REVIEW: Properties of Bacterial and Archaeal Branched-Chain Amino Acid Aminotransferases
E. Yu. Bezsudnova1*, K. M. Boyko1,2, and V. O. Popov1,2
1Bach Institute of Biochemistry, The Federal Research Centre “Fundamentals of Biotechnology”, Russian Academy of Sciences, 119071 Moscow, Russia; E-mail: eubez@inbi.ras.ru2National Research Center “Kurchatov Institute”, 123098 Moscow, Russia
* To whom correspondence should be addressed.
Received June 16, 2017; Revision received July 5, 2017
Branched-chain amino acid aminotransferases (BCATs) catalyze reversible stereoselective transamination of branched-chain amino acids (BCAAs) L-leucine, L-isoleucine, and L-valine. BCATs are the key enzymes of BCAA metabolism in all organisms. The catalysis proceeds through the ping-pong mechanism with the assistance of the cofactor pyridoxal 5′-phosphate (PLP). BCATs differ from other (S)-selective transaminases (TAs) in 3D-structure and organization of the PLP-binding domain. Unlike other (S)-selective TAs, BCATs belong to the PLP fold type IV and are characterized by the proton transfer on the re-face of PLP, in contrast to the si-specificity of proton transfer in fold type I (S)-selective TAs. Moreover, BCATs are the only (S)-selective enzymes within fold type IV TAs. Dual substrate recognition in BCATs is implemented via the “lock and key” mechanism without side-chain rearrangements of the active site residues. Another feature of the active site organization in BCATs is the binding of the substrate α-COOH group on the P-side of the active site near the PLP phosphate group. Close localization of two charged groups seems to increase the effectiveness of external aldimine formation in BCAT catalysis. In this review, the structure-function features and the substrate specificity of bacterial and archaeal BCATs are analyzed. These BCATs differ from eukaryotic ones in the wide substrate specificity, optimal temperature, and reactivity toward pyruvate as the second substrate. The prospects of biotechnological application of BCATs in stereoselective synthesis are discussed.
KEY WORDS: branched-chain amino acid aminotransferases (BCATs), pyridoxal 5′-phosphate catalysis, bacterial and archaeal enzymes, substrate specificity, 3D structure, dual substrate recognition, characteristic sequence motifsDOI: 10.1134/S0006297917130028
Abbreviations: AAT, aspartate aminotransferase; BCAA, branched-chain amino acid; BCAT, branched-chain amino acid aminotransferase; DAAT, D-amino acid transaminase; eBCAT, BCAT from E. coli; GDH, glutamate dehydrogenase; MtBCAT, BCAT from Mycobacterium tuberculosis; PLP, pyridoxal 5′-phosphate; PMP, pyridoxamine 5′-phosphate; TA, transaminase; Ts-BcAT, BCAT from Thermococcus sp. CKU-1; TUZN1299, BCAT from Thermoproteus uzoniensis; VMUT0738, BCAT from Vulcanisaeta moutnovskia.
Among organic cofactors, the pyridine derivative
pyridoxal-5′-phosphate (PLP) is characterized by exceptional
polyfunctionality; in the diversity of its functions in cell processes,
it inferior only to zinc ion [1] and, obviously,
nucleotides and polynucleotides as chemical energy carriers, allosteric
effectors, structural components of enzymes, and functional templates
of ribonucleoprotein complexes [2, 3]. PLP-dependent enzymes in cells connect carbon,
lipid and nitrogen metabolisms via the synthesis of acyl-CoA,
transamination of acyl-CoA derivatives, and synthesis of amino acids
and biogenic amines with the formation of α-ketoglutarate and
pyruvate as a secondary product [2]. PLP-dependent
enzymes perform about 160 catalytic functions [4].
In prokaryotes, they are involved in 4% of all catalytic cellular
processes, and genes encoding PLP-dependent enzymes make up 1.5% of the
genome [5].
The PLP-dependent enzymes that have been characterized thus far are divided into seven fold types (I-VII) supposedly corresponding to five evolutionary lineages of PLP enzymes [4, 6, 7]. Irrespective of the fold type, PLP-dependent enzymes are α/β-globular proteins with a functional lysine group in the active site that is covalently bound to PLP in the holoenzyme. Enzymes of different fold types differ in the spatial organization of the protein globule and of its active site, cofactor orientation in the active site, and the manner of cofactor coordination. In addition, PLP enzymes of different fold types and, importantly, enzymes of different classes with the same fold type, differ in the composition and arrangement of amino acid residues, forming the substrate-binding site, and in the mutual arrangement of the asymmetric cofactor molecule and the substrate, which determine the specificity of the catalyzed reaction and the substrate specificity of the PLP enzyme. Therefore, the entire diversity of functions is implemented in a limited set of tertiary structures (via seven fold types), on one hand. On the other hand, the diversity of orientations of the functional groups involved in catalysis and in binding of substrate and cofactor provides the specificity of PLP enzymes and the diversity of their functions.
Among PLP-dependent enzymes, enzymes of nitrogen metabolism are most widely represented in all organisms. Such enzymes catalyze a variety of transformations of compounds with primary amino groups, namely, the transfer of amino groups of amino acids and amines, racemization of amino acids, β-, γ-addition/elimination, decarboxylation and removal of the side group of amino acids [5, 8-10]. The specificity toward the amino group results from the ability of PLP to bind covalently the substrate primary amino group with the formation of a Schiff base and, further, as an electrophilic catalyst, to stabilize carbanions – the key intermediates of the above catalytic processes [9, 10]. The specificity of catalyzed reaction is determined by the combination of functional groups of amino acid residues in the active site of PLP enzymes. Toney provides a deeper insight into the fine regulation of reaction specificity of PLP enzymes in [10].
Transaminases (TAs; aminotransferases; EC 2.6.1.) catalyze reversible stereoselective amino group transfer from the amino substrate to ketone/keto acid/aldehyde with the formation of a chiral amine/amino acid and a new keto compound [10-15]. In all organisms, TAs are the key enzymes of amino acid metabolism. With regard to the 3D structure, TAs are PLP enzymes of fold types I or IV. Fold type I TAs, also referred to as aspartate aminotransferase superfamily [7, 16], are most numerous and diverse in their substrate and reaction specificity. Some of them are currently used in the synthesis of optically active amines and unnatural amino acids, for stereoselective amination of organic compounds [16, 17]. Fold type IV TAs, or D-amino acid superfamily, are less studied and represented by three families with different properties: D-amino acid aminotransferases, branched-chain amino acid TAs (BCATs), and (R)-amine TAs. This review includes analysis of literature on the properties and structure of BCATs (EC 2.6.2.42) from archaea and bacteria. The earlier review by Hutson [18] was devoted to the properties of two human BCATs, mitochondrial and cytoplasmic, while the data on bacterial BCATs have been presented only in the context of comparison.
Branched-chain amino acids (BCAAs) – L-leucine, L-isoleucine, and L-valine – are natural hydrophobic α-amino acids with a branched aliphatic side chain. The hydrophobic properties of BCAAs determine their most important role in protein globule folding, maintaining protein native state, and formation of a number of characteristic structural motifs, including superhelices and leucine zippers [19, 20]. Four enzymes are required for the biosynthesis of L-valine and L-isoleucine in bacteria and archaea from pyruvate or pyruvate and 2-oxobutyrate. L-leucine is formed from 2-ketoisovalerate – an intermediate of valine biosynthesis [20-22]. The final stage of the biosynthesis of each BCAA and the first stage of BCAA catabolism in a cell are catalyzed by a single enzyme – BCAT. The reversible stereoselective deamination of L-leucine, L-isoleucine, and L-valine proceeds with the formation of the respective α-keto acid; the amino group acceptor, like in most transamination reactions in a cell, is α-ketoglutarate (2-oxoglutarate) or, less frequently, pyruvate:
L-leucine + α-ketoglutarate ↔ 4-methyl-2-oxovalerate + L-glutamic acid,
L-isoleucine + α-ketoglutarate ↔ 3-methyl-2-oxovalerate + L-glutamic acid,
L-valine + α-ketoglutarate ↔ 2-oxoisovalerate + L-glutamic acid.
The keto derivatives of BCAAs are then decarboxylated, and the reaction products are utilized in the tricarboxylic acid cycle. L-glutamic acid is involved further in other metabolic processes that provide nitrogen transport in a cell [2]. BCAAs biosynthesis and catabolism take place in bacteria, archaea, yeasts, and lower eukaryotes. In higher eukaryotes, BCATs perform only the catabolic function and, therefore, bacterial BCATs and BCAA biosynthesis in general are potential targets in the development of antibacterial drugs [9-18, 23-25].
This review is devoted to:
BCATs in transaminase classification. TAs are currently classified based on the multiple alignment of their sequences in the PFAM Database [26]. This classification also takes into account the enzymes’ substrate specificity and combines TAs into classes and subclasses (families) according to the pairs of reacting substrates, i.e., amino group donor and amino group acceptor (see below for the mechanism of transamination). There are six main classes of TAs (Table 1). All TAs of classes I, II, III, V, and VI are fold type I PLP enzymes that catalyze the transfer of only (S)-amino group of L-amino acids and (S)-amines (see Table 1 for typical family-forming representatives of these classes). According to the results of structural alignment, the main classes also include some PLP-dependent lyases, synthases, racemases, mutases, etc., which comprise separate families. The more detailed analysis of TA classification and characteristics of fold type I TAs are given in the reviews [3, 16]. Fold type IV TAs comprise a single separate class IV that includes four subclasses: (i) D-amino acid TAs; (ii) BCATs; (iii) (R)-amine TAs catalyzing the synthesis and deamination of (R)-primary amines, and (iv) 4-amino-4-deoxychorismate lyases. The lyases do not participate in nitrogen metabolism in a cell but catalyze the cleavage of 4-amino-4-deoxychorismate with the formation of pyruvate and p-aminobenzoate in the biosynthesis of salicylic acid [27, 28]. Thus, fold type IV TAs can catalyze the stereoselective transformation of both (S)-substrates (BCAAs) and (R)-substrates (D-amino acids and (R)-primary amines).
