REVIEW: Novel HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors: A Combinatorial Approach
V. T. Valuev-Elliston* and S. N. Kochetkov
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia; E-mail: gansfaust@mail.ru* To whom correspondence should be addressed.
Received July 24, 2017; Revision received August 1, 2017
Highly active antiretroviral therapy (HAART) is one of the most effective means for fighting against HIV-infection. HAART primarily targets HIV-1 reverse transcriptase (RT), and 14 of 28 compounds approved by the FDA as anti-HIV drugs act on this enzyme. HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) hold a special place among HIV RT inhibitors owing to their high specificity and unique mode of action. Nonetheless, these drugs show a tendency to decrease their efficacy due to high HIV-1 variability and formation of resistant virus strains tolerant to clinically applied HIV NNRTIs. A combinatorial approach based on varying substituents within various fragments of the parent molecule that results in development of highly potent compounds is one of the approaches aimed at designing novel HIV NNRTIs. Generation of HIV NNRTIs based on pyrimidine derivatives explicitly exemplifies this approach, which is discussed in this review.
KEY WORDS: HIV, HIV reverse transcriptase, inhibitors, NNRTIsDOI: 10.1134/S0006297917130107
Abbreviations: DABO, dihydroalkoxybenzyloxopyrimidine; DAPY, diarylpyrimidine; DLV, delavirdine; DPV, dapivirine; EC50, effective concentration (concentration of a substance which causes 50% viral particle reduction); EFV, efavirenz; ETV, etravirine; FDA, Food and Drug Administration; HEPT, 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine; HIV, human immunodeficiency virus; HAART, highly active antiretroviral therapy; IC50, half maximal inhibitory concentration; NNRTIs, non-nucleoside reverse transcriptase inhibitors; NVP, nevirapine; RPV, rilpivirine; RT, reverse transcriptase; WT, wild type (virus or protein).
Since the human immunodeficiency virus (HIV) was described in 1983,
tremendous efforts aimed at prevention, prophylaxis, and suppression of
HIV-infection have been made. This significantly lowered the incidence
of HIV-infection in countries of Europe and North America, making it a
chronic, non-lethal infection, which is quite efficiently restrained by
drugs used in highly active antiretroviral therapy (HAART).
Nonetheless, the global number of HIV carriers generally continues to
increase, now reaching ~40 million people worldwide [1].
In Russia, the situation remains disappointing. According to monitoring data presented by the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing, 1,114,815 HIV-infection cases (preliminary data) were reported among citizens of the Russian Federation by December 31, 2016, including 243,883 persons who died from various causes. In 2016, territorial centers for AIDS Prevention and Control reported 103,438 new HIV-infection cases, 5.3% higher than in 2015.
Since 2005, an increase in newly identified HIV-cases has been recorded in Russia, in 2011-2016 comprising 10% annual average increase. In 2016, HIV morbidity rate was 70.6 per 100,000 people [2]. In Russia, one of the negative factors is poor coverage with treatment interventions. Among 675,000 HIV-infected persons subject to dispensary check-up, 286,000 (42.3%) patients received HAART. This treatment coverage does not serve as a preventive intervention and allow drastically lower rate of HIV-infection spread [2], due, in part, to high cost of therapy because of insufficient availability of domestic anti-HIV drugs.
By 2016, around 30 drugs were approved by the FDA to fight against HIV-infection [3], primarily including inhibitors of reverse transcriptase (RT), a key enzyme involved in HIV replication, as well as inhibitors blocking entry of the virus into cells (fusion and binding to co-receptors), maturation of virus particles (protease inhibitors), and insertion of HIV DNA provirus into the host genome (integrase inhibitors). Such drugs are applied separately and as a part (in most cases) of HAART combination cocktails containing at least three drugs simultaneously targeting several stages in the HIV life cycle, thereby most efficiently limiting disease progression.
Administration of drug cocktails is one of the most successful strategies for treatment of HIV-infection, limiting disease progression especially at early stages. HAART lowers plasma viremia level below the detection limit within six months [4].
However, some issues are still far from solved. While applying HAART, the main problem is rapidly developing drug resistance in HIV strains, thus requiring periodical change in anti-HIV drugs. According to WHO, in 2010 in developing versus developed countries ~7% versus 10-20% of HIV patients at the onset of antiretroviral therapy (ART) were found to develop drug resistance. Recently, some countries report 15% or greater drug resistance rate in patients at the onset of anti-HIV therapy and up to 40% in patients restarting treatment [5]. Adverse effects and toxicity issues often resulting in non-adherence to ART regimens are still of high significance [6]. Thus, development of novel anti-HIV drugs with improved resistance profile and increased tolerability remains relevant. Among anti-HIV drugs, so-called non-nucleoside reverse transcriptase inhibitors, the development of which is reviewed here, hold a special place.
HIV RT INHIBITION
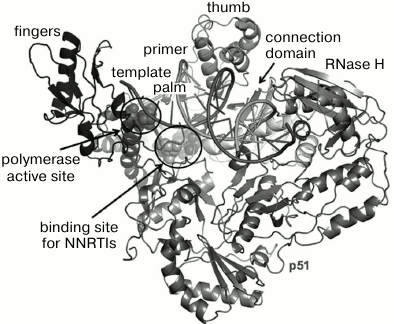
Properties of HIV and features of its life cycle have been described in textbooks and numerous reviews [7-10]. Reverse transcription converting genetic information of single-stranded viral RNA into its double-stranded DNA copy is a key stage in the HIV life cycle [11]. Reverse transcription is catalyzed by HIV reverse transcriptase (RT), which is an asymmetric heterodimer consisting of the two structurally dissimilar subunits p66 and p51, the latter being formed from proteolytically cleaved p66. RT exhibits two catalytic activities mediated through active sites located in the p66 subunit: (i) it acts as a polymerase, i.e. it can complete DNA chain synthesis on an RNA or DNA template, and (ii) it can act as RNase H, degrading RNA within RNA-DNA hybrids. The structure of the HIV RT (p66/p51) heterodimer resembles a right-hand shape [12] typical of several nucleotide polymerase types. Accordingly, several subdomains are distinguished within the RT structure, such as “fingers” (residues 1-85, 118-155), “palm” (residues 86-117, 156-236), “thumb” (residues 237-318), “connection” (residues 319-426), and RNase H (residues 427-560) (Fig. 1) [13, 14]. A catalytic site responsible for polymerase activity is located within the “palm” subdomain, which contains a highly conserved YMDD motif. In the RNase H subdomain, the active center is located 17-18 bp from the polymerase active center.
Fig. 1. HIV RT spatial organization [15].
