-1032.
11.Goodey, N. M., and Benkovic, S. J. (2008)
Allosteric regulation and catalysis emerge via a common route, Nat.
Chem. Biol., 4, 474-482.
12.Monod, J., Wyman, J., and Changeux, J.-P. (1965)
On the nature of allosteric transitions: a plausible model, J. Mol.
Biol., 12, 88-118.
13.Koshland, D. E., Nemethy, G., and Filmer, D.
(1966) Comparison of experimental binding data and theoretical models
in proteins containing subunits, Biochemistry, 5,
365-385.
14.Saibil, H. R., Fenton, W. A., Clare, D. K., and
Horwich, A. L. (2013) Structure and allostery of the chaperonin GroEL,
J. Mol. Biol., 425, 1476-1487.
15.Hilser, V. J., Wrabl, J. O., and Motlagh, H. N.
(2012) Structural and energetic basis of allostery, Ann. Rev.
Biophys., 41, 585-609.
16.Hol, T. C., Cox, M. B., Bryant, H. U., and
Draper, M. W. (1997) Selective estrogen receptor modulators and
postmenopausal women’s health, J. Womens Health, 6,
523-531.
17.Katzenellenbogen, B. S., Montano, M. M., Ekena,
K., Herman, M. E., and McInerney, E. M. (1997) Antiestrogens:
mechanisms of action and resistance in breast cancer, Breast Cancer
Res. Treat., 44, 23-38, doi: 10.1023/A:1005835428423.
18.Gunasekaran, K., Ma, B., and Nussinov, R. (2004)
Is allostery an intrinsic property of all dynamic proteins?
Proteins, 57, 433-443.
19.Feher, V. A., Durrant, J., Van Wart, A. T., and
Amaro, R. E. (2014) Computational approaches to mapping allosteric
pathways, Curr. Opin. Struct. Biol., 25, 98-103.
20.Holliday, M. J., Camilloni, C., Armstrong, G. S.,
Vendruscolo, M., and Eisenmesser, E. Z. (2017) Networks of dynamic
allostery regulate enzyme function, Structure, 25,
276-286.
21.Daily, M. D., and Gray, J. J. (2009) Allosteric
communication occurs via networks of tertiary and quaternary motions in
proteins, PLoS Comput. Biol., 5, e1000293.
22.Tsai, C. J., Del Sol, A., and Nussinov, R. (2008)
Allostery: absence of a change in shape does not imply that allostery
is not at play, J. Mol. Biol., 378, 1-11.
23.Cooper, A., and Dryden, D. T. F. (1984) Allostery
without conformational change. A plausible model, Eur. Biophys.
J., 11, 103-109.
24.Panjkovich, A., and Daura, X. (2012) Exploiting
protein flexibility to predict the location of allosteric sites,
BMC Bioinformatics, 13, 273.
25.Chan, Y. L., Dresios, J., and Wool, I. G. (2006)
A pathway for the transmission of allosteric signals in the ribosome
through a network of RNA tertiary interactions, J. Mol. Biol.,
355, 1014-1025.
26.Burakovsky, D. E., Sergiev, P. V., Steblyanko, M.
A., Konevega, A. L., Bogdanov, A. A., and Dontsova, O. A. (2011) The
structure of helix 89 of 23S rRNA is important for peptidyl transferase
function of Escherichia coli ribosome, FEBS Lett.,
585, 3073-3078.
27.Rheinberger, H. J., Sternbach, H., and Nierhaus,
K. H. (1986) Codon–anticodon interaction at the ribosomal E site,
J. Biol. Chem., 261, 9140-9143.
28.Gnirke, A., Geigenmuller, U., Rheinberger, H. J.,
and Nierhaus, K. H. (1989) The allosteric three-site model for the
ribosomal elongation cycle. Analysis with a heteropolymeric mRNA, J.
Biol. Chem., 264, 7291-7301.
29.Nierhaus, K. H. (1990) The allosteric three-site
model for the ribosomal elongation cycle: features and future,
Biochemistry, 29, 4997-5008.
30.Marquez, V., Wilson, D. N., Tate, W. P.,
Triana-Alonso, F., and Nierhaus, K. H. (2004) Maintaining the ribosomal
reading frame: the influence of the E site during translational
regulation of release factor 2, Cell, 118, 45-55.