Table 1. Classification of transaminases
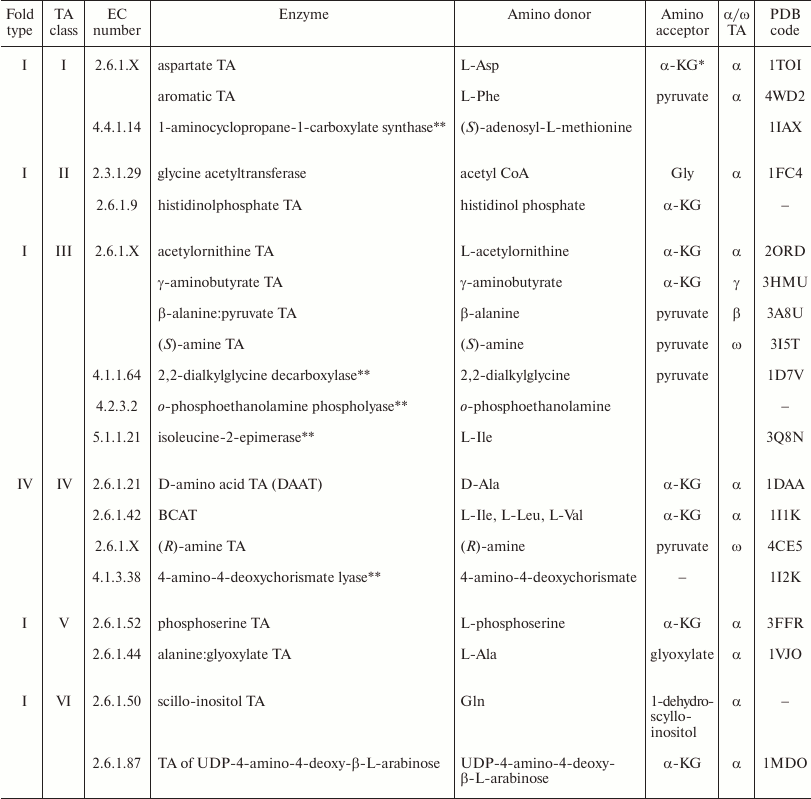
* α-KG, α-ketoglutarate.
** Not transaminases.
Though most of transamination reactions implement the transfer of the α-amino group of amino acids, some transaminases are active toward β-, γ-, ε-amino groups of amino acids and amino groups of primary amines lacking the COOH group; therefore, classification of TAs could be done based on the distance of the amino group from the COOH group in the substrate. Based on this criterion, TAs could be divided into α-, β-, γ-, and ω-TAs. BCATs belong to α-TAs. Note that ω-TAs active toward (S)- and (R)-primary amines are considered in modern biotechnology as potential biocatalysts of stereoselective amination of ketones and keto groups in organic compounds and kinetic resolution of racemates. Applications of TAs are described in reviews [16, 29-31].
Mechanism of transaminase catalysis. Formally, any TA-catalyzed reaction is oxidative deamination of the donor substrate followed by reductive amination of the acceptor substrate. Transamination proceeds by the ping-pong mechanism. The mechanism of transamination has been ascertained for aspartate aminotransferase (AAT) from E. coli [32-36]. The identification of key intermediates in the reactions catalyzed by different TAs, including BCATs, has led to a conclusion about the universal character of this mechanism [9, 10, 37] (Fig. 1). The overall reaction is a sum of two consecutive half-reactions. In the initial holo form of TA, PLP is covalently bound to the ε-amino group of lysine, forming a Schiff base (the so-called internal aldimine). This form of the cofactor is more reactive than the unbound PLP, because protonated imine is more electrophilic than aldehyde (R2C=NH2+ >> R2C=O) in the subsequent reaction of nucleophilic substitution. Amino acid (amine) is the first substrate that enters the reaction, with the formation of external aldimine by the mechanism of nucleophilic substitution of the substrate α-amino group for the ε-amino group of lysine at the C4′ atom of the cofactor. The next reaction stage is a stereo-specific 1,3-proton transfer catalyzed by the ε-amino group of lysine by the general-base catalysis mechanism. The kinetic isotope effect studies have shown that this is a rate-limiting step [35]. The 1,3-proton transfer is a two-step reaction: it starts with the removal of α-proton from the external aldimine with the formation of the carbanion, one of its resonance forms being called “quinonoid intermediate” [10, 37], and then the proton from the ε-amino group of lysine is transferred to the C4′ atom of the cofactor with the formation of ketimine. The existence of quinonoid intermediate for different TAs has been shown by spectrophotometry; it is accumulated, e.g., as a result of introduction of mutations in the active site of the enzyme [37] or in the reaction with “slow” substrates [38]. The isotope effect studies have also shown that the cleaved α-proton is not necessarily attached to the C4′ carbon atom of the cofactor ̶ any of the four protons of the catalytic lysine ε-amino group could be bound to the cofactor [39]. Then the water molecule is attached to ketimine at the double C=N bond via the general-base catalysis by the lysine ε-amino group with the formation of carbinolamine, followed by the release of keto acid and the cofactor in a form of pyridoxamine-5′-phosphate (PMP). The second half-reaction proceeds in the reverse order strictly via the same intermediate compounds with the formation of a new amino acid and regeneration of the cofactor to the initial PLP form. Each step of the process is stereospecific and reversible and, therefore, both the substrates and products inhibit transamination, so that the extent of transformation is increased by withdrawing the products from the reaction.
Fig. 1. The mechanism of transamination (absorption maxima of intermediate compounds are shown).
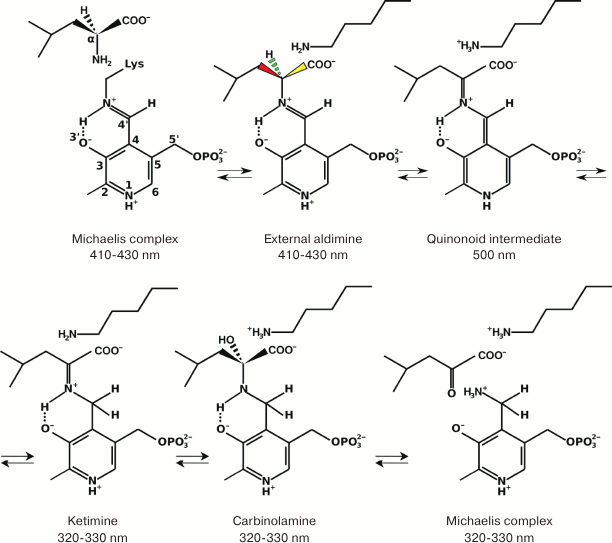
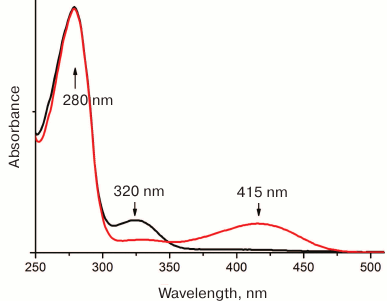
The intermediate compounds and the PLP and PMP forms of the cofactor have different absorption maxima; therefore, the half-reaction or, to be more exact, the half-turnover of the enzyme, can be monitored spectrophotometrically (Fig. 2). Since the shift of the absorption maxima happens at the stage of 1,3-proton transfer, the rates of 1,3-transfer for different substrates can be assessed by the rapid kinetics technique. However, 1,3-proton transfer is not always a rate-limiting step: some studies of class I TAs from different organisms demonstrated that hydrolysis of ketimine might be the limiting step or that the entire process is partially limited by several reaction steps [32, 40, 41].
Fig. 2. The spectra of PLP (red) and PMP (black) forms of BCAT from T. uzoniensis. Reproduced with modifications from [79].
Specific features of BCAT-catalyzed transamination. The mechanism of BCAT catalysis has not been studied in detail, although kinetic analysis confirmed the ping-pong mechanism [22, 42, 43]. It is believed that BCAT transamination proceeds via the same intermediate compounds as transamination by the thoroughly studied AAT from E. coli [44]. However, it has been supposed recently that in the half-reaction catalyzed by BCAT from Mycobacterium tuberculosis, 1,3-proton transfer occurs not successively, in two steps, but in a concerted fashion, without the formation of quinonoid intermediate. This assumption was based on the analysis of the influence of the solvent kinetic isotope effect on the half-reaction kinetic isotope effects [22].
Let us define the key properties of the BCAT catalytic mechanism:
1) Transamination is a two-substrate process; both substrates are bound sequentially at the same substrate-binding site. For most TAs, substrates differ in hydrophobicity and charge (Table 1). Therefore, the principle of dual substrate recognition is realized by different mechanisms in different classes and even subclasses of TAs. Dual substrate recognition in BCATs is implemented by the “lock and key” mechanism, the details of which will be discussed in the “Structural analysis of BCAT” section of this review.
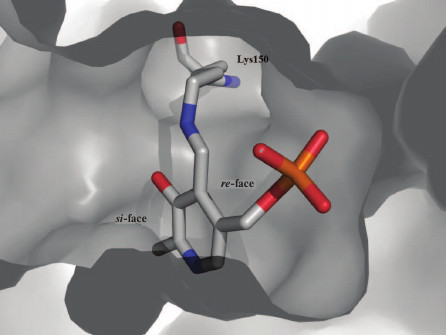
2) Transamination is a stereoselective process: L-amino acid is deaminated and new L-amino acid is formed [10]. In fold type I (S)-selective TAs, proton transfer occurs on the si-face of the cofactor; in class IV TAs (fold type IV), including BCATs, the other side of the cofactor faces the functional group of lysine and protein transfer occurs on the re-face of the cofactor [45] (Fig. 3). Thus, BCATs are the only L-amino acid-specific TAs with the re-specificity of proton transfer. Moreover, among fold type IV TAs, only BCATs exhibit (S)-selectivity.
Fig. 3. PLP position in BCAT. The model of internal aldimine of BCAT from T. uzoniensis (PDB ID: 5CE8); the si-face of the cofactor is turned toward the substrate channel entry.
BCAT-catalyzed transamination is not accompanied by the side reactions of decarboxylation or racemization, which also proceed via the formation of carbanions (Fig. 1). The principle that explains the dominance of one process in the active site over another was postulated for the first time by Dunathan [46]. According to the Dunathan’s hypothesis, preference is given to the cleavage of the substrate Cα bond that is perpendicular to the plane of the cofactor pyridine ring. Such geometry leads to the most effective resonance stabilization of the ρ-orbital of carbanion formed as a result of break of any of the three Cα bonds [46]. In addition, according to the structural data, the N1 atom of the pyridine ring in PLP in the BCAT holo form is protonated. It enhances the electrophilicity of the pyridine ring and, consequently, promotes carbanion stabilization [10]. In the holo forms of PLP-dependent racemases, PLP N1 atom is deprotonated, which supposedly results in the diminished stabilization of the carbanion and higher efficiency of racemization [44, 47]. The structural data have also shown that BCATs have a hydrogen bond between N4 atom of the aldimine imino group and O3′ of the cofactor. This hydrogen bond stabilizes the planar geometry of external aldimine, which also increases resonance stabilization of the carbanion and contributes to the polarization of αC–H bond in external aldimine [9, 10, 44, 45].
PHYSICOCHEMICAL PROPERTIES AND SUBSTRATE SPECIFICITY OF BACTERIAL
AND ARCHAEAL BCATs
Bacterial BCATs. BCAA transamination in bacteria was discovered in 1953 [48, 49]. The total BCAT activity was first described in the cell extract of E. coli and attributed to three TAs: A, B, and C [48, 50, 51]. Later, transaminase A proved to be a mixture of two TAs: AAT and aromatic aminotransferase. Transaminase C proved to be a unique alanine-valine transaminase, a product of the avtA gene [52], that does not belong to the PLP-dependent class IV of TAs. Transaminase B, the first bacterial BCAT, a product of the ilvE gene expression, catalyzed deamination of BCAAs and L-glutamic acid. Later on, a universal name of ilv genes was assigned to the genes encoding BCATs in bacteria [53, 54]. Analysis of currently available bacterial genomes confirms the assumption on the presence of BCATs in all organisms. However, since that time, the number of structurally and functionally characterized BCATs has hardly exceeded ten.