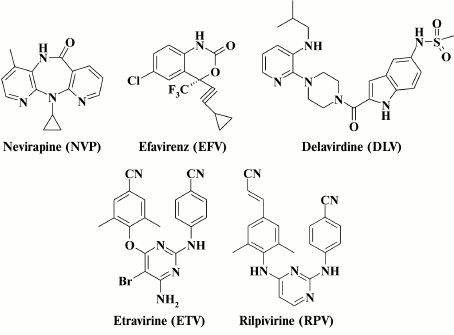
Generation of a set of specific inhibitors that can serve as effective pharmaceutical agents was motivated by the key role that RT plays in the HIV life cycle. These inhibitors belong to two major classes: (i) nucleoside (nucleotide) reverse transcriptase inhibitors (NRTIs); (ii) non-nucleoside RT inhibitors (NNRTIs). The NRTIs entering infected cells, convert nucleosides into relevant NTPs, and after being inserted into a nascent viral DNA result in its termination, as their structure does not allow further DNA chain elongation. Nucleotide inhibitors must be phosphorylated at two sites, as they already contain the 5′-phosphate residue. The NNRTIs are organic compounds belonging to various classes containing substantial amounts of aromatic hydrophobic radicals (Fig. 2). The NNRTIs are noncompetitive RT inhibitors binding to its allosteric site, which affects mobility and plasticity of the polymerization site, thereby resulting in profoundly downregulated RT activity. The binding site for NNRTIs is located 10 Å from the polymerase site in the p66 subunit, which is mainly constructed from hydrophobic amino acid residues (p66 subunit: L100, K101, K103, V106, T107, V108, V179, Y181, Y188, V189, G190, F227, W229, L234, Y318; p51 subunit: E138 [13, 16]). Examination of RT HIV crystals complexed with NNRTIs demonstrated that the hydrophobic pocket is formed only when NNRTIs are bound, and they are not observed in the inhibitor-free structure [17-20].
Fig. 2. HIV NNRTIs approved by the FDA [25].
NNRTIs exhibit inhibitory activity differently. For instance, binding of nevirapine (NVP; see Fig. 3) changes the position of hydrophobic residues Y181 and Y188, and, consequently, move apart two components of protein tertiary structure such as β-sheets β6-β10-β9 and β12-β13-β14 containing catalytic amino acid residues D110-D185-D186 and the primer grip region for its 3′-OH terminus in polymerase reaction, respectively. As a result, the primer is displaced relative to the enzyme catalytic site by 4 Å, so the conserved catalytic YMDD motif loses interaction with the primer terminus [21].
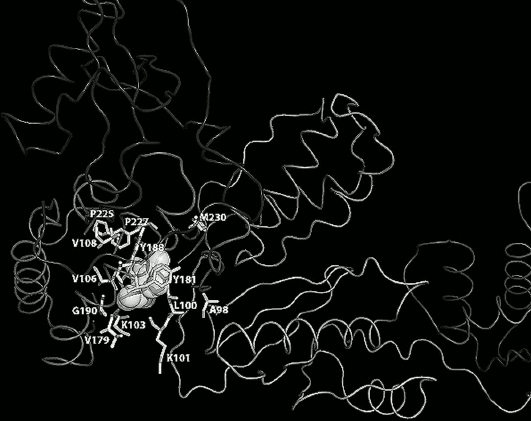
Fig. 3. Amino acid residues in the hydrophobic pocket (shown as gray balls), responsible for resistance to NNRTIs. E138 in the p51 and L234, P236, K238, L318 in the extended hydrophobic pocket are not shown.
On the other hand, NNRTIs may also influence dynamic interactions between RT and a nucleic acid template [22, 23]. In particular, in the absence of the inhibitor, HIV RT can slide along a nucleic acid, whereas binding to NNRTIs promotes this process [23, 24]. For efavirenz (EFV; see Fig. 3), it was demonstrated that after binding it contributes to opening of the hand and extended the distance between HIV RT thumb and fingers subdomains. Displacement towards a sliding conformation profoundly reduces binding to dNTPs. Formation of the E138–K101 salt bridge between the p51 and p66 subunits of RT results in emergence of the opened hand structure typical for sliding conformation, whereas EFV in the hydrophobic pocket stabilizes it. Amino acid residue K103 contributes to orienting K101 towards E138, and its substitution in the K103N mutant disrupts the E138–K101 salt bridge as well as stabilizing the enzyme in a closed hand position, thus enhancing polymerase activity. This explains resistance of the K103N mutant to a whole range of HIV NNRTIs [22].
Emergence of resistance to all anti-HIV drugs occurring in patients undergoing antiretroviral therapy is the most acute issue [26, 27]. In addition, HIV NNRTIs is not an exception; two drug generations are distinguished depending on the resistance profile – the first-generation drugs include nevirapine (NVP), efavirenz (EFV), and delavirdine (DLV), whereas etravirine (ETV) and rilpivirine (RPV) belong to the second-generation drugs [28, 29]. However, despite a broad range of resistance profiles observed in the second-generation HIV NNRTIs, patients with mutations in HIV RT have been already reported in clinical practice, which makes HIV resistant to this class of agents [26].
High HIV genetic variability results in multiple amino acid substitutions within the RT, leading, in part, to its resistance to NNRTIs. Most resistance mutations are related to substitution of amino acids forming the hydrophobic pocket (Fig. 3), but also there are substitutions found in the connection subdomain that result in resistance to NNRTIs [30, 31]. Several mechanisms underlying RT resistance to NNRTIs have been proposed: steric hindrance for positioning of NNRTIs inside the hydrophobic pocket (L100I, G190A) [32]; loss of interactions with amino acid residues within the pocket (Y181C, Y188L) [33]; hindered access of NNRTIs to the hydrophobic pocket (K103N and K101E) [34, 35]; disrupted K101-E138 salt bridge stabilizing the opened RT conformation (K103N, K101E/H/P, E138A/G/K/Q/R) [22]; indirectly reduced association rate of NNRTI with RT (mutations in the connection subdomain N348I, T369I/V, T376S) [30, 31].
It should be noted that the impact of different mechanisms of NNRTI resistance varies greatly depending on the class of the agents. Due to a rigid structure of the first-generation drugs, NVP and EFV had a very low genetic barrier to resistance, so that even a single mutation was able to sharply reduce their efficacy, thus resulting in elimination of the drug from clinical practice [36]. More advanced second-generations drugs etravirine and rilpivirine lack this drawback and are suitable for the treatment of patients with resistance to the first-generation NNRTIs [37-39]. Nonetheless, a combination of three or more mutations also results in resistance to the second-generation NNRTIs [40]. For patients unresponsive to ETV therapy, it was found that they typically contain HIV strains with mutations K103N, Y181C, G190A [41] combined with another 15 mutations in the hydrophobic pocket – V90I, A98G, L100I, K101E/H/P, V106I, E138A, V179D/F/T, Y181I/V, G190S, M230L [37, 38] as well as mutation N348I within the connection subdomain in p66 [42]. Patients strongly resistant to RPV therapy were found to usually contain mutations K101P, Y181I/V [41] in the HIV strains in combination with another 14 mutations L100I, K101E, E138A/G/K/Q/R, V179L, Y181C, Y188L, H221Y, F227C, and M230I/L [43-45]. Compared to ETV, RPV is resistant to mutation N348I in the connection subdomain [31].