31.Geigenmuller, U., and Nierhaus, K. H. (1990)
Significance of the third tRNA binding site, the E site, on E.
coli ribosomes for the accuracy of translation: an occupied E site
prevents the binding of non-cognate aminoacyl-tRNA to the A site,
EMBO J., 9, 4527-4533.
32.Di Giacco, V., Marquez, V., Qin, Y., Pech, M.,
Triana-Alonso, F. J., Wilson, D. N., and Nierhaus, K. H. (2008)
Shine–Dalgarno interaction prevents incorporation of noncognate
amino acids at the codon following the AUG, Proc. Natl. Acad. Sci.
USA, 105, 10715-10720.
33.Nierhaus, K. H. (2006) Decoding errors and the
involvement of the E-site, Biochimie, 88, 1013-1019.
34.Leger, M., Dulude, D., Steinberg, S. V., and
Brakier-Gingras, L. (2007) The three transfer RNAs occupying the A, P,
and E sites on the ribosome are involved in viral programmed –1
ribosomal frameshift, Nucleic Acids Res., 35,
5581-5592.
35.Liao, P. Y., Gupta, P., Petrov, A. N., Dinman, J.
D., and Lee, K. H. (2008) A new kinetic model reveals the synergistic
effect of E-, P- and A-sites on –1 ribosomal frameshifting,
Nucleic Acids Res., 36, 2619-2629.
36.Semenkov, Y. P., Rodnina, M. V., and Wintermeyer,
W. (1996) The “allosteric three-site model” of elongation
cannot be confirmed in a well-defined ribosome system from
Escherichia coli, Proc. Natl. Acad. Sci. USA, 93,
12183-12188.
37.Chen, C., Stevens, B., Kaur, J., Smilansky, Z.,
Cooperma, B. S., and Goldman, Y. E. (2011) Allosteric vs. spontaneous
exit-site (E-site) tRNA dissociation early in protein synthesis,
Proc. Natl. Acad. Sci. USA, 108, 16980-16985.
38.Schmeig, T. M., Moore, P. B., and Steitz, T. A.
(2003) Structures of deacylated tRNA mimics bound to the E site of the
large ribosomal subunit, RNA, 9, 1345-1352.
39.Rakauskaite, R., and Dinman, J. D. (2008) rRNA
mutants in the yeast peptidyltransferase center reveal allosteric
information networks and mechanisms of drug resistance, Nucleic
Acids Res., 36, 1497-1507.
40.Schmeing, T. M., Voorhees, R. M., Kelley, A. C.,
Yong-Gui, G., Murphy, F. V., 4th, Weir, J. R., and Ramakrishnan, V.
(2009) The crystal structure of the ribosome bound to EF-Tu and
aminoacyl-tRNA, Science, 326, 688-694.
41.Schmeing, T. M., Voorhees, R. M., Kelley, A. C.,
and Ramakrishnan, V. (2011) How mutations in tRNA distant from the
anticodon affect the fidelity of decoding, Nat. Struct. Mol.
Biol., 18, 432-436.
42.Hirsh, D. (1970) Tryptophan tRNA of
Escherichia coli, Nature, 228, 57.
43.James, M. O., Frank, V., Murphy, I. V., Michael,
J. T., and Ramakrishnan, V. (2002) Selection of tRNA by the ribosome
requires a transition from an open to a closed form, Cell,
111, 721-732.
44.Lodmell, J. S., and Dahlberg, A. E. (1997) A
conformational switch in Escherichia coli 16S ribosomal RNA
during decoding of messenger RNA, Science, 277,
1262-1267.
45.Kamath, D., Gregory, S. T., and O’Connor,
M. (2017) Selection of tRNA by the ribosome requires a transition from
an open to a closed form, Antimicrob. Agents Chemother.,
61, e01186-16.
46.Kamath, D., Allgeyer, B. B., Gregory, S. T.,
Bielski, M. C., Roelofsz, D. M., Sabapathypillai, S. L., Vaid, N., and
O’Connor, M. (2017) The C-terminus of ribosomal protein uS4
contributes to small ribosomal subunit biogenesis and the fidelity of
translation, Biochimie, 138, 194-201.