Before proceeding to the detailed analysis of BCATs, we should note that the methods for determining BCAT activity are very complicated and mostly indirect. In majority of these techniques, concentrations of the product or, less frequently, substrate are determined using the second coupled enzymatic reaction that could be monitored by an increase/decrease in the optical density in the UV-Vis range. The methods for determining BCAT activity developed before 2000 are described in [55]. They include detection of L-glutamic acid or α-ketoglutarate by the second reaction with glutamate dehydrogenase (GluDH) or in the GluDH-diaphorase coupled system [56] and detection of pyruvate or L-alanine accumulation by the coupled reaction with lactate dehydrogenase or L-alanine dehydrogenase, respectively. There are also nonenzymatic methods for assessing BCAT activity, e.g. from the reaction with 2,4-dinitrophenylhydrazine [57, 58]. Hence, comparison of BCAT enzyme activities should be done with care.
TA from Escherichia coli (eBCAT), a product of the ilv gene, forms hexamers in solution (according to gel filtration data) [59, 60]. The absorption spectra of the PLP and PMP forms of the enzyme have the maxima at 330 and 410 nm, respectively; the PLP maximum in the holo form spectrum is not shifted at pH-titration, as in class I TAs [61]. The optimal conditions for the eBCAT-catalyzed reactions are 25-37°C and pH 8.0. The specificity of the enzyme to amino donors and keto substrates is shown in Table 2. The minimum value of the Michaelis constant (Km, 0.42 mM) was observed for leucine; the maximum values, 19 and 72 mM, were observed for methionine and tryptophan, respectively; for α-ketoglutarate, Km was 2.6 mM. The catalytic constant kcat for the best substrates (L-leucine and L-isoleucine) was 48 s–1. The eBCAT activity was determined spectrophotometrically by measuring the concentration of keto acids (products) at 310 and 315 nm; the extinction coefficient was determined for each keto acid separately. The authors of [62] determined the kinetic parameters of eBCAT-catalyzed amination of keto analogs of BCAA and their unnatural isomers, such as L-norleucine, L-norvaline, L-neopentylglycine, and L-tert-leucine. The eBCAT activity was measured from the product (α-ketoglutarate) accumulation using a unique technique of coupled enzymatic reduction of α-ketoglutarate to 2-hydroxyglutarate catalyzed by (R)-hydroxyglutarate dehydrogenase. The Km values for the amination of keto acids with L-glutamic acid as an amino group donor were 0.07 mM for 3-methyl-2-oxovalerate (keto analog of L-Ile), 0.08 mM for 4-methyl-2-oxovalerate (keto analog of L-Leu), 0.08 mM for 4,4-dimethyl-2-oxovalerate (keto analog of L-neopentylglycine), 0.22 mM for 2-oxohexanoate (keto analog of L-norleucine), 0.2 mM for 2-oxoisovalerate (keto analog of L-Val), 0.6 mM for 2-oxovalerate (keto analog of L-norvaline), 0.15 mM for trimethyl pyruvate (keto analog of L-tert-Leu), 56 mM for pyruvate, and 3.37 mM for 2-oxobutyrate. The kcat for the best substrates – keto analogs of L-leucine and isomers– was 23-25 s–1. The obtained kinetic parameters of the forward and reverse reactions for different substrates have led to a conclusion that in vitro eBCAT catalyzes both amination of BCAA keto analogs and deamination of BCAAs with equal efficiently. Yu et al. [62] showed inhibition of transamination by the keto substrates 4-methyl-2-oxovalerate, 4,4-dimethyl-2-oxovalerate, and 2-oxohexanoate in concentrations below 10 mM; the inhibition by the amino substrate L-glutamic acid was observed at concentrations above 200 mM.
Table 2. The substrate specificity of
BCATs

* Co-substrate α-ketoglutarate (α-KG).
** Co-substrate L-glutamic acid.
*** Co-substrate pyruvate.
**** Co-substrate L-alanine.
BCAT from Pseudomonas aeruginosa [63] is a tetramer in a working buffer (according to gel filtration data). The specificity of this enzyme toward amino donors is shown in Table 2. The enzyme activity was calculated from the changes in the concentration of the product (L-glutamic acid), that was separated from other amino and keto acids by TLC of the reaction mixture with preliminary protein precipitation, followed by quantitative extraction with a methanol–acetic acid mixture. The Km vales for the keto substrates 2-oxovalerate and α-ketoglutarate were 0.26 and 0.4 mM, respectively; Km for amino substrates in the reaction with 2-oxovalerate was 1.0 and 1.2 mM for leucine and isoleucine, respectively, and 18 mM for L-glutamic acid. The enzyme showed no activity with L-alanine, L-aspartate, L-glycine, L-serine, L-threonine, L-tryptophan and L-tyrosine. Another wild-type BCAT from a Pseudomonas bacterium was characterized by Koide et al. [43]. The enzyme activity was calculated from the changes in the substrate and product concentrations using TLC followed by quantitative extraction and spectrophotometric measurement of the components in the solution. The specificity of the enzyme toward amino donors and keto substrates is presented in Table 2. The inhibition of the enzyme by its keto substrates in a concentration above 2 mM was shown for the transamination reaction with 4-methyl-2-oxovalerate and L-glutamic acid.
At approximately the same time, BCAT from Gluconobacter oxydans (Acetobacter suboxydans) was characterized [64, 65] (Table 2). According to the results of gel filtration and sedimentation analysis, the enzyme exists as a dimer in solution. Its pH optimum is shifted to the alkaline region (pH 8.8-9.0). The Km values for the transamination reaction between keto acids and L-glutamic acid are 2.5 mM for 3-methyl-2-oxovalerate (Ile), 0.91 mM for 4-methyl-2-oxovalerate (Leu), and 0.33 mM for 2-oxoisovalerate (Val). The Km value for α-ketoglutarate in the reverse reaction varied from 4.57 to 6.67 mM, depending on the amino donor. The activity with L-aspartic acid, L-arginine, L-citrulline, L-lysine, L-ornithine, L-alanine, β-alanine, and γ-aminobutyrate was not detected; the activity with pyruvate was 1% of the activity with α-ketoglutarate. The enzyme affinity to the cofactor was determined, the binding constants being 0.53 and 0.62 µM for PLP and PMP, respectively.
The properties of BCATs from the Lactococcus lactis and Lactobacillus paracasei bacteria have been given close attention, because these microorganisms synthesize aromatic substances, such as isobutyrate, isovalerate, 3-methylbutanal, 3-methylbutanic acid, 2-methylbutanal, and 3-methylpropanal from the keto analogs of BCAAs in the process of cheese ripening [66, 67]. According to the results of gel filtration, the active forms of BCATs from L. lactis and L. paracasei are a dimer and a monomer, respectively. The specificity of BCATs from L. lactis and L. paracasei toward amino donors and amino acceptors is shown in Table 2; no activity toward other natural amino acids has been detected. These BCATs were completely inhibited by Hg2+ ions, carboxymethoxyamine, hydroxylamine, and phenylhydrazine. In the presence of 4% NaCl, the activity of these enzymes decreased by no more than 20%.
Kanda et al. [68] isolated and characterized BCAT from the Gram-positive bacterium Bacillus brevis. According to the data of gel filtration, the enzyme is a dimer. The pH optimum for the reaction with leucine and α-ketoglutarate was 8.3. In addition to BCAAs, the activity with aromatic amino acids L-phenylalanine and L-tryptophan was detected (Table 2). The enzyme showed no activity toward the keto analogs of glycine and aspartate or pyruvate and was inhibited by semicarbazide, hydroxylamine, phenylhydrazine, and hydrazine. The Km values for amino donors in the reaction of transamination with α-ketoglutarate were 0.22 mM for L-valine, 0.25 mM for L-leucine, 0.43 mM for L-isoleucine, 2.0 mM for L-phenylalanine, and 2.9 mM for L-tryptophan; Km for α-ketoglutarate and L-glutamic acid in the reaction with leucine and its keto analog was 0.77 and 0.56 mM, respectively. The PLP binding constant was 6.5 µM.
Saito et al. isolated and characterized BCAT from the bacterium Helicobacter pylori [42]. The enzyme activity was calculated from the change in the L-glutamic acid concentration as determined by TLC and HPLC techniques. According to gel filtration data, the enzyme is a dimer; the pH optimum of the transamination reaction with L-isoleucine and α-ketoglutarate is 8.0. The specificity of this enzyme toward amino donors in the reaction with α-ketoglutarate is given in Table 2.
The recombinant form of BCAT (MtBCAT), which was characterized in the study of the metabolic pathway of regeneration of L-methionine consumed in polyamine biosynthesis in the bacterium Mycobacterium tuberculosis, catalyzes not only the final step of BCAA biosynthesis and the first step of BCAA catabolism, but also the transfer of an amino group from any of the BCAAs to 4-methylthio-2-oxobutyrate, the keto precursor of L-methionine, i.e. the final step of L-methionine biosynthesis [69]. It has been established that, in addition to BCAAs, L-phenylalanine could be the amino group donor in this process. The Km values for L-isoleucine, L-leucine, L-valine, and L-phenylalanine in the reaction of amination of 4-methylthio-2-oxobutyrate are within 1.77-7.44 mM; kcat varies within a range of 1.22-3.23 s–1. The Km values for L-isoleucine, L-leucine, and L-valine in the reaction of deamination with α-ketoglutarate as the amino acceptor vary within 5.79-6.95 mM; the kcat values are within a range of 6.7-8.1 s–1. Thus, the catalytic efficiency of 4-methylthio-2-oxobutyrate amination by leucine is two times lower than the catalytic efficiency of α-ketoglutarate amination by leucine, and the most efficient process is the synthesis of BCAAs from keto precursors [22]. In addition, the mixed-type inhibition of MtBCAT by hydroxylamines was shown; the inhibition constants for o-benzylhydroxylamine, o-tret-butylhydroxylamine, and o-allylhydroxylamine were 8.20, 11.0, and 21.61 µM, respectively. MtBCAT was proposed as a potential target in the development of antituberculosis drugs.
BCATs from the bacteria Staphylococcus carnosus [70], Bacillus subtilis [71], Pseudomonas cepacia [72], Pseudomonas putida [73], Deinococcus radiodurans [74], Salmonella typhimurium [75, 76] have been mentioned in publications in the context of BCAA metabolism but without detailed biochemical characterization.