High activity of NNRTIs combined with their low toxicity makes this class of inhibitors of great promise both for the future search for novel compounds [46], refining a mode of application for already approved drugs as stand-alone agents [47], a part of inhibitor cocktails [48], as well as drugs preventing HIV infection [49].
DIARYLPYRIMIDINE (DAPY)-BASED NNRTIs
In 2008 and 2011, the two second-generation HIV NNRTIs etravirine and rilpivirine were approved for clinical application, both being diarylpyrimidines (DAPY). Conformational flexibility is the major feature found in DAPYs [50] allowing them to better adapt to amino acid substitutions occurring within the hydrophobic pocket, which is the reason for resistance to the first-generation HIV NNRTIs.
Diarylpyrimidines were developed from studies identifying HIV NNRTIs among α-anilinophenylacetamides (α-APA) [51], which led to compound R15345 (1; Table 1) exhibiting submicromolar inhibitory activity in HIV-infected cells [52]. Studies aimed at optimizing R15345 produced another HIV NNRTI known as R89439 (loviride) (2), which is more potent than R15345 (1), but it was neglected during clinical trials due to lack of clear benefit compared to the first-generation HIV NNRTIs delavirdine and nevirapine. Using R89439 (2), imidoyl thiourea derivatives (ITU) were generated, among which a compound R100943 (3) displayed the strongest activity [53]. However, R100943 (3) also failed during clinical trials due to hydrolytic instability of its imidoyl thiourea moiety. To improve the pharmacokinetic properties of R100943 (3), a thioketo group was replaced by an iminocyano group, which resulted in an unexpected ring closure, producing the diaryl triazine R106168 (4) having nanomolar inhibitory activity against WT HIV (EC50 = 0.0063 µM [54]) and a high genetic barrier to resistance (Table 1) [55].
Table 1. Inhibitory activity for compounds
1-5 against WT and NNRTI-resistant HIV mutants [51, 52]
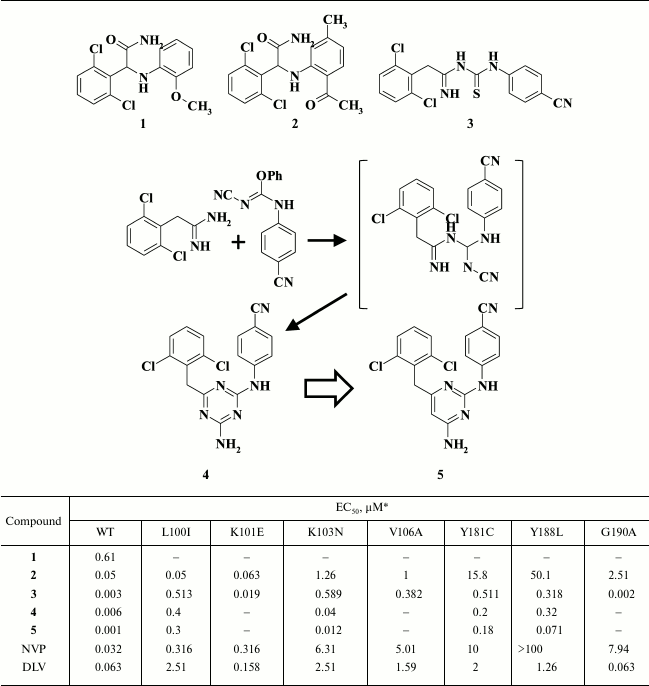
* MT-4 cell line was used in the experiments.
During further investigations, the central triazine moiety was replaced by a pyrimidine ring [56]. Studies with three isomers of diarylpyrimidine (DAPY) and a triazine derivative demonstrated that compound 5 showed the most promise, not only exhibiting the highest antiviral potency against WT HIV RT, but a broad resistance profile as well (Table 1) [39].
Molecular modeling of compound 5 [57] predicted that the amino group between the central pyrimidine ring and the 4-cyanobenzene fragment might form a hydrogen bond with the main chain K101 carbonyl oxygen, which was fully confirmed by obtaining HIV RT crystalized with various DAPYs: TMC120 [39], etravirine [58], and rilpivirine [58, 59].
Further description of modifications in various moieties of DAPYs will be done in accordance with the abbreviations shown in Fig. 4.
Fig. 4. Scheme for modifications in the DAPY scaffold based on compound 5.
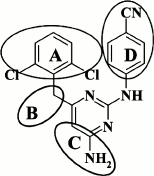
Modified DAPY A-, B-, and C-rings. By substituting an alkyl linker between the left aromatic ring and the central ring with an oxygen, sulfur, or amino group [57], it was demonstrated that a nature of these substituents insignificantly affected the antiviral activity of the corresponding compounds against both WT and resistant mutant HIV strains.
Examining the impact of substituents in the left aromatic ring of the molecule revealed some patterns showing that the most potent DAPYs should have substituents at the 2,4,6-positions of the left aromatic ring (Table 2) [57].
Table 2. Inhibitory activity for compounds
6-11 against WT and mutant HIV-1 RT [56, 57]
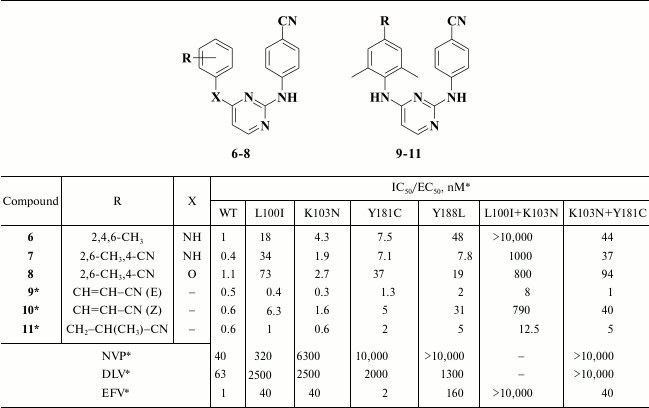
* MT-4 cell line was used in the experiments.
Compound 6 (R147681, TMC120, dapivirine) was developed as a microbicidal agent [60, 61]; it displayed paradoxically good bioavailability for such a hydrophobic molecule, and it was even tested in phase III clinical trial, but was withdrawn [62]. Compounds 7 and 8 became the basis for further designing various DAPY derivatives. An important feature of such compounds is the presence of a cyano group at the para-position of the aromatic ring. According to molecular modeling data for analogs of compound 7 [63] and X-ray structural analysis of dapivirine (6) crystalized with HIV RT [64], a cyano group at the para-position of the aromatic ring should interact with a conserved amino acid residue W229, which is of great value in fighting against resistant mutations due to extreme HIV variability. To strengthen binding to W229, analogs of compound 7 containing a linker between the cyano group at the para-position and the aromatic ring (Table 2) were synthesized [65].