47.McClory, S. P., Leisring, J. M., Qin, D., and
Fredrick, K. (2010) Missense suppressor mutations in 16S rRNA reveal
the importance of helices h8 and h14 in aminoacyl-tRNA selection,
RNA, 16, 1925-1934.
48.Fagan, C. E., Dunkle, J. A., Maehigashi, T.,
Dang, M. N., Devaraj, A., Miles, S. J., Daoming, Q., Fredrick, K., and
Dunham, C. M. (2013) Reorganization of an intersubunit bridge induced
by disparate 16S ribosomal ambiguity mutations mimics an EF-Tu-bound
state, Proc. Natl. Acad. Sci. USA, 110, 9716-9721.
49.Smith, M. W., Meskauskas, A., Wang, P., Sergiev,
P. V., and Dinman, J. D. (2011) Saturation mutagenesis of 5S rRNA in
Saccharomyces cerevisiae, Mol. Cell. Biol., 21,
8264-8275.
50.O’Connor, M., and Dahlberg, A. E. (1995)
The involvement of two distinct regions of 23S ribosomal RNA in tRNA
selection, J. Mol. Biol., 254, 838-847.
51.Polacek, N., Gomez, M. J., Ito, K., Xiong, L.,
Nakamura, Y., and Mankin, A. (2003) The critical role of the
universally conserved A2602 of 23S ribosomal RNA in the release of the
nascent peptide during translation termination, Mol. Cell,
11, 103-112.
52.Youngman, E. M., Brunelle, J. L., Kochaniak, A.
B., and Green, R. (2004) The active site of the ribosome is composed of
two layers of conserved nucleotides with distinct roles in peptide bond
formation and peptide release, Cell, 117, 589-599.
53.Simonovic, M., and Steitz, T. A (2008)
Cross-crystal averaging reveals that the structure of the
peptidyl-transferase center is the same in the 70S ribosome and the 50S
subunit, Proc. Natl. Acad. Sci. USA, 105, 500-505.
54.Long, K. S., and Vester, B. (2012) Resistance to
linezolid caused by modifications at its binding site on the ribosome,
Antimicrob. Agents Chemother., 56, 603-612.
55.Tenson, T., and Ehrenberg, M. (2002) Regulatory
nascent peptides in the ribosomal tunnel, Cell, 108,
591-594.
56.Bogdanov, A. A., Sumbatyan, N. V., Shishkina, A.
V., Karpenko, V. V., and Korshunova, G. A. (2010) Ribosomal tunnel and
translation regulation, Biochemistry (Moscow), 75,
1501.
57.Vázquez-Laslop, N., and Mankin, A. S.
(2014) Triggering peptide-dependent translation arrest by small
molecules: ribosome stalling modulated by antibiotics, in Regulatory
Nascent Polypeptides (Ito, K., ed.), Springer, New York.
58.Ito, K., and Chiba, S. (2013) Arrest peptides:
cis-acting modulators of translation, Annu. Rev.
Biochem., 82, 171-202.
59.Vazquez-Laslop, N., Ramu, H., and Mankin, A.
(2011) Nascent peptide-mediated ribosome stalling promoted by
antibiotics, in Ribosomes: Structure, Function, and Dynamics,
Section V, pp. 377-392.
60.Sothiselvam, S., Liu, B., Han, W., Ramu, H.,
Klepacki, D., Atkinson, G. C., Brauer A., Remm, M., Tenson, T.,
Schulten, K., Vazquez-Laslop, N., and Mankin, A. S. (2014) Macrolide
antibiotics allosterically predispose the ribosome for translation
arrest, Proc. Natl. Acad. Sci. USA, 111, 9804-9809.
61.Seidelt, B., Innis, C. A., Wilson, D. N.,
Gartmann, M., Armache, J.-P., Villa, E., Leonardo G. T., Becker, T.,
Mielke, T., Schulten, K., Steitz, T. A., and Beckmann, R. (2009)
Structural insight into nascent polypeptide chain-mediated
translational stalling, Science, 326, 1412-1415.
62.Makarov, G. I., Golovin, A. V., Sumbatyan, N. V.,
and Bogdanov, A. A. (2015) Molecular dynamics investigation of a
mechanism of allosteric signal transmission in ribosomes,
Biochemistry (Moscow), 80, 1047-1056.