Archaeal BCATs. The first BCAT from the moderately thermophilic archaeon Methanococcus aeolicus was characterized in 1992 [77, 78]. The wild-type enzyme is a hexamer, according to the results of gel filtration; the pH optimum of the reaction with BCAAs is 7.5. The enzyme activity was determined at 37°C by nonenzymatic measurement of the concentration of keto acids in the reaction with 2,4-dinitrophenylhydrazine [57]. The enzyme showed the maximum activity with L-leucine and L-valine; the activity with L-isoleucine was two times lower; and the activity with aromatic acids L-tyrosine, L-tryptophan and L-phenylalanine was less than 5-10% of the activity with BCAAs (Table 2). The amino group acceptor was α-ketoglutarate. The enzyme did not catalyze reactions with L-aspartate and L-alanine. The Km values for amino donors were 1.4 mM for L-isoleucine, 1.1 mM for L-leucine, 2.8 mM for L-valine, 0.1 mM for L-phenylalanine, and 2.8 mM for L-tryptophan; Km for α-ketoglutarate in the reaction with BCAAs was within a range of 0.3-0.6 mM. Deamination rate was 0.7-1.5 µmol/min per mg enzyme for BCAAs and 0.1-0.23 µmol/min per mg enzyme for aromatic amino acids. Substrate inhibition studies have shown that 5 mM α-ketoglutarate decreases the enzyme activity with valine and isoleucine by more than 50%; at the same time, the activity with leucine decreases by 10%, while the activity with the nonspecific substrate L-tyrosine remains unchanged.
In 2016, two recombinant BCATs from the hyperthermophilic archaea Thermoproteus uzoniensis (TUZN1299) and Vulcanisaeta moutnovskia (VMUT0738) were characterized in detail [79-81]. The similarity between the TUZN1299 and VMUT0738 amino acid sequences is 54%. According to the results of gel filtration, TUZN1299 is a dimer and VMUT0738 is a tetramer in solution. The unique property of both enzyme is the absence of activity toward α-ketoglutarate and L-glutamic acid and the high activity with pyruvate. The temperature optimum of the reaction of transamination between leucine and pyruvate was above 90°C for both BCATs; the pH optimum of the reaction was 8.0. The enzyme activities were determined by the noncoupled enzymatic assay: the transamination reaction was performed at 65°C; the concentrations of pyruvate and L-alanine in the aliquots were determined further enzymatically at 25°C. The specificities of TUZN1299 and VMUT0738 toward keto substrates and amino donors are shown in Table 2. Both enzymes are characterized by the high activity toward L-methionine, L-threonine, and L-2-aminobutyric acid. The activity toward L-alanine and L-phenylalanine is comparable to the activity toward leucine for VMUT0738 and two times lower for TUZN1299. Another unique feature of both enzymes is high activity toward positively charged L-amino acids. For example, the specific activity of TUZN1299 with leucine was 1.70 µmol/min per mg enzyme, while the specific activity toward L-ornithine, L-arginine, L-histidine and (D,L)-lysine was 4.4, 3.5, 2.3, and 0.65 µmol/min per mg enzyme, respectively. The specific activity of VMUT0738 toward L-leucine was 1.72 µmol/min per mg enzyme, while the specific activity toward L-ornithine, L-arginine, L-histidine and (D,L)-lysine was 3.9, 2.64, 1.76, and 3.93 µmol/min per mg enzyme, respectively. At the same time, both enzymes were inactive toward L-aspartic acid, L-serine, D-alanine, and β-L-alanine. The Km values for L-valine and L-norvaline in the VMUT0738-catalyzed reaction of transamination with pyruvate were 1.17 and 0.84 mM, respectively. The Km values for L-leucine and pyruvate in the TUZN1299-catalyzed transamination reaction were 0.21 and 16.0 mM, respectively; the kcat value was 1.31 s–1. For TUZN1299, analysis of kinetic parameters of the (E-PLP + amino substrates)- and (E-PMP + keto substrate)-half-reactions showed that the most efficient process was amination of BCAA keto analogs; the specificity constant of the (E-PMP + 4-methyl-2-oxovalerate)-half-reaction was up to 1,113,000 s–1·M–1 at 25°C. For comparison, the specificity constants of the (E-PLP + L-leucine)- and (E-PLP + L-methionine)-half-reactions were 176,000 and 9600 s–1·M–1, respectively. It means that for TUZN1299, BCAA synthesis is more preferable that BCAA deamination. The keto substrates 4-methyl-2-oxovalerate and 3-methyl-2-oxovalerate in concentrations above 1 mM inhibited TUZN1299 in the reaction of transamination with the second substrate L-alanine. Therefore, archaeal BCATs demonstrate broader substrate specificity but lower specific activities than their bacterial homologues.
One of the articles published in 2014 contained detailed characterization of the fold type I BCAT from the archaeon Thermococcus sp. CKU-1, Ts-BcAT [82], that is homologous both to the hypothetical fold type I AAT (sequence similarity, 91-95%; the degree of overlap, 100%) and to the characterized AAT from Sulfolobus solfataricus MT4 (sequence similarity, 26%; the degree of overlap, 91%). At the same time, its identity to eBCAT was 29%, with the degree of sequence overlap being only 15%. According to the data of gel filtration, recombinant Ts-BcAT forms dimers in solution. The enzyme showed an exceptional thermal stability: its activity did not decrease after the 5-h incubation at 90°C. Like the archaeal BCATs described above, Ts-BcAT showed broad substrate specificity (Table 2) and activity with α-ketoglutarate, but was inactive with positively charged amino acids. The analysis of kinetic parameters of the half-reaction has shown that the maximum catalytic activity of Ts-BcAT is achieved in the processes of amination of keto acids: the specificity constant of the (E-PMP + 4-methyl-2-oxovalerate)-half-reaction is 11,000,000 s–1·M–1 at 25°C. Oxo-amines, semicarbazide, and hydroxylamine insignificantly inhibited the enzyme activity. The change in pH was accompanied by a shift in the PLP maximum in the holo form spectrum, similarly to the known class I TAs, so that it was possible to calculate pKa of ε-amino group of the catalytic lysine in internal aldimine (pKa = 5.5). The enzyme seems to be an aromatic aminotransferase with a marked activity toward hydrophobic amino acids, including BCAAs. The low activity toward valine, isoleucine, 2-aminooctanoate, and 2-aminobutyrate has been shown previously for the aromatic aminotransferases from E. coli K-12 [83] and Paracoccus denitrificans IFO 12442 [84]. It is still unclear whether Thermococcus sp. CKU-1 has the true fold type IV BCAT; at the same time, it is known that this microorganism grows only in the presence of L-leucine, L-methionine, L-phenylalanine, and some other amino acids.
Generalization of the above characteristics leads to the following conclusion about the properties of bacterial and archaeal BCATs. (i) BCATs in solution exist in different oligomeric states. (ii) The pH optimum of BCATs is 7.5-8.0, up to 9.0 in some enzymes. (iii) In vitro, bacterial and archaeal BCATs do not show preference for amination or deamination, while kinetic parameters of half-reactions indicate that amination of BCAA keto precursors is the most efficient process. (iv) The specificity of bacterial BCATs is not limited to BCAAs and their keto analogs, as in mammalian BCATs [18, 85]; bacterial BCATs are active toward methionine, aromatic amino acids, and threonine. Archaeal BCATs have even broader substrate specificity. (v) The affinity to keto substrates is higher than the affinity to amino substrates. (vi) The preferential amino acceptor is α-ketoglutarate; the activity toward pyruvate is much lower, with the exception of TUZN1299 and VMUT0738, which are inactive with α-ketoglutarate. (vii) The BCAT activity is inhibited by keto substrates, which seems to be the mechanism for the regulation of their activity in cells. (viii) The BCAT activity is inhibited by oximes, hydrazine and its derivatives, and semicarbazide. Inhibition by oximes is supposedly implemented via the reaction with PLP [86, 87]. (ix) PLP is bound in the BCAT active site 100- to 1000-fold more tightly than the substrate. (x) The absorption spectra of BCATs, in addition to the protein maximum, show two supplementary maxima at 320-330 and 410-420 nm, that correspond to the PMP and PLP forms of the bound cofactor, respectively. The pH-titration is not accompanied by a shift of the PLP maximum, unlike in AAT, where this shift corresponds to the deprotonation of imine nitrogen of the internal aldimine [9, 61].
Potential biotechnological applications of BCATs. Unlike the development of biotechnological processes involving ω-TAs, which are active toward amines and ketones/aldehydes, i.e. capable of stereospecific synthesis of primary amines, the use of BCATs in the biotechnology is still uncommon [31, 88]. The authors of [89] considered the possibility of using BCATs in the synthesis of L-homophenylalanine, L-2-aminobutyrate, and L-tert-leucine from the respective keto acid and 2-oxoglutarate paired with the second enzymatic reaction catalyzed by ornithine-δ-aminotransferase, where the second enzyme is used to withdraw the product (L-glutamic acid) from the first reaction. As a result of such approach, the product yield in the first reaction was increased to 80-90%. In the work [66], it was proposed to use eBCAT in the asymmetric synthesis of some unnatural amino acids, such as L-norleucine, L-norvaline, L-neopentylglycine, and L-tert-leucine, that are potential synthons for a number of pharmaceutical (including antitumor) preparations, as well as therapeutic anti-HIV agents [90, 91]. The use of eBCAT in the synthesis of L-glutamic acid derivatives with substituents in positions 3 and/or 4 for studying the specificity of glutamate receptors was shown in [92]. Four stereoisomers of L-2-(2-carboxycyclobutyl)glycine (cyclic analogs of L-glutamate for studying L-glutamate transport in the central nervous system) were synthesized from cis- and trans-2-oxalyl-cyclobutane carboxylic acids using AAT and eBCAT [93]. Therefore, potential applications of BCATs are based on the enzyme specificity toward both hydrophobic aliphatic amino acids and L-glutamic acid derivatives.
STRUCTURAL ANALYSIS OF BCATs
The structure of functional BCAT dimer. The first structure of BCAT from E. coli, the holo form with PLP, was solved in 1997 (PDB code 1A3G) [94]. Later, eBCAT structures with inactive substrate analogs 4-methylvalerate (PDB code 1I1M) and glutaric acid (PDB code 1IYD), as well as with the substrates 2-methylleucine (PDB code 1I1L) and L-glutamic acid (PDB code 1IYE) [44, 95] were also obtained. Structures of some other bacterial BCATs were determined: the holo form and the complex with the inhibitor o-benzylhydroxylamine of BCATs from M. smegmatis (PDB code 3DTF, 3JZ6) [96], and the holo forms of BCATs from M. tuberculosus (PDB code 3HT5) [97], Burkholderia pseudomallei (PDB code 3U0G, 4WHX), and Streptococcus mutans (PDB code 4DQN). In addition, structures of BCATs from several extremophilic bacteria and archaea have been elucidated: the holo form and the complexes with 4-methyl-2-oxovalerate and L-glutamate of BCAT from D. radiodurans (PDB code 3UYY, 3UZB, 3UZO) [74]; the holo forms of BCATs from Thermotoga maritima (PDB code 3CSW) and the archaeon T. uzoniensis (PDB code 5CE8); the holo form and the complex with the inhibitor gabapentin of BCAT from Thermus thermophiles Hb8 (PDB code 1WRV, 2EIY, 2EJ0, 2EJ2, and 2EJ3); and the holo form and the complex with α-ketoglutarate of BCAT from the archaeon Geoglobus acetivorans (PDB code 5E25, 5CM0).