It turned out that the most potent were compounds 9 and 11, and the Z-isomer of compound 10, as expected according to computer modeling data, was less potent due to a weaker interaction between the cyano group and W229. Due to a high selectivity index exceeding 60,000, it directed further investigation to compound 9 [65]. After varying substituents at the ortho-position of the aromatic A-ring as well as a linker between the left and central aromatic moieties, no evident benefits for analogs of compound 9 were found [66]. Compound 9 was patented as TMC278 (rilpivirine) [67], and based on the results of clinical trials in 2011 it was approved by the FDA as HIV NNRTI [68].
Trying to enhance inhibitory activity of the compounds, an amino group between the central and left aromatic rings was substituted for a hydroxyimino group [69].
Despite exhibiting high inhibitory activity against WT HIV (for the lead compound EC50 = 13 nM), in the case of double mutants K103N+Y181C, for these compounds it was reduced by several orders of magnitude. Among those studied by Feng et al. [69], special attention should be given to compound 12 displaying inhibitory activity against HIV-2 (EC50 = 8.3 µM), which might suggest several potential mechanisms of its action on HIV-1.
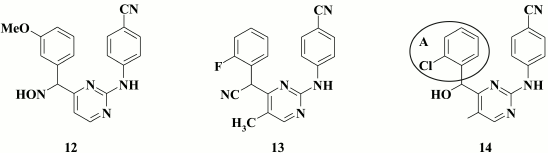
A similar effect was observed by incorporating a cyano group into a linker [70]. The lead compound 13 (EC50 = 1.8 nM) exhibited inhibitory activity in the nanomolar range against WT HIV, but it completely lacked activity against double-mutant K103N+Y181C virus.
Like hydroxyimino derivatives, hydroxymethyl analogs of DAPY demonstrated several potential mechanisms for inhibiting HIV, such as strong inhibitory activity against WT HIV, which was profoundly reduced against double mutants K103N+Y181C, as well as maintaining inhibitory activity against HIV-2 [71].
A thorough examination was conducted for optical isomers of the most potent compound 14 (EC50 = 5 nM), and it revealed that the R-isomer was more potent against WT HIV-1, whereas the S-isomer better acted against the K103N+Y181C double mutant and HIV-2.
Computer modeling of both isomers revealed a difference in antiviral activity. It was found that the left aromatic A-ring of the R-isomer 14 could make a stacking interaction with amino acids Y181, Y188, and W229 similarly to that in case of the DAPY TMC120 (6) crystal [72]. S-isomer 14 is positioned in the hydrophobic pocket of the HIV RT differently, thus hindering a stacking interaction of aromatic A-ring and the side chain of Y181, which positively influences the inhibitory activity in the case of mutation Y181C [73].
Another approach to improve the properties of DAPY is replacement of the benzene ring in the A-ring with a naphthyl moiety (pioneers in this field were researchers led by F. E. Chen [74, 75]). Originally, it was assumed that by substituting the benzene ring by a naphthyl moiety might enhance stacking interaction between the aromatic A-ring of the inhibitor and side chains of amino acids Y181, Y188, and W229, typical of the most promising DAPY TMC278 (9) [58].
An increased affinity of DAPY to the hydrophobic pocket by substituting the benzene ring in the A-position with a 1- or 2-naphthyl moiety resulted in a series of compounds displaying high activity against WT HIV, which was substantially reduced in the case of the K103N+Y181C double mutation in RT [74]. The most potent compound 15 (EC50 = 8.4 nM) exhibited only modest activity against the double mutant (EC50 = 4.5 µM). In addition, changes in the linker structure between aromatic A- and C-rings did not improve the properties of the DAPY derivatives. Replacement of oxygen by sulfur [74], incorporation of a cyano group [70], or a hydroxyimino group [69] merely reduced the inhibitory activity of the compounds, as well as negatively acted on the genetic barrier to resistance.
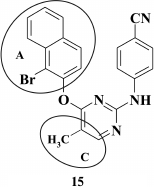
Computer modeling of naphthyl-substituted compounds based on rilpivirine (9) crystalized with HIV RT [59] demonstrated that 2-naphthyl-substituted DAPY analogs could make a stacking interaction with side chains of amino acids Y181, Y188, and F227, but did not interact with the conserved amino acid W229 as in compound 9, thus resulting in sharply reduced activity against the Y181C mutant [74].
According to the modeling data, incorporation of a cyano group at the 6-position in the naphthyl moiety should promote interaction with conserved amino acid W229 similar to benzene analogs, as confirmed previously [75]. It turned out that these compounds were more resistant to the RT K103N+Y181C double mutation compared to non-substituted analogs of compound 15 [74].
Based on these data, compounds were generated containing substituents at the 1- and 3-position in the naphthyl moiety that exhibited submicromolar activity against the K103N+Y181C mutant (for lead compound 16, EC50 = 0.16 µM) [76].
Moreover, even more potent were compounds where oxygen was substituted by an amino group in the linker between naphthyl and pyrimidine rings [77]. It was demonstrated that the most potent compound 17 (EC50 = 2.9 nM) displayed inhibitory activity within the nanomolar range both against WT and NNRTI-resistant HIV strains. According to X-ray structure analysis, the naphthyl moiety in the compounds interacts with side chains of amino acids Y181, Y188, and F227, as earlier proposed in computer modeling of naphthyl analogs [75]. Compared to naphthyl analogs, compounds with an indolizine moiety, with the most potent compound 18 (EC50 = 1 nM), were more potent against WT HIV, but was inferior to the former for the K103N+Y181C double mutant. In addition, in terms of solubility, compound 18 was less efficient than naphthyl analog 17 (12.2 and 8.2 µg/ml for 17 and 18, respectively) [77].
Gu et al. [78] attempted the drastic substitution of the left aromatic ring by a cyclohexyl moiety. Although the most potent compound 19 (EC50 = 55 nM) was highly potent against WT HIV-1, it completely lost potency against the double mutant K103N+Y181C.
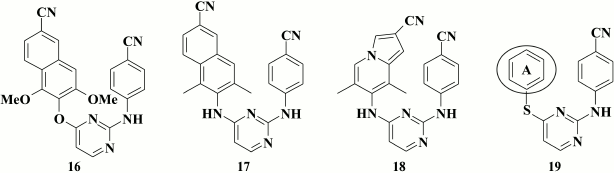
These obtained fully agree with the results of computer modeling noted earlier [65, 75, 76], which demonstrated that the aromatic ring at the A-position is of crucial importance for DAPY, as it can be involved in stacking interaction with the side chains of amino acids Y181, Y188, F227, and W229.
Substituents in the pyrimidine moiety can significantly affect inhibitory activity. A series of halogen-substituted compounds was synthesized using HIV-1 NNRTIs 6-8. Compounds with halogen substituents in the pyrimidine moiety had superior inhibitory activity against RT with both point and double L100I+K103N and K103N+Y181C mutations (Table 3) [56]. Moreover, analogs of rilpivirine (9) and dapivirine (6) containing fluorine in the pyrimidine moiety exhibited much lower cytotoxicity, showing a selectivity index of about 42,000 [80].