63.Rhodin, M. H. J., and Dinman, J. D. (2011) An
extensive network of information flow through the B1b/c intersubunit
bridge of the yeast ribosome, PLoS One, 6, e20048.
64.Khaitovich, P., and Mankin, A. S. (1999) Effect
of antibiotics on large ribosomal subunit assembly reveals possible
function of 5S rRNA, J. Mol. Biol., 291,
1025-1034.
65.Fischer, N., Konevega, A. L., Wintermeyer, W.,
Rodnina, M. V., and Stark, H. (2010) Ribosome dynamics and tRNA
movement by time-resolved electron cryomicroscopy, Nature,
466, 329-333.
66.Frank, J., and Agrawal, R. K. (2000) A
ratchet-like inter-subunit reorganization of the ribosome during
translocation, Nature, 406, 318-322.
67.Blanchard, S. C., Kim, H. D., Gonzalez, R. L.,
Puglisi, J. D., and Chu, S. (2004) tRNA dynamics on the ribosome during
translation, Proc. Natl. Acad. Sci. USA, 101,
12893-12898.
68.Fei, J., Kosuri, P., MacDougall, D. D., and
Gonzalez, R. L. (2008) Coupling of ribosomal L1 stalk and tRNA dynamics
during translation elongation, Mol. Cell, 30,
348-359.
69.Cornish, P. V., Ermolenko, D. N., Noller, H. F.,
and Ha, T. (2008) Spontaneous intersubunit rotation in single
ribosomes, Mol. Cell, 30, 578-588.
70.Cornish, P. V., Ermolenko, D. N., Staple, D. W.,
Hoang, L., Hickerson, R. P., Noller, H. F., and Ha, T. (2009) Following
movement of the L1 stalk between three functional states in single
ribosomes, Proc. Natl. Acad. Sci. USA, 106,
2571-2576.
71.Liu, Q., and Fredrick, K. (2013) Contribution of
intersubunit bridges to the energy barrier of ribosomal translocation,
Nucleic Acids Res., 41, 565-574.
72.Fei, J., Bronson, J. E., Hofman, J. M., Srinivas,
R. L., Wiggins, C. H., and Gonzalez, R. L. (2009) Allosteric
collaboration between elongation factor G and the ribosomal L1 stalk
directs tRNA movements during translation, Proc. Natl. Acad. Sci.
USA, 106, 15702-15707.
73.Shoji, S., Walker, S. E., and Fredrick, K. (2009)
Ribosomal translocation: one step closer to the molecular mechanism,
ACS Chem. Biol., 4, 93-107.
74.Tsai, A., Uemura, S., Johansson, M., Puglisi, E.
V., Marshall, R. A., Aitken, C. E., Korlach, J., Ehrenberg, M., and
Puglisi, J. D. (2013) The impact of aminoglycosides on the dynamics of
translation elongation, Cell Rep., 3, 497-508.
75.Wang, L., Pulk, A., Wasserman, M. R., Feldman, M.
B., Altman, R. B., Cate, J. H. D., and Blanchard, S. C. (2012)
Allosteric control of the ribosome by small-molecule antibiotics,
Nat. Struct. Mol. Biol., 19, 957-963.
76.Ning, W., Fei, J., and Gonzalez, R. L. (2014) The
ribosome uses cooperative conformational changes to maximize and
regulate the efficiency of translation, Proc. Natl. Acad. Sci.
USA, 111, 12073-12078.
77.Shoji, S., Walker, S. E., and Fredrick, K. (2006)
Reverse translocation of tRNA in the ribosome, Mol. Cell,
24, 931-942.
78.Fredrick, K., and Noller, H. F. (2003) Catalysis
of ribosomal translocation by sparsomycin, Science, 300,
1159-1162.
79.Ermolenko, D. N., Cornish, P. V., Ha, T., and
Noller, H. F. (2013) Antibiotics that bind to the A site of the large
ribosomal subunit can induce mRNA translocation, RNA, 19,
158-166.
80.Zavialov, A. V., and Ehrenberg, M. (2003)
Peptidyl-tRNA regulates the GTPase activity of translation factors,
Cell, 114, 113-122.