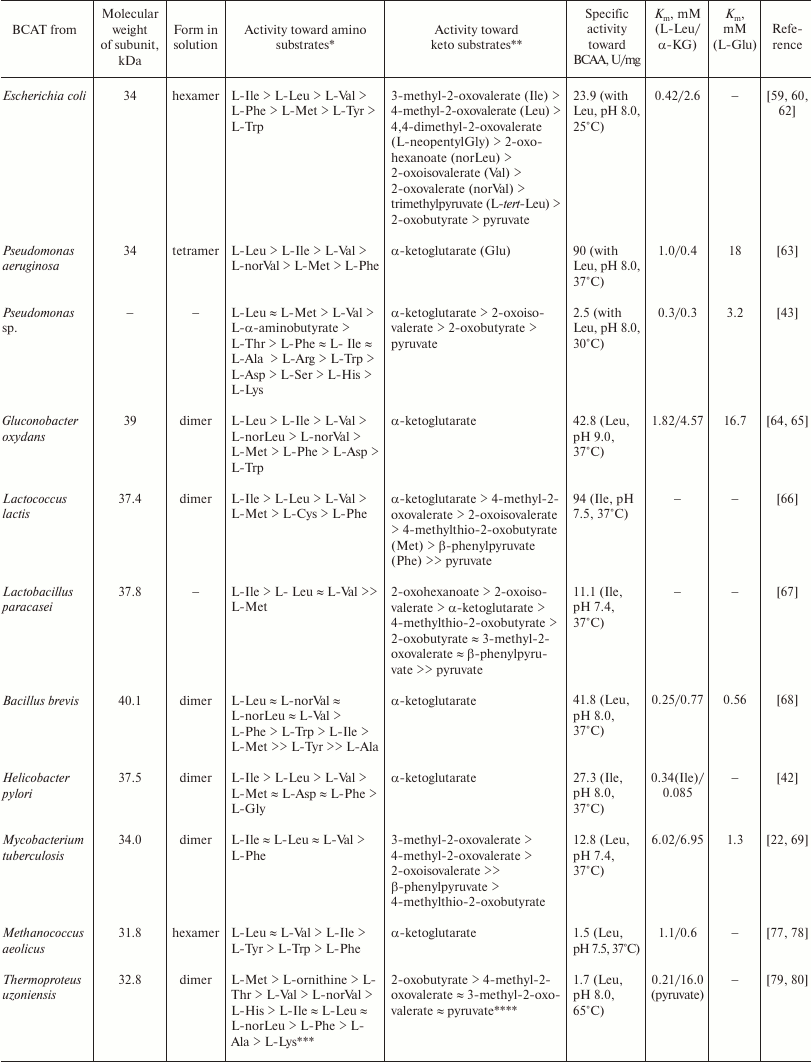
Most structurally characterized BCATs crystallize as dimers; the known exceptions are eBCAT and BCAT from T. thermophiles, which crystallize as hexamers, and BCAT from D. radiodurans that in a complex with the substrate forms the tetrameric crystal. Though hexameric and tetrameric forms have been shown for some enzymes by gel filtration (Table 2), the functional unit of BCAT is a dimer (Fig. 4). This conclusion follows from the structural analysis of the active site of BCATs [44, 74, 94-97]. All BCAT dimers, including those of mammalian BCATs [18], are characterized by the high structural similarity revealed by structural alignment and by analysis of the arrangement of functionally important residues and superposition of the secondary structure elements [18, 47, 79, 96].
Fig. 4. The superposition of dimers of BCATs from E. coli (gray) and T. uzoniensis (the dimer subunits are blue and green). The intersubunit loop in the BCAT from T. uzoniensis is red; PLP is pink.
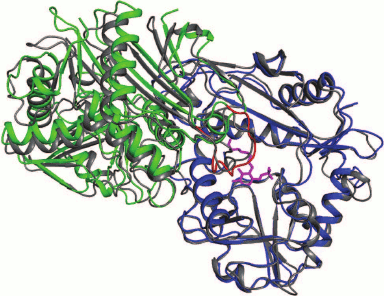
The subunit of the dimer in all BCATs consists of two α/β domains (small and large) and the interdomain loop essential for the enzyme function (Fig. 5). Comparison of the models of holo forms of BCATs and substrate complexes shows the absence of mutual movement of domains during substrate binding. The relative positions of domains are fixed by the C-terminal helix (residues 279-293 in eBCAT, residues 267-280 in TUZN1299). The interdomain loop isolates the active site from the solvent after substrate binding [105]; the lability of the loop is reflected in its absence from the electron density maps of the BCAT holo forms [74, 95] and its identification in the enzyme complexes with substrates and their analogs [44, 74]. The interdomain loop in the holo form of TUZN1299 (PDB code 5CE8) is characterized by an increased value of the temperature factor (B-factor) of its residues [79], which confirms its relative mobility in the absence of bound substrate. Interestingly, the interdomain loop is well seen in the models of holo forms of archaeal BCATs and BCAT from D. radiodurans, which probably correlates with higher rigidity of the structures of thermostable enzymes.
Fig. 5. The superposition of subunits of BCATs from E. coli (gray) and T. uzoniensis. The domains are green and blue; the C-terminal α-helix is red; the interdomain loop in the T. uzoniensis BCAT is yellow.
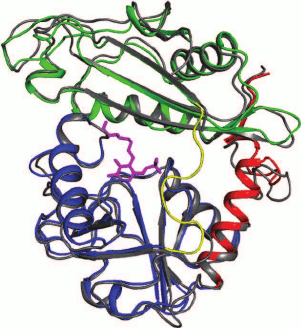
The functional dimer contains two active sites. The active site is formed by the large and small domains of one subunit and the small domain of the other subunit. In the models of BCAT holo forms, the active site contains PLP molecules covalently bound to the catalytic lysine (internal aldimine). PLP is turned to the protein globule with the re-face (Fig. 3); it is this (isolated from the solvent) side of the cofactor where the 1,3-proton transfer occurs in BCATs and other TAs of IV fold type. PLP in the active site of BCAT (Fig. 6a) forms a C4′=N double bond with lysine and is fixed by: (i) phosphate group via hydrogen bonding and salt bridges with residues R59, I220, T221, T257, and A258 (in eBCAT) or R54, I211, T212, and T248 (in TUZN1299); (ii) oxygen atom of the PLP phenol group via hydrogen bonding with Y164 (in eBCAT) or Y155 (in TUZN1299); and (iii) nitrogen atom of the pyridine ring via hydrogen bonding with E193 (in eBCAT) or E184 (in TUZN1299). The identical residues are involved in PLP coordination in all structurally and biochemically characterized BCATs, including eukaryotic enzymes [18, 44, 95, 98]. Additional hydrogen bonds between PLP and non-conservative residues have been described for BCAT from D. radiodurans [74]. Comparative analysis of the structures of holo forms and substrate complexes of BCATs demonstrated rotation of the PLP molecule along the N–C6 or N–C4 axes during transition from the internal aldimine to the external aldimine. The torsion angle (C3–C4–C4′–N) varies from 30° in the models of holo forms to 0° in the models of substrate complexes, which is indicative of the presence of a hydrogen bond between the phenol group of PLP and the imine nitrogen. The protonated form of the imine nitrogen at neutral and weakly alkaline media seems to be a characteristic feature of BCATs [9, 10, 95]. In class I TAs, the pKa value of the imine nitrogen is 5.0-7.5, as determined by pH titration of the enzyme holo form. The BCAT holo form, as has been discussed above, is not titrated [60, 80].
Fig. 6. a) PLP binding in T. uzoniensis BCAT (the figure was created using LigPlot). b) Binding of L-glutamate external aldimine in E. coli BCAT (PDB ID: 1IYE). The large pocket is pink; the small pocket is blue (the figure was created using LigPlot).
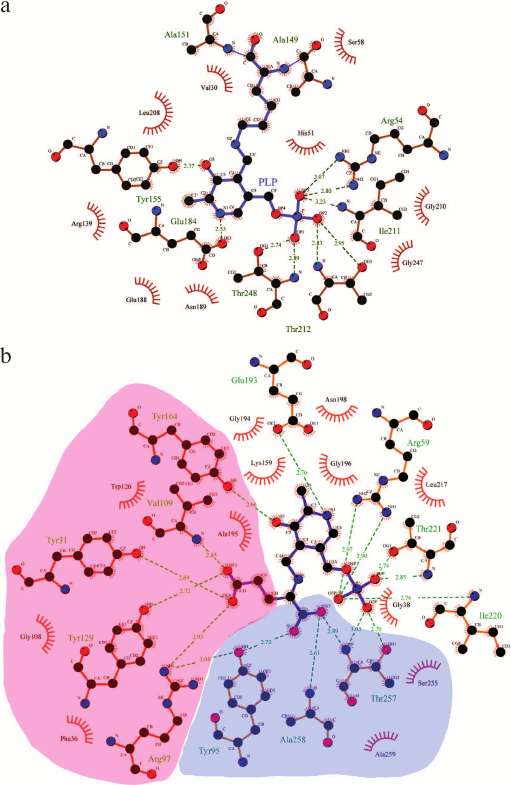
Substrate binding; dual substrate recognition. In BCAT complexes, a substrate or its analog are located on the si-face of the cofactor and can be covalently bound to PLP (external aldimine) (Fig. 6b). α-COOH groups of the amino donor and the amino acceptor are located in the small pocket on the side of the PLP phosphate group (the P-side), and the BCAA hydrophobic residue is located in the large pocket on the side of the PLP phenol group (the O-side). In complexes with substrate of eBCAT and BCAT from T. thermophilus, the interdomain loop (residues 126-137 in eBCAT) is structured; in complex of BCAT from D. radiodurans, the loop (residues 173-179) takes another conformation relative to the holo form. In all the complexes, the interdomain loop blocks the access of the solvent molecules to the active site, thereby preventing the side process of racemization that occurs in the PLP-dependent alanine racemase with the solvent-accessible active site [45, 99, 100]. The conformation of BCAT molecule with this particular structure of the active site is called closed, in contrast to the open conformation of the holo form. The amino acid sequence analysis of the eBCAT interdomain loop performed by Okada et al. [95] showed high similarity of this region among BCATs with sequence identity over 30%. The authors believe that it indicates the similar loop ordering and interaction with substrates in these BCATs. The loop mobility in the open conformation provides substrate access to the active site. As has been mentioned above, the interdomain loop is structured in the models of enzyme holo forms from thermophilic organisms: TUZN1299, BCATs from G. acetivorans and D. radiodurans. It is possible that the loop gets disordered at high (optimal) reaction temperatures, and the enzyme acquires the open conformation. Substrate binding leads to the changes in the positions of residues in the small pocket. The rotation of R40 towards the conservative β-turn (G256-T257-A258-A259) is observed in eBCAT; in this case, R40 forms hydrogen bonds with carbonyl oxygens of T257 and A258. The nitrogen atoms of these residues fix via hydrogen bonds the α-COOH group of the substrate, which is additionally coordinated by a conservative hydrogen bond with the hydroxyl group of the tyrosine residue (Y95 in eBCAT and Y91 in TUZN1299) from the Y(I/L/V)R triad, which is conservative in BCATs. It has been shown that arginine in this triad activates tyrosine through the formation of a conservative hydrogen bond with its hydroxyl group [101]. The R35 residue in TUZN1299, which is identical to R40 in eBCAT, is already in the fixed conformation in the holo form. Conservative residues G122 and G127 of the ordered interdomain loops in the TUZN1299 holo form and in the eBCAT–substrate complex form hydrogen bonds with the R35 and R40 residues, respectively, thereby completing optimal conformation of the small pocket. Comparative analysis of substrate binding in the active sites of related fold type IV TAs (BCATs, DAATs and (R)-amine TAs) showed that it is precisely the small pocket organization that determines the reaction specificity of these enzymes [102-104].