Table 3. Inhibitory activity of compounds
6, 20, 21 against WT and NNRTI-resistant mutant
HIV RT [56, 79]
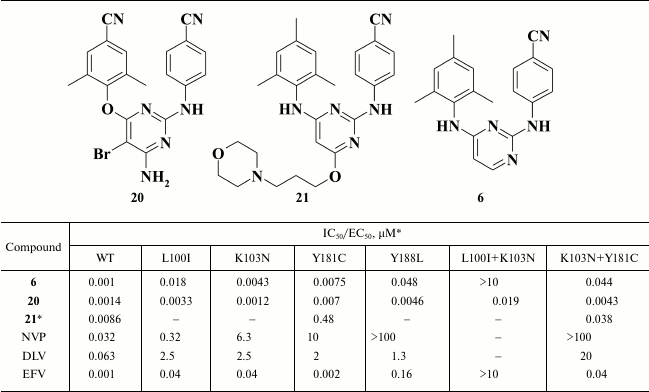
* MT-2 cell line was used in the experiments.
A high genetic barrier to resistance along with high inhibitory activity led to clinical trials with compound 20 named TMC125 (etravirine), which in 2008 was approved by the FDA as an HIV NNRTI. According to the clinical trials, TMC125 (20) had EC50 < 100 nM in 98% of samples and in 97% of samples of NNRTI-resistant strains, as well as having EC50 < 10 nM in 77% of samples of NNRTI-resistant strains [81]. Nonetheless, due to a serious pharmacokinetic drawback [82], etravirine should be taken twice a day.
The majority of DAPY-based HIV NNRTIs have low water solubility, which complicates their application in clinical practice [39, 83, 84]. To solve this problem, Huang et al. [79] proposed DAPY derivatives containing a morpholine moiety attached to the pyrimidine group. Hence, the water solubility for compound 43 was 27.3 µg/ml, which is superior to DPV (6) by 180-fold. However, inhibitory activity of the compound was decreased both against WT and NNRTI-resistant HIV strains.
Zeng et al. [85], in contrast, incorporated a rigid moiety instead of substituents at the 5- and 6-position in the pyrimidine moiety. To generate new promising HIV NNRTIs, molecular hybridization of the template DPC083 and etravirine (20) was used [87, 88]. The final compounds displayed strong inhibitory activity against HIV-1 and had a high genetic barrier to resistance (Table 4) [85]. High potency of the lead compound 22 was due to its interaction with amino acids E138, V179, K101, and K103, which should expand the genetic barrier [85]. Nonetheless, in terms of activity, compound 22 was inferior to etravirine (20), a structural motif of which was used as a template.
Table 4. Inhibitory activity of compounds
22, 23, and 9 against WT and NNRTI-resistant HIV
mutants [85, 86]
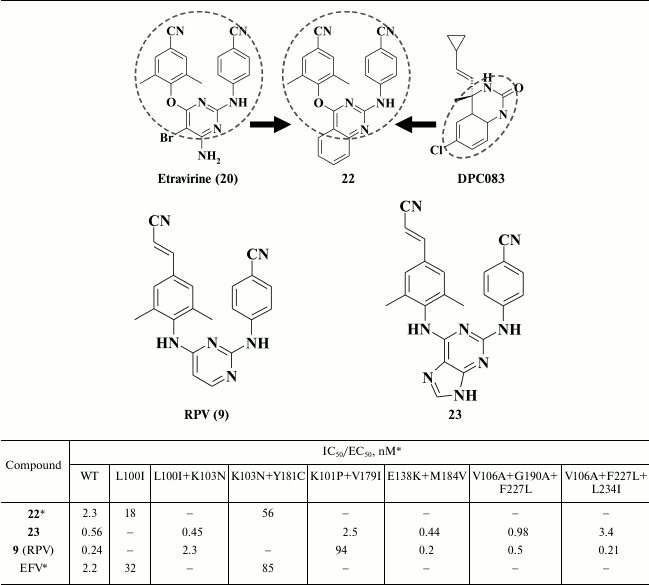
* MT-4 cell line was used in the experiments.
Discovery of highly potent patented HIV NNRTIs [89] that are analogs of etravirine (ETV, 20), where the pyrimidine moiety is substituted by a purine ring, gave rise to the idea of modifying the pyrimidine moiety to generate analogs of rilpivirine (RPV, 9) [86]. It was shown that the most potent compounds exhibited subnanomolar activity against WT HIV RT and a whole range of mutant HIV strains resistant to the first- and second-generation NNRTIs as well as the promising HIV NNRTI doravirine [90], which is currently undergoing phase III clinical trial. During the study, compound 23 was identified, which has a broader resistance profile compared to rilpivirine (9), thus explaining its preserved high potency even against rilpivirine-resistant double mutant K101P+V179I of HIV RT (Table 4). By computer modeling of compound 23 inside the hydrophobic pocket of HIV-1 RT [86] based on rilpivirine crystalized with HIV RT [58, 59], it was demonstrated that a bulkier purine ring shifts the cyanovinyl and benzonitrile moieties deeper into the hydrophobic pocket by 0.5 and 1 Å, respectively. Because in compound 23 all three aromatic rings were shifted, the double mutation K101P+Y179I in HIV RT made no steric hindrance, in contrast to the loss of affinity to the hydrophobic pocket in the case of rilpivirine. High potency of compound 23 against all mutant HIV RT variants makes it a third-generation HIV NNRTI.
Modifications in the DAPY D-ring. According to X-ray structure analysis, a cyano group at the ortho-position of the aromatic D-ring in etravirine (20) and rilpivirine (9) crystalized with HIV-1 RT makes a dipole–dipole interaction with the backbone at H235 [58]. Heeres and Lewi [57] demonstrated that by incorporating chlorine or amide substitutions, it not only negatively affects inhibitory activity of various compounds, but creates a genetic barrier to resistance as well.
Nonetheless, it turned out to be perhaps possible to make successful modifications of the right aromatic D-ring and the linker between the D-ring and the pyrimidine ring. X-ray structure analysis of etravirine crystalized with HIV-1 RT [58] showed that the amino group between the central and right aromatic rings of etravirine forms a hydrogen bond to the backbone at K101. An attempt to enhance interactions of the linker with hydrophobic pocket amino acids via formation of new hydrogen bonds was made in a study [33], where the linker and the right aromatic ring of compound 20 were replaced with a fragment of the GW678248 molecule, another well-known HIV RT inhibitor currently in clinical trial. Crystals of GW678248 with HIV-1 RT demonstrate that the amide moiety of the compound forms a hydrogen bond with K103 and interacts with residues P236 and V106 at the exit from the hydrophobic pocket [91]. Compound 55 generated by combining structural motifs from both molecules (Fig. 5) exhibited moderate activity against WT HIV RT (IC50 = 0.9 µM), but it acutely lost activity in the presence of mutations in the hydrophobic pocket of the enzyme typical of HIV-1 RT resistant to the first-generation HIV-1 NNRTIs. This was a reason to substitute the amide linker with a piperidine-methyl moiety [33].