81.Chen, C., Stevens, B., Kaur, J., Cabral, D., Liu,
H., Wang, Y., Zhang, H., Rosenblum, G., Smilansky, Z., Goldman, Y. E.,
and Cooperman, B. S. (2011) Single molecule fluorescence measurements
of ribosomal translocation dynamics, Mol. Cell, 42,
367-377.
82.Sergiev, P. V., Lesnyak, D. V., Kiparisov, S. V.,
Burakovsky, D. E., Leonov, A. A., Bogdanov, A. A., Brimacombe, R., and
Dontsova, O. A. (2005) Function of the ribosomal E-site: a mutagenesis
study, Nucleic Acids Res., 33, 6048-6056.
83.Sergiev, P. V., Kiparisov, S. V., Burakovsky, D.
E., Lesnyak, D. V., Leonov, A. A., Bogdanov, A. A., and Dontsova, O. A.
(2005) The conserved A-site finger of the 23S rRNA: just one of the
intersubunit bridges or a part of the allosteric communication pathway,
J. Mol. Biol., 353, 116-123.
84.Sergiev, P. V., Lesnyak, D. V., Burakovsky, D.
E., Kiparisov, S. V., Leonov, A. A., Bogdanov, A. A., Brimacombe, R.,
and Dontsova, O. A. (2005) Alteration in location of a conserved
GTPase-associated center of the ribosome induced by mutagenesis
influences the structure of peptidyltransferase center and activity of
elongation factor G, J. Biol. Chem., 280,
31882-31889.
85.Sergiev, P. V., Bogdanov, A. A., Dahlberg, A. E.,
and Dontsova, O. A. (2000) Mutations at position A960 of E. coli
23S ribosomal RNA influence the structure of 5S ribosomal RNA and the
peptidyltransferase region of 23S ribosomal RNA, J. Mol. Biol.,
299, 379-389.
86.Lancaster, L., Lambert, N. J., Maklan, E. J.,
Horan, L. H., and Noller, H. F. (2008) The sarcin-ricin loop of 23S
rRNA is essential for assembly of the functional core of the 50S
ribosomal subunit, RNA, 14, 1999-2012.
87.Petrov, A., Meskauskas, A., and Dinman, J. D.
(2004) Ribosomal protein L3: influence on ribosome structure and
function, RNA Biol., 1, 59-65.
88.Belousoff, M. J., Eyal, Z., Radjainia, M., Ahmed,
T., Bamert, R. S., Matzov, D., Bashan, A., Zimmerman, E., Mishra, S.,
Cameron, D., Elmlund, H., Peleg, A. Y., Bhushan, S., Lithgow, T., and
Yonath, A. (2017) Structural basis for linezolid binding site
rearrangement in the Staphylococcus aureus ribosome,
mBio, 8, e00395-17.
89.Blaha, G., Gurel, G., Schroeder, S. J., Moore, P.
B., and Steitz, T. A. (2008) Mutations outside the anisomycin binding
site can make ribosomes drug-resistant, J. Mol. Biol.,
379, 505-519.
90.Stagno, J. R., Liu, Y., Bhandari, Y. R., Conrad,
C. E., Panja, S., Swain, M., Fan, L., Nelson, G., Li, C., Wendel, D.
R., White, T. A., Coe, J. D., Wiedorn, M. O., Knoska, J., Oberthuer,
D., Tuckey, R. A., Yu, P., Dyba, M., Tarasov, S. G., Weierstall, U.,
Grant, T. D., Schwieters, C. D., Zhang, J., Ferre-D’Amare, A. R.,
Fromme, P., Draper, D. E., Liang, M., Hunter, M. S., Boutet, S., Tan,
K., Zuo, X., Ji, X., Barty, A., Zatsepin, N. A., Chapman, H. N.,
Spence, J. C., Woodson, S. A., and Wang, Y. X. (2017) Structures of
riboswitch RNA reaction states by mix-and-inject XFEL serial
crystallography, Nature, 541, 242-246.
91.Makarov, G. I., Makarova, T. M., Sumbatyan, N.
V., and Bogdanov, A. A. (2016) Investigation of ribosomes using
molecular dynamics simulation methods, Biochemistry (Moscow),
81, 1579-1588.
92.Achenbach, J., and Nierhaus, K. H. (2013)
Translocation at work, Nat. Struct. Mol. Biol.,
20, 1019-1022.