The unique feature of BCATs is the binding of the substrate α-COOH group on the P-side of the active site (Fig. 6b). In other fold type I and IV TAs, negatively charged phosphate group of PLP and the substrate α-COOH group are separated in the active site. Goto [44] suggested that, since the neighboring of two negatively charged groups increases the total energy of the system, the presence of a positively charged particle between them would favor system stabilization. Probably, close location of two negatively charged groups serves for trapping/accepting the proton of the substrate α-amino group during the Michaelis complex formation. As has been mentioned above, the imine nitrogen of the internal aldimine of BCATs is apparently protonated and cannot accept a proton, as it is assumed in transaminases of fold type I; therefore, there must be an additional proton acceptor site for the substrate α-amino group. It is also possible that this negatively charged site is required only for the activation of BCAAs, which are insufficient as nucleophiles during the internal aldimine–external aldimine transition, so the additional measures should be taken to remove the proton from the substrate α-amino group.
The side groups of the amino donor and the amino acceptor are successively bound in the large pocket at the O-side of the active site. When binding residues with different properties (such as hydrophobic side groups of BCAAs and the γ-COOH group of α-ketoglutarate), BCATs, similarly to all TAs, realize the principle of dual substrate recognition. In BCATs, this recognition proceeds via the “lock and key” mechanism and has been described in detail for eBCAT [44, 105]. The large pocket of eBCAT dimer is formed by amino acid residues of both subunits: the side groups of F36, R97, W126, Y129, Y164 of one subunit and of Y31*, V109* of the other subunit. These amino acids form a hydrophobic surface of the pocket with the inclusion of four hydrophilic sites “submerged” in it: these are guanidine group of R97, hydroxyl groups of Y31* and Y129, and nitrogen atom of the V109* main chain (Fig. 6b). The hydrophobic group of BCAA interacts with the hydrophobic surface of the pocket, while α-ketoglutarate, which is by one -CH2 unit longer than BCAA, reaches the “submerged” hydrophilic sites with its γ-COOH group, forming with them a salt bridge and three hydrogen bonds [44]. Dual substrate recognition occurs via the “lock and key” mechanism in other fold type IV TAs and in some fold type I TAs, e.g. in glutamine phenylpyruvate TA from T. thermophilus HB8 [106]. “Induced fit” is another mechanism of dual substrate recognition, which is implemented in fold type I aromatic aminotransferases and fundamentally differs in the rotation of the conservative arginine residue in the large pocket [105, 107, 108], allowing the formation of a positively charged binding site for the γ-COOH group of α-ketoglutarate and its removal during aromatic residue binding.
CHARACTERISTIC MOTIFS OF BCAT SEQUENCES
As has been mentioned above, BCATs are fold type IV PLP enzymes, together with DAATs, isochorismate lyases, and (R)-amine TAs. Detailed analysis of amino acid sequences of these structurally similar, though differing in reaction and substrate specificity [101], TAs has shown that the specificity of fold-type IV TAs is determined by two characteristic motifs (Fig. 7a). The alignment of BCAT sequences discussed in this review clearly reveals the following two characteristic motifs of BCATs (Fig. 7b):
motif_1 31-Y(G/A)XXXF(E,D)GX(K/R)-40;motif_2 95-Y(I/L/V)Rxx…xx(M/I)G(V/L)-109 (here, the numeration of eBCAT).
Fig. 7. a) Characteristic motifs of representatives of four families of fold type IV TAs.

Fig. 7. b) Alignment of characterized BCATs. Black squares, conservative residues of characteristic motifs; gray squares, residues involved in PLP binding; B and S, residues forming large and small pockets, respectively.

The sequence alignment and structural superposition of BCATs and substrates [44, 74, 95] have shown that all characteristic residues form a substrate binding site. The functions of identical residues are as follows: Y31 provides the binding of the γ-COOH group of α-ketoglutarate and L-glutamic acid, R38/K40 form a space for binding the substrate α-COOH group in the small pocket; these residues interact with nitrogen atoms of the main chain of the 257-TAA-259 triad (conservative in BCATs) that also forms the small pocket. The role of the conservative triad 95-Y(I/L/V)R-97 in α-COOH coordination has been discussed above. As follows from the currently available biochemical data, these residues and R40 are the key elements of the BCAT sequence and their substitution results in the impairment of the BCAT-specific binding of the substrate α-COOH group in the small pocket [101, 109]. The F36 residue and the 107-XGX-109 motif, where X is one of hydrophobic amino acids (I, L, V, M), are involved in the formation of the hydrophobic surface of the large substrate pocket. The 107-XGX-109 motif is located in the intersubunit loop, which thereby not only forms the large pocket but also determines the substrate specificity of fold type IV TAs (Fig. 4). The flexibility of the intersubunit loop probably enables bacterial and archaeal BCATs to bind differently shaped side groups of amino substrates [97], in other words, determines the broad substrate specificity of these enzymes. Interestingly, the substitution of residues in the characteristic motif_2 found in some archaeal BCATs has a dramatic effect on the substrate specificity of these enzymes, e.g., substitution of serine and asparagine for glycine in the conservative triad 107-XGX-109 in TUZN1299 from T. uzoniensis and VMUT0738 from V. moutnovskia, respectively. Modeling of the enzyme structure demonstrated that this very substitution determines the absence of productive α-ketoglutarate binding by both BCATs [80, 81]. The substitution of asparagine or serine for G108 residue has been also found in other sequences of potential BCATs from archaea of the genera Vulcanisaeta, Pyrobaculum, and Thermoproteus [81].
In conclusion, it should be noted that adjustment of substrate specificity in BCATs is considered as the finest (and most complex) adjustment in fold type IV PLP enzymes. The functional groups listed above and the active site organization serve for fixing the substrate relative to the cofactor and the catalytic lysine, thereby determining the specificity of proton removal from the carbon α-atom in BCAAs. BCATs are unique among TAs of different fold types and families in their structural and functional properties. However, it is yet unclear why BCAA transamination is catalyzed by fold type IV TAs only. As one can see from the example of Thermococcus sp. CKU-1 TA, family I aromatic aminotransferases can effectively convert BCAAs; however, there appears to be no widespread occurrence of this BCAT form.
Acknowledgments
The authors are grateful to Evgeny Osipov for his assistance in making figures.
The work was partially supported by the Russian Science Foundation (project No. 14-24-00172).
REFERENCES
1.Vallee, B. L., and Falchuk, K. H. (1993) The
biochemical basis of zinc physiology, Physiol. Rev., 73,
79-118.
2.Chubukov, V., Gerosa, L., and Kochanowski, K., and
Sauer, U. (2014) Coordination of microbial metabolism, Nat. Rev.
Microbiol., 12, 327-340.
3.Schiroli, D., and Peracchi, A. (2015) A subfamily
of PLP-dependent enzymes specialized in handling terminal amines,
Biochim. Biophys. Acta, 1854, 1200-1211.
4.Percudani, R., and Peracchi, A. (2009) The B6
database: a tool for the description and classification of vitamin
B6-dependent enzymatic activities and of the corresponding protein
families, BMC Bioinformatics, 10, 1-8.
5.Percudani, R., and Peracchi, A. (2003) A genomic
overview of pyridoxal-phosphate-dependent enzymes, EMBO Rep.,
4, 850-854.
6.Grishin, N. V., Phillips, M. A., and Goldsmith, E.
J. (1995) Modeling of the spatial structure of eukaryotic ornithine
decarboxylases, Protein Sci., 4, 1291-1304.
7.Schneider, G., Kack, H., and Lindqvist, Y. (2000)
The manifold of vitamin B6 dependent enzymes, Structure,
8, 1-6.
8.Rudat, J., Brucher, B. R., and Syldatk, C. (2012)
Transaminases for the synthesis of enantiopure beta-amino acids, AMB
Express, 2, 1-10.
9.Eliot, A. C., and Kirsch, J. F. (2004) Pyridoxal
phosphate enzymes: mechanistic, structural, and evolutionary
considerations, Annu. Rev. Biochem., 73, 383-415.
10.Toney, M. D. (2011) Controlling reaction
specificity in pyridoxal phosphate enzymes, Biochim. Biophys.
Acta, 1814, 1407-1418.
11.John, R. A. (1995) Pyridoxal phosphate-dependent
enzymes, Biochim. Biophys. Acta, 1248, 81-96.
12.Hayashi, H. (1995) Pyridoxal enzymes mechanistic
diversity and uniformity, J. Biochem., 118, 463-473.
13.Jansonius, J. N. (1998) Structure evolution and
action of vitamin B6-dependent enzymes, Curr. Opin. Struct.
Biol., 8, 759-769.
14.Braunstein, A. E., and Kritzmann, M. G. (1937)
Formation and breakdown of amino acids by inter-molecular transfer of
amino group, Nature, 140, 503-504.
15.Cooper, A. J., and Meister, A. (1989) An
appreciation of Professor Alexander E. Braunstein. The discovery and
scope of enzymatic transamination, Biochimie, 71,
387-404.
16.Steffen-Munsberg, F., Vickers, C., Kohls, H.,
Land, H., Mallin, H., Nobili, A., Skalden, L., Van den Bergh, T.,
Joosten, H.-J., Berglund, P., Hohne, M., and Bornscheuer, U. T. (2015)
Bioinformatic analysis of a PLP-dependent enzyme superfamily suitable
for biocatalytic applications, Biotechnol. Adv., 33,
566-604.
17.Fuchs, M., Farnberger, J. E., and Kroutil, W.
(2015) The industrial age of biocatalytic transamination, Eur. J.
Org. Chem., 32, 6965-6982.
18.Hutson, S. (2001) Structure and function of
branched-chain aminotransferases, Prog. Nucl. Acid. Res. Mol.
Biol., 70, 175-206.
19.Deng, Y., Liu, J., Zheng, Q., Li, Q., Kallenbach,
N. R., and Lu, M. (2008) A heterospecific leucine zipper tetramer,
Chem. Biol., 15, 908-919.
20.Duan, Y., Li, F., Li, Y., Tang, Y., Kong, X.,
Feng, Z., Anthony, T. G., Watford, M., Hou, Y., Wu, G., and Yin, Y.
(2016) The role of leucine and its metabolites in protein and energy
metabolism, Amino Acids, 48, 41-51.
21.Danson, M. J., Lamble, H. J., and Hough, D. W.