Fig. 5. Strategy for generation of highly potent NNRTIs by combining structural motifs from GW678248 and etravirine (20).
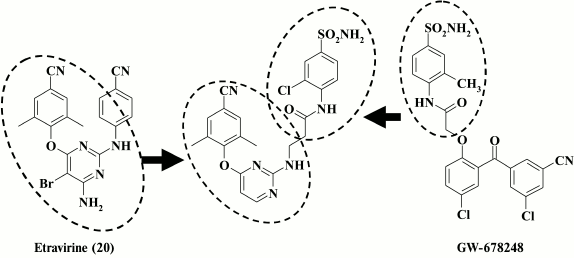
The most potent compound 24 was superior to the second-generation NNRTI etravirine both against Y188L and the common double-mutant K103N+Y181C RT. Compound 25 shows great promise due to the important impact of the sulfoamino group on the solubility of the compound. Compound 25 demonstrated a high genetic barrier to resistance, comparable with that of the second-generation HIV NNRTI etravirine [33] (Table 5).
Table 5. Inhibitory activity of DAPY
piperidine derivatives against WT and NNRTI-resistant HIV mutants [33, 92-94]
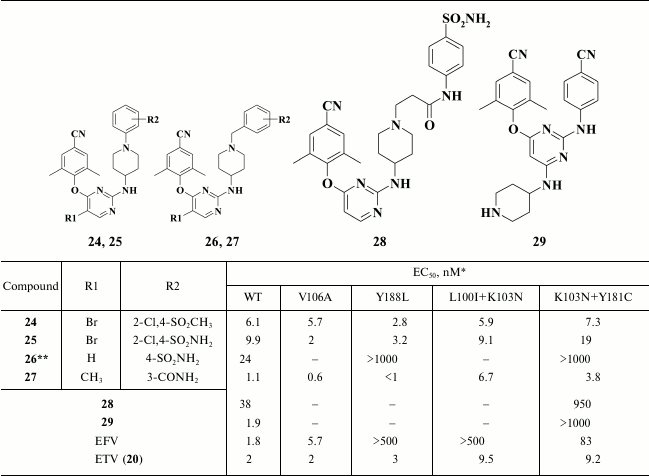
* MT-4 cell line was used in the experiments.
** IC50 (nM) is shown.
Piperidine derivatives comprised the second subclass of compounds synthesized based on compound 55 (Table 5). By optimizing compound 61, it was found that compared to N-benzyl piperidines (24, 25), a substituent at the meta- rather than at the para-position of the benzene moiety in the D-ring was preferable (26, 27). A series of highly potent HIV RT inhibitors was generated based on compound 27, among which the most potent compounds exceeded the inhibitory activity of the second-generation HIV NNRTI etravirine as well as exhibiting a high genetic barrier to resistance [92].
In addition, compounds 24 [33] and 26 [92] were crystalized with WT HIV RT. Data of X-ray structure analysis showed that both compounds were similarly positioned inside the hydrophobic pocket [92]: an aromatic moiety in D-position was located near the exit from the hydrophobic pocket, which interacts with P236, whereas the polar substituent makes a hydrogen bond to the backbone of V106; the piperidine moiety interacts with K103, whereas an amino group between it and the central aromatic ring makes a hydrogen bond to the main chain of K101. A finding showing an interaction between the aromatic A-ring and the conserved amino acid W229 was of great importance [92]. In all likelihood, it was a mere interaction supporting a high genetic barrier to resistance for this subclass of compounds, previously found only in rilpivirine (9), which also interacts with W229 [58].
A high genetic barrier to resistance in piperidine derivatives of DAPY promoted the idea of generating new-generation HIV NNRTIs able to adopt several different positions within the hydrophobic pocket of HIV RT via multiple interactions between various moieties of the inhibitor molecule and amino acid residues of the pocket, thereby more readily adopting to its shape in response to appearing mutations, and successfully overriding their impact by preserving high inhibitory activity [93]. To solve this issue, an approach combining structural motifs from compound 26 and etravirine (20) was selected (Fig. 6).
Fig. 6. Strategy for generation of highly potent NNRTIs by combining structural motifs from compound 26 and etravirine (20).
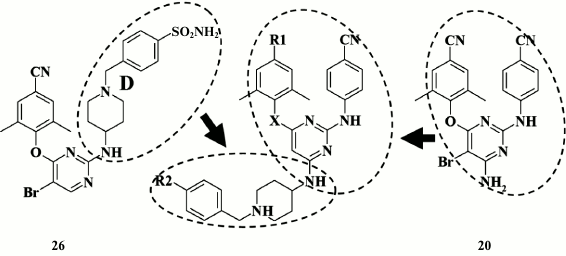
However, this approach failed, as the inhibitory activity of the new subclass of piperidine-substituted DAPYs turned out to be lower compared to the initial analogs (24, 25) and etravirine (20) (Table 5). Virtually all compounds were found to lose or have no potency against double mutant K103N+Y181C HIV strains that are usually found in clinical practice. Nonetheless, Chen et al. [93] consider that this is a promising approach, and assumed that it will be further developed by using derivatives of compound 28, which demonstrated the highest potency against the double mutant K103N+Y181C [93].
To enhance the affinity of piperidin-DAPY derivatives to the hydrophobic pocket by formation of additional interactions with the piperidin moiety, particularly to the conserved amino acid residue L234, the approach of combining structural motifs from piperidine-DAPY derivatives and VRX-480773 was applied [94]. The new compounds displayed high inhibitory activity up to the nanomolar range of concentrations. A compound with a hydrophilic substituent at the 4-position of the distal benzene ring (29) was found to be the most potent analog, although incorporation of a hydroxyl group in the para-position also positively influenced inhibitory activity. It should be noted that the compound containing a fluorine atom at the ortho-position of the distal benzene ring exhibited very low cytotoxicity and hence selectivity index exceeding 16,000, although it was less potent compared to compounds bearing substituents at the para-position. For all the compounds of this series, a common drawback was the complete loss of inhibitory activity with regard the double mutant K103N+Y181C HIV RT.
HIV-1 NNRTIs BASED ON
1-[(2-(HYDROXYETHOXY)METHYL]-6-(PHENYLTHIO)THYMINE (HEPT)
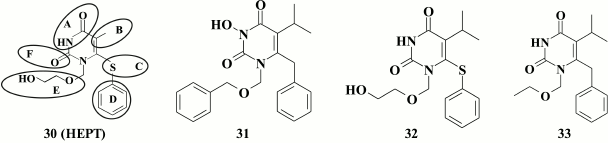
The second extensive pyrimidine-based NNRTI class consists of derivatives of 1-[(2-(hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT, 30). HEPT (EC50 = 7 µM) was discovered in 1989 at the dawn of antiretroviral anti-HIV therapy [95]. Since then, a variety of highly potent HEPT-based compounds has been generated, thus allowing to consider it as one of the most popular and extensive NNRTI classes [96].