(2007) Archaea: Molecular and Cellular Biology, ASM Press,
Washington DC, pp. 260-287.
22.Amorim Franco, T. M., Hegde, S., and Blanchard,
J. S. (2016) Chemical mechanism of the branched-chain aminotransferase
IlvE from Mycobacterium tuberculosis,
Biochemistry, 55, 6295-6303.
23.Zhang, S., Zeng, X., Ren, M., Mao, X., and Qiao,
S. (2017) Novel metabolic and physiological functions of branched chain
amino acids: a review, J. Anim. Sci. Biotechnol., 8,
10.
24.Murima, P., McKinney, J. D., and Pethe, K. (2014)
Targeting bacterial central metabolism for drug development, Chem.
Biol., 21, 1423-1432.
25.Amadasi, A., Bertoldi, M., Contestabile, R.,
Bettati, S., Cellini, B., Di Salvo, M. L., Borri-Voltattorni, C.,
Bossa, F., and Mozzarelli, A. (2007) Pyridoxal 5′-phosphate
enzymes as targets for therapeutic agents, Curr. Med. Chem.,
14, 1291-1324.
26.Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy,
S. R., Mistry, J., Mitchell, A. L., Potter, S. C., Punta, M., Qureshi,
M., Sangrador-Vegas, A., Salazar, G. A., Tate, J., and Bateman, A.
(2016) The Pfam protein families database: towards a more sustainable
future, Nucleic Acids Res., 44, D279-D285.
27.Zaitseva, J., Lu, J., Olechoski, K. L., and Lamb,
A. L. (2006) Two crystal structures of the isochorismate pyruvate lyase
from Pseudomonas aeruginosa, J. Biol. Chem., 281,
33441-33449.
28.Wildermuth, M. C., Dewdney, J., Wu, G., and
Ausubel, F. M. (2001) Isochorismate synthase is required to synthesize
salicylic acid for plant defense, Nature, 414,
562-565.
29.Hwang, B.-Y., Cho, B.-K., Yun, H., Koteshwar, K.,
and Kim, B.-G. (2005) Revisit of aminotransferase in the genomic era
and its application to biocatalysis, J. Mol. Catal. B Enzym.,
37, 47-55.
30.Koszelewski, D., Lavandera, I., Clay, D.,
Rozzell, D., and Kroutil, W. (2008) Asymmetric synthesis of optically
pure pharmacologically relevant amines employing ω-transaminases,
Adv. Synth. Catal., 350, 2761-2766.
31.Littlechild, J. A. (2015) Enzymes from extreme
environments and their industrial applications, Front. Bioeng.
Biotechnol., 161, 1-9.
32.Goldberg, J. M., and Kirsch, J. F. (1996) The
reaction catalyzed by Escherichia coli aspartate
aminotransferase has multiple partially rate-determining steps, while
that catalyzed by the Y225F mutant is dominated by ketimine hydrolysis,
Biochemistry, 35, 5280-5291.
33.Kirsch, J. F., Eichele, G., Ford, G. C., Vincent,
M. G., Jansonius, J. N., Gehring, H., and Christen, P. (1984) Mechanism
of action of aspartate aminotransferase proposed on the basis of its
spatial structure, J. Mol. Biol., 174, 497-525.
34.Toney, M. D., and Kirsch, J. F. (1989) Direct
Brensted analysis of the restoration of activity to a mutant enzyme by
exogenous amines, Science, 243, 1485-1488.
35.Toney, M. D., and Kirsch, J. F. (1992)
Brønsted analysis of aspartate aminotransferase via exogenous
catalysis of reactions of an inactive mutant, Protein Sci.,
1, 107-119.
36.Liao, R. Z., Ding, W. J., Yu, J. G., Fang, W. H.,
and Liu, R. Z. (2008) Theoretical studies on pyridoxal
5′-phosphate-dependent transamination of alpha-amino acids, J.
Comput. Chem., 29, 1919-1929.
37.Toney, M. D., and Kirsch, J. F. (1991) The K258R
mutant of aspartate aminotransferase stabilizes the quinonoid
intermediate, J. Biol. Chem., 266, 23900-23903.
38.Karsten, W. E., and Cook, P. F. (2009) Detection
of a gem-diamine and a stable quinonoid intermediate in the reaction
catalyzed by serine-glyoxylate aminotransferase from Hyphomicrobium
methylovorum, Biochim. Biophys. Acta, 1790,
575-580.
39.Gehring, H. (1984) Transfer of C alpha-hydrogen
of glutamate to coenzyme of aspartate aminotransferase during
transamination reaction, Biochemistry, 23, 6335-6340.
40.Rishavy, M. A., and Cleland, W. W. (2000)
13C and 15N kinetic isotope effects on the
reaction of aspartate aminotransferase and the tyrosine-225 to
phenylalanine mutant, Biochemistry, 39, 7546-7551.
41.Spies, M. A., and Toney, M. D. (2003) Multiple
hydrogen kinetic isotope effects for enzymes catalyzing exchange with
solvent: application to alanine racemase, Biochemistry,
42, 5099-5107.
42.Saito, M., Nishimura, K., Wakabayashi, S.,
Kurihara, T., and Nagata, Y. (2007) Purification of branched-chain
amino acid aminotransferase from Helicobacter pylori NCTC 11637,
Amino Acids, 33, 445-449.
43.Koide, Y., Honma, M., and Shimomura, T. (1977)
Branched chain amino acid aminotransferase of Pseudomonas sp.,
Agric. Biol. Chem., 41, 1171-1177.
44.Goto, M., Miyahara, I., Hayashi, H., Kagamiyama,
H., and Hirotsu, K. (2003) Crystal structures of branched-chain amino
acid aminotransferase complexed with glutamate and glutarate: true
reaction intermediate and double substrate recognition of the enzyme,
Biochemistry, 42, 3725-3733.
45.Soda, K., Yoshimura, T., and Esaki, N. (2001)
Stereospecificity for the hydrogen transfer of pyridoxal enzyme
reactions, Chem. Rec., 1, 373-384.
46.Dunathan, H. C. (1966) Conformation and reaction
specificity in pyridoxal phosphate enzymes, Proc. Natl. Acad. Sci.
USA, 55, 712-716.
47.Ruan, J., Hu, J., Yin, A., Wu, W., Cong, X.,
Feng, X., and Li, S. (2012) Structure of the branched-chain
aminotransferase from Streptococcus mutants, Acta
Crystallogr. Sect. D, 68, 996-1002.
48.Rudman, D., and Meister, A. (1953) Transamination
in Escherichia coli, J. Biol. Chem., 200,
591-604.
49.Feldman, L. I., and Gunsalus, I. C. (1950) The
occurrence of a wide variety of transaminases in bacteria, J. Biol.
Chem., 187, 821-830.
50.Gelfand, D. H., and Steinberg, R. A. (1977)
Escherichia coli mutants deficient in the aspartate and aromatic
amino acid aminotransferases, J. Bacteriol., 130,
429-440.
51.Jensen, R. A., and Calhoun, D. H. (1981)
Intracellular roles of microbial aminotransferases: overlap enzymes
across different biochemical pathways, Crit. Rev. Microbiol.,
8, 229-266.
52.Wang, M. D., Liu, L., Wang, B. M., and Berg, C.
M. (1987) Cloning and characterization of Escherichia coli K-12
alanine-valine transaminase (avtA) gene, J. Bacteriol.,
169, 4228-4234.
53.Kline, E. L., Brown, C. S., Coleman, W. G., Jr.,
and Umbarger, H. E. (1974) Regulation of isoleucine-valine biosynthesis
in an ilvDAC deletion strain of Escherichia coli K-12,
Biochem. Biophys. Res. Commun., 57, 1144-1151.
54.Kline, E. L., Manross, D. N., Jr., and Warwick,
M. L. (1977) Multivalent regulation of isoleucine-valine transaminase
in an Escherichia coli K-12 ilvA deletion strain, J.
Bacteriol., 130, 951-953.
55.Harris, R. A., and Sokatch, J. R. (2000)
Branched-chain amino acids (Part B) N324, in Methods in
Enzymology, Academic Press, London, p. 535.
56.Rej, R. (1982) A convenient continuous-rate
spectrophotometric method for determination of amino acid substrate
specificity of aminotransferases: application to isoenzymes of
aspartate aminotransferase, Anal. Biochem., 119,
205-210.
57.Duggan, D. E., and Wechsler, J. A. (1973) An
assay for transaminase B enzyme activity in Escherichia
coli K-12, Anal. Biochem., 51, 67-79.
58.Taylor, R. T., and Jenkins, W. T. (1966) Leucine
aminotransferase I. Colorimetric assays, J. Biol. Chem.,
241, 4391-4395.
59.Lee-Peng, F.-C., Hermodson, M. A., and Kohlhaw,
G. B. (1979) Transaminase B from Escherichia coli: quaternary
structure, amino-terminal sequence, substrate specificity, and absence
of a separate valine-α-ketoglutarate activity, J.
Bacteriol., 139, 339-345.
60.Inoue, K., Kuramitsu, S., Aki, K., Watanabe, Y.,
Takagi, T., Nishigai, M., Ikai, A., and Kagamiyama, H. (1988)
Branched-chain amino acid aminotransferase of Escherichia coli:
overproduction and properties, J. Biochem., 104,
777-784.
61.Toney, M. D. (2014) Aspartate aminotransferase:
an old dog teaches new tricks, Arch. Biochem. Biophys.,
544, 119-127.
62.Yu, X., Wang, X., and Engel, P. C. (2014) The
specificity and kinetic mechanism of branched-chain amino acid
aminotransferase from Escherichia coli studied with a new
improved coupled assay procedure and the enzyme’s potential for
biocatalysis, FEBS J., 281, 391-400.
63.Norton, J. E., and Sokatch, J. R. (1970)
Purification and partial characterization of the branched chain amino
acid transaminase of Pseudomonas aeruginosa, Biochim.
Biophys. Acta, 206, 261-269.
64.Tachiki, T., and Tochikura, T. (1973) Separation
of L-leucine-pyruvate and L-leucine-alpha-ketoglutarate transaminases
in Acetobacter suboxydans and identification of their reaction
products, Agric. Biol. Chem., 37, 1439-1448.
65.Tachiki, T., and Tochikura, T. (1976)
Purification and characterization of L-leucine-alpha-ketoglutarate
transaminase from Acetobacter suboxydans, Agric. Biol.
Chem., 40, 2187-2192.
66.Yvon, M., Chambellon, E., Bolotin, A., and
Roudot-Algaron, F. (2000) Characterization and role of the
branched-chain aminotransferase (BcaT) isolated from Lactococcus
lactis subsp. cremoris NCDO 763, Appl. Environ.
Microbiol., 66, 571-577.
67.Thage, B. V., Rattray, F. P., Laustsen, M. W.,
Ardo, Y., Barkholt, V., and Hourberg, U. (2004) Purification and
characterization of a branched-chain amino acid aminotransferase from
Lactobacillus paracasei subsp. paracasei CHCC 2115, J.
Appl. Microbiol., 96, 593-602.