Structurally, HEPT molecule (30) can be divided into the six moieties, which focused studies on their modifications to create highly potent NNRTIs.
Positions N-3 and C-4 are conserved in the HEPT molecule, as hydrogen at N-3 position makes a hydrogen bond to the main chain of amino acid K101, whereas oxygen at C-4 position creates hydrogen bonds to the main chain of E138 and the side chain of K101 [97, 98]. It was confirmed experimentally that no substituents at C-4 position are required [99]: incorporating various substituents at this position reduced inhibitory activity against HIV RT.
Alkylation [99] and incorporation of an amino group [100] at N-3 position resulted in complete loss of inhibitory activity, whereas hydroxylation, in contrast, had a positive effect, likely related to preserving the hydrogen bond between the hydroxyl group at N-3 position of the inhibitor molecule and the main chain of K101 [100]. Moreover, compound 31 was even active against HIV integrase, which was the starting point for creating a dual-mode HIV inhibitor [100-102].
In HEPT derivatives, the presence of a C-2 hydrogen is considered rather conserved. According to X-ray structure analysis of HEPT derivatives crystalized with HIV RT [97], the C-2 oxygen in the pyrimidine ring makes a hydrogen bond to the hydroxyl group of Y318. Substitution of oxygen by sulfur at the C-2 position in the pyrimidine moiety did not result in evident increase in inhibitory activity against the WT or NNRTI-resistant HIV strains bearing mutations L100I, K103N, V106A, Y181C, Y181I, and Y188H [103, 104].
Alkyl substituents incorporated at C-5 position increased the inhibitory activity of HEPT derivatives, with the most prominent effect using the isopropyl substituent (32; EC50 = 0.063 µM) [105].
Inhibitory activity increases in the series Pr < Me < Et < i-Pr, and similar dependency was observed for multiple HEPT analogs containing in the linker between aromatic rings sulfur [106], selenium [107], a methylene moiety [103, 108], or a keto group [109].
While examining HEPT derivatives containing various alkyl substituents at the C-5 position, compound 33 (EC50 = 0.008 µM) known as emivirine (MKC-442), which reached phase III clinical trial [110], became one of the first clinically relevant NNRTIs. It was noted that compound 33 had a synergistic effect with NRTI azidothymidine (AZT) [111] and could activate cytochrome P450 involved in metabolism of HIV RT inhibitors [112].
Unfortunately, clinical trials with compound 33 were discontinued due to the rapidly developing mutation Y181C in HIV strains [110] (common in patients with HIV strains resistant to other first-generation NNRTIs nevirapine and delavirdine), as well as financial inexpediency after efavirenz appeared on the market.
Using X-ray structure analysis of NNRTIs crystalized with HIV RT, high potency for HEPT derivatives with an isopropyl substituent at C-5 position was found [97, 98].
The isopropyl moiety in the inhibitor molecule is surrounded by hydrophobic amino acid residues V106, V179, Y181, Y188, V189, and G190, being tightly bound to this region of the hydrophobic pocket in HIV RT and strongly interacting with Y181. The bulky isopropyl moiety triggers changes in the position of the side chain of Y181, thereby strengthening stacking interactions of the latter with the benzene ring of the inhibitor molecule [97]. Such conformational restructuring might not occur due to insufficient size of the methyl and ethyl group at C-5 position [106, 107]. Incorporation of bulkier substituents in the C-5 position, such as phenylthio, benzene, benzoyl, vinyl, or diphenylvinyl groups, reduced inhibitory activity of the compounds against HIV RT and sharp growth of cytotoxicity [113]. Moreover, C-5 halogen-substituted derivatives also had profoundly decreased activity [102, 114].
The aromatic ring at the C-6 position of the pyrimidine moiety is another conserved region in HEPT compounds. According to X-ray structure analysis data of NNRTIs crystalized with HIV RT, the aromatic ring is involved in stacking interaction with residues Y181 or Y188 [98]. Moreover, substitution of the benzene moiety by an alkenylene or a cyclohexenyl group, which also retain the ability to interact with the π-system of the side chains of aromatic amino acids in the pocket, was unsuccessful, as all these compounds were less potent compared to the relevant aromatic analogs [115].
On the other hand, due to the rapidly developing mutation Y181C, stacking interaction of the benzene moiety with Y181 weakens applicability of this class of compounds. Hopkins et al. [98] tried to override this issue by removing the aromatic moiety in the distal ring via incorporating a cyclohexyl group (34; EC50 = 3 nM). X-ray structure analysis demonstrated that compounds 33 and 34 crystalized with HIV RT were differently positioned inside the hydrophobic pocket. In particular, the cyclohexyl group in compound 34 interacts with highly conserved residue W229 compared to the aromatic moiety of compounds 33 located near Y188 and Y181. In turn, in compound 34 it imparted resistance to amino acid substitution Y181C.
Other studies also reported successful ways to overcome the negative impact of amino acid substitution Y181C on potency of HEPT-based NNRTIs, which can be arbitrarily divided into two groups: (i) related to substitution of the benzene moiety with another aromatic system [116-121], and (ii) related to incorporation of substituents into the benzene ring [114, 116].
Substitution of the benzene ring by a pyridine moiety resulted in sustained high inhibitory activity of the compounds [116]. Examination of inhibitory activity regarding mutant NNRTI-resistant HIV revealed a decline in potency of the lead compound 35 by more than 50-fold in the case of amino acid substitutions Y181C and P236L, as well as complete disappearance of activity in the case of the double mutation K103N/Y181C [117].
Based on computer modeling and X-ray structure data for HEPT (30) crystalized with HIV RT [118], it was found that the benzene moiety was far from occupying the entire free space, therefore suggesting that it might be replaced by a naphthyl ring to enhance interaction with Y181, Y188, F227, and W229. Compounds with submicromolar inhibitory activity were found among a series of naphthyl-substituted HEPT derivatives [119-121], and naphthylmethyl derivatives (36) turned out to be more potent than naphthylthio- (37) or naphthoyl- (38) derivatives, whereas inhibitory activity against WT HIV was higher in α-naphthyl versus β-naphthyl HEPT derivatives [119-121].
In addition, apart from substituting the distal benzene group at the C-6 position in pyrimidine [116-121], inhibitory activity against HIV RT was additionally achieved by incorporating substituents at the meta-position in the benzene moiety in various HEPT analogs containing a sulfur [122], selenium [107], or methylene moiety [108] in the C linker.
Compound 39 showed high potency against WT (EC50 = 4.7 nM) and mutant NNRTI-resistant HIV strains, had low toxicity, and thus high selectivity index (SI = 52,500), that destined it for preclinical studies [107].