68.Kanda, M., Hori, K., Kurotsu, T., Ohgishi, K.,
Hanawa, T., and Saito, Y. (1995) Purification and properties of
branched chain amino acid aminotransferase from gramicidin S-producing
Bacillus brevis, J. Nutr. Sci. Vitaminol., 41,
51-60.
69.Venos, E. S., Knodel, M. H., Radford, C. L., and
Berger, B. J. (2004) Branched-chain amino acid aminotransferase and
methionine formation in Mycobacterium tuberculosis, BMC
Microbiol., 4, 1-14.
70.Madsen, S. M., Beck, H. C., Rawn, P., Vrang, A.,
Hansen, A. M., and Israelsen, H (2002) Cloning and inactivation of a
branched-chain-amino-acid aminotransferase gene from Staphylococcus
carnosus and characterization of the enzyme, Appl. Environ.
Microbiol., 68, 4007-4014.
71.Berger, B. J., English, S., Chan, G., and Knodel,
M. H. (2003) Methionine regeneration and aminotransferases in
Bacillus subtilis, Bacillus cereus, and Bacillus
anthracis, J. Bacteriol., 185, 2418-2431.
72.Wong, H. C., and Lessie, T. G. (1979) Branched
chain amino acid aminotransferase isoenzymes of Pseudomonas
cepacia, Arch. Microbiol., 120, 223-229.
73.Massey, L. K., Conrad, R. S., and Sokatch, J. R.
(1974) Regulation of leucine catabolism in Pseudomonas
putida, J. Bacteriol., 118, 112-120.
74.Chen, C. D., Lin, C. H., Chuankhayan, P., Huang,
Y. C., Hsieh, Y. C., Huang, T. F., Guan, H. H., Liu, M. Y., Chang, W.
C., and Chen, C. J. (2012) Crystal structures of complexes of the
branched-chain aminotransferase from Deinococcus radiodurans
with α-ketoisocaproate and L-glutamate suggest the radiation
resistance of this enzyme for catalysis, J. Bacteriol.,
194, 6206-6216.
75.Lipscomb, E. L., Horton, H. R., and Armstrong, F.
B. (1974) Molecular weight, subunit structure, and amino acid
composition of the branched chain amino acid aminotransferase of
Salmonella typhimurium, Biochemistry, 13,
2070-2077.
76.Feild, M. J., Nguyen, D. C., and Armstrong, F. B.
(1989) Amino acid sequence of Salmonella typhimurium
branched-chain amino acid aminotransferase, Biochemistry,
28, 5306-5310.
77.Xing, R. Y., and Whitman, W. B. (1991)
Characterization of enzymes of the branched-chain amino acid
biosynthetic pathway in Methanococcus spp., J.
Bacteriol., 173, 2086-2092.
78.Xing, R. Y., and Whitman, W. B. (1992)
Characterization of amino acid aminotransferases of Methanococcus
aeolicus, J. Bacteriol., 174, 541-548.
79.Boyko, K. M., Stekhanova, T. N., Nikolaeva, A.
Y., Mardanov, A. V., Rakitin, A. L., Ravin, N. V., Bezsudnova, E. Yu.,
and Popov, V. O. (2016) First structure of archaeal branched-chain
amino acid aminotransferase from Thermoproteus uzoniensis
specific for L-amino acids and R-amines, Extremophiles,
20, 215-225.
80.Bezsudnova, E. Yu., Stekhanova, T. N., Suplatov,
D. A., Mardanov, A. V., Ravin, N. V., and Popov, V. O. (2016)
Experimental and computational studies on the unusual substrate
specificity of branched-chain amino acid aminotransferase from
Thermoproteus uzoniensis, Arch. Biochem. Biophys.,
607, 27-36.
81.Stekhanova, T. N., Rakitin, A. L., Mardanov, A.
V., Bezsudnova, E. Yu., and Popov, V. O. (2017) A novel highly
thermostable branched-chain amino acid aminotransferase from the
crenarchaeon Vulcanisaeta moutnovskia, Enzyme Microb.
Technol., 96, 127-134.
82.Uchida, Y., Hayashi, H., Washio, T., Yamasaki,
R., Kato, S., and Oikawa, T. (2014) Cloning and characterization of a
novel fold-type I branched-chain amino acid aminotransferase from the
hyperthermophilic archaeon Thermococcus sp. CKU-1,
Extremophiles, 18, 589-602.
83.Hayashi, H., Inoue, K., Nagata, T., Kuramitsu,
S., and Kagamiyama, H. (1993) Escherichia coli aromatic amino
acid aminotransferase: characterization and comparison with aspartate
aminotransferase, Biochemistry, 32, 12229-12239.
84.Oue, S., Okamoto, A., Nakai, Y., Nakahira, M.,
Shibatani, T., Hayashi, H., and Kagamiyama, H. (1997) Paracoccus
denitrificans aromatic amino acid aminotransferase: a model enzyme
for the study of dual substrate recognition mechanism, J.
Biochem., 121, 161-171.
85.Conway, M. E., and Hutson, S. M. (2000) Mammalian
branched-chain aminotransferases, Methods Enzymol., 324,
355-365.
86.Braunstein, A. E. (1973) The Enzymes
(Boyer, P. D., ed.) 3rd Edn., Academic Press, New York, pp.
379-471.
87.Beeler, T., and Churchich, J. E. (1976)
Reactivity of the phosphopyridoxal groups of cystathionase, J. Biol.
Chem., 251, 5267-5271.
88.Ward, J., and Wohlgemuth, R. (2010) High-yield
biocatalytic amination reactions in organic synthesis, Curr. Org.
Chem., 14, 1-12.
89.Li, T., Kootstra, A. B., and Fotheringham, I. G.
(2002) Nonproteinogenic α-amino acid preparation using
equilibrium shifted transamination, Org. Proc. Res. Dev.,
6, 533-538.
90.Bommarius, A. S., Schwarm, M., and Drauz, K.
(1998) Biocatalysis to amino acid-based chiral
pharmaceuticals – examples and perspectives, J. Mol.
Catal. B Enzym., 5, 1-11.
91.Krix, G., Bommarius, A. S., Drauz, K.,
Kottenhahn, M., Schwarm, M., and Kula, M. R. (1997) Enzymatic reduction
of alpha-keto acids leading to L-amino acids, D- or L-hydroxy acids,
J. Biotech., 53, 29-39.
92.Xian, M., Alaux, S., Sagot, E., and Gefflaut, T.
(2007) Chemoenzymatic synthesis of glutamic acid analogues: substrate
specificity and synthetic applications of branched chain
aminotransferase from Escherichia coli, J. Org. Chem.,
72, 7560-7566.
93.Faure, S., Jensen, A. A., Maurat, V., Gu, X.,
Sagot, E., Aitken, D. J., Bolte, J., Gefflaut, T., and Bunch, L. (2006)
Stereoselective chemoenzymatic synthesis of the four stereoisomers of
L-2-(2-carboxy-cyclobutyl)glycine and pharmacological characterization
at human excitatory amino acid transporter subtypes 1, 2, and 3, J.
Med. Chem., 49, 6532-6538.
94.Okada, K., Hirotsu, K., Sato, M., Hayashi, H.,
and Kagamiyama, H. (1997) Three-dimensional structure of Escherichia
coli branched-chain amino acid aminotransferase at 2.5 Å
resolution, J. Biochem., 121, 637-641.
95.Okada, K., Hirotsu, K., Hayashi, H., and
Kagamiyama, H. (2001) Structures of Escherichia coli
branched-chain amino acid aminotransferase and its complexes with
4-methylvalerate and 2-methylleucine: induced fit and substrate
recognition of the enzyme, Biochemistry, 40,
7453-7463.
96.Castell, A., Mille, C., and Unge, T. (2010)
Structural analysis of mycobacterial branched-chain aminotransferase:
implications for inhibitor design, Acta Crystallogr. Sect. D,
66, 549-557.
97.Tremblay, L. W., and Blanchard, J. S. (2009) The
1.9 Å structure of the branched-chain amino-acid transaminase
(IlvE) from Mycobacterium tuberculosis, Acta Crystallogr.
Sect. F, 65, 1071-1077.
98.Yennawar, N., Dunbar, J., Conway, M., Hutson, S.,
and Farber, G. (2001) The structure of human mitochondrial
branched-chain aminotransferase, Acta Crystallogr. Sect. D,
57, 506-515.
99.Kochhar, S., and Christen, P. (1992) Mechanism of
racemization of amino acids by aspartate aminotransferase, Eur. J.
Biochem., 203, 563-569.
100.Shaw, J. P., Petsko, G. A., and Ringe, D.
(1997) Determination of the structure of alanine racemase from
Bacillus stearothermophilus at 1.9 Å resolution,
Biochemistry, 36, 1329-1342.
101.Hohne, M., Schatzle, S., Jochen, H., Robins,
K., and Bornscheuer, U. T. (2010) Rational assignment of key motifs for
function guides in silico enzyme identification, Nat. Chem.
Biol., 6, 807-813.
102.Skalden, L., Thomsen, M., Hohne, M.,
Bornscheuer, U. T., and Hinrichs, W. (2015) Structural and biochemical
characterization of the dual substrate recognition of the (R)-selective
amine transaminase from Aspergillus fumigatus, FEBS J.,
282, 407-415.
103.Peisach, D., Chipman, D. M., Van Ophem, P. W.,
Manning, J. M., and Ringe, D. (1998) Crystallographic study of steps
along the reaction pathway of D-amino acid aminotransferase,
Biochemistry, 37, 4958-4967.
104.Iwasaki, A., Matsumoto, K., Hasegawa, J., and
Yasohara, Y. (2012) A novel transaminase, (R)-amine:pyruvate
aminotransferase, from Arthrobacter sp. KNK168 (FERM BP-5228):
purification, characterization, and gene cloning, Appl. Microbiol.
Biotechnol., 93, 1563-1573.
105.Hirotsu, K., Goto, M., Okamoto, A., and
Miyahara, I. (2005) Dual substrate recognition of aminotransferases,
Chem. Rec., 5, 160-172.
106.Goto, M., Omi, R., Miyahara, I., Hosono, A.,
Mizuguchi, H., Hayashi, H., Kagamiyama, H., and Hirotsu, K. (2004)
Crystal structures of glutamine:phenylpyruvate aminotransferase from
Thermus thermophilus HB8: induced fit and substrate recognition,
J. Biol. Chem., 279, 16518-16525.
107.Koshland, D. E. (1995) The key-lock theory and
the induced fit theory, Angew. Chem., 33, 2375-2378.
108.Karsten, W. E., Reyes, Z. L., Bobyk, K. D.,
Cook, P. F., and Chooback, L. (2011) Mechanism of the aromatic
aminotransferase encoded by the Aro8 gene from Saccharomyces
cerevisiae, Arch. Biochem. Biophys., 516, 67-74.
109.Jiang, J., Chen, X., Zhang, D., Wu, Q., and
Zhu, D. (2015) Characterization of (R)-selective amine transaminases
identified by silico motif sequence blast, Appl. Microbial.
Biotechnol., 99, 2613-2621.