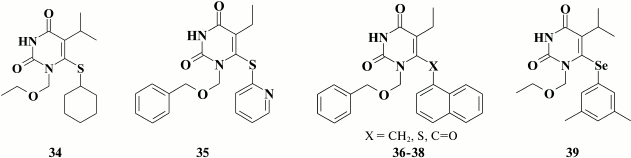
X-ray structure analysis of HIV RT crystalized with HEPT-based NNRTIs containing substituents at the meta-position in benzene ring explained sustained high inhibitory activity against mutations Y181C and Y188C [98]. In particular, substituents at the meta-position of the aromatic D-ring were found to sit deeper within the hydrophobic pocket, where they made van der Waals contacts with the side chain of the conserved amino acid W229, thereby compensating for the loss of interactions with Y181 and Y188 in cases when the latter two residues might be mutated. In contrast, substituents at ortho- and para-position in the benzene moiety negatively influenced potency, as they sterically hindered interaction of the compounds with the hydrophobic pocket of HIV RT, which is consistent with QSAR studies of HEPT analogs [123-125].
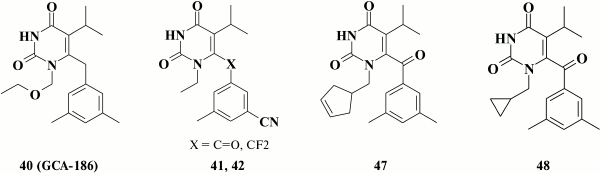
A positive effect of substituents at the meta-position in the benzene ring led to the highly potent HIV NNRTI GCA-186 (40) [122]. For this, two methyl groups were incorporated at the meta-position of the benzene moiety of the previously investigated inhibitor emivirine (MKC-442) (33) that was studied in clinical trial [110]. This compound exhibited high inhibitory activity and broad resistance profile [98].
Later, numerous analogs of compound 40 containing a linear substituent at the N-1 position were synthesized [103], such that the most promising compounds had nanomolar inhibitory activity against WT HIV and a high genetic barrier to resistance. Relatively rare mutations Y181I and Y188H of HIV isolated in clinical practice were found to result in severely decreased inhibitory activity in many analogs of compound 40.
Nonetheless, preclinical studies with compound 40 and its analogs demonstrated that compounds with methyl groups at meta-position in the benzene ring were robustly metabolized by cytochrome P450 [126]. Analogs of compound 40 containing chlorine [126] and fluorine [127] as well as trifluoromethyl [127] substituents at meta-position in the benzene moiety were synthesized to increase their stability. Such analogs were found to abruptly lose their inhibitory activity against Y181C mutant HIV, thus making this approach pointless.
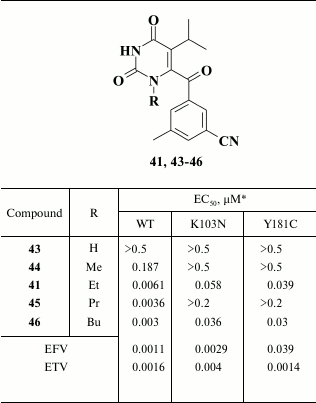
It turned out that a more promising approach was to substitute one of the methyl groups in meta-position for a cyano group in the benzene ring. It was found that these compounds had high potency against WT HIV and remained active in the case of mutation Y181C. The most potent were compounds with a keto- (41; EC50 = 6.1 nM) or difluoromethylene (42; EC50 = 4.2 nM) group in the linker between the pyrimidine and benzene rings, which, however, turned out to be metabolically unstable. A solubility issue was the major drawback of the entire series of compounds that caused their poor bioavailability [109].
While generating highly potent HEPT derivatives (30), the greatest number of changes was made in the structure of substituents at the N-1 position in the pyrimidine moiety. It was found that branched substituents at N-1 position in the pyrimidine had an extremely negative effect on inhibitory activity against WT HIV-1 [128], whereas incorporation of sulfur instead of oxygen into the linker resulted in insignificantly increased potency, but sharply elevated cytotoxicity [108, 128]. Moreover, also generated were compounds with an alkyl substituent at the N-1 position in the pyrimidine, which, however, were still inferior to ethoxymethyl analogs [104, 108].
In 2010, after highly potent compounds with methyl substituents at meta-position in the benzene ring were found, it was again decided to incorporate an alkyl group at the N-1 position in the pyrimidine [109].
Increased inhibitory activity of the compounds correlated with increased length of the alkyl group; however, examination of the compounds and HIV strains containing amino acid substitutions K103N and Y181C in HIV RT revealed that only compounds with ethyl and butyl substituents remained active (Table 6) [109]. X-ray structure analysis of HIV RT crystalized with compound 45 containing a propyl substituent helped to clarify the reason for its low activity. It was found that compounds 41 and 46 are differently positioned inside the hydrophobic pocket: the propyl group in compound 45 interacts with Y181; therefore, this mutation profoundly reduces the activity of the inhibitory agent. In contrast, the ethyl group interacts less with Y181, and thus its substitution does not play a crucial role for this compound, whereas the butyl moiety in compound 46 makes no contacts with Y181C due to the distinct position of the inhibitor in the hydrophobic pocket of the HIV RT [109].
Table 6. Inhibitory activity of
alkyl-substituted pyrimidines against WT and NNRTI-resistant HIV
mutants [109]
* MT-2 cell line was used in the experiments.
A large-scale screening study in 2001 identified compound 47 (EC50 = 0.9 nM) containing a cyclopentenyl moiety at the N-1 position [129]. It was demonstrated that compound 47 lost its activity against mutations K101E, K103N, and Y188C, which also completely disappeared in the case of double mutant K103N+Y181C HIV-1. Additionally, compound 47 might inhibit viral fusion, thus making it active against HIV-2 [129].
Using compound 47, a broad series of compounds was synthesized containing cyclopropylmethyl, cyclobutylmethyl, cyclohexylmethyl, 1- and 3-cyclopentenemethyl substituents, as well as compounds with extended linker between the pyrimidine moiety and a non-aromatic ring such as cyclopentyl or 1-, 2-, 3-cyclopentenyl substituent [130]. No clear dependence between inhibitory activity against HIV-1 and structure of the cycloalkyl moiety at the N-1 position in the pyrimidine ring of several dozens of compounds of this series was found. Activity of compounds containing even three-, four-, and five-membered rings was observed after changing the type of linker between the pyrimidine ring and the benzene moiety at C-6 position. In this series of HEPT derivatives, the most promising was compound 48, due to its low cytotoxicity and selectivity index exceeding 2,500,000 [130]. Like compound 47, all these compounds were found to exert another mode of action in inhibiting HIV-1, i.e. that aimed at suppressing viral binding to cell receptors [129, 130].
In addition, it was found that incorporating a third aromatic ring at the N-1 position in the pyrimidine (49) efficiently enhanced potency of HEPT derivatives [131]. Compound 49 exhibited submicromolar activity (Table 7), which together with incorporation of efficient substituents at the C-5 and C-6 positions in the pyrimidine resulted in HIV NNRTIs having nanomolar inhibitory activity against WT HIV (50, 51) [104]. Modification of the linker between the pyrimidine ring and the benzene moiety at the C-6 position produced compound 52, which was tested as TNK-651 in clinical trial [96, 97].
Table 7. Comparison of inhibitory activity
for HEPT-containing compounds 49-52 against WT HIV [97, 104, 131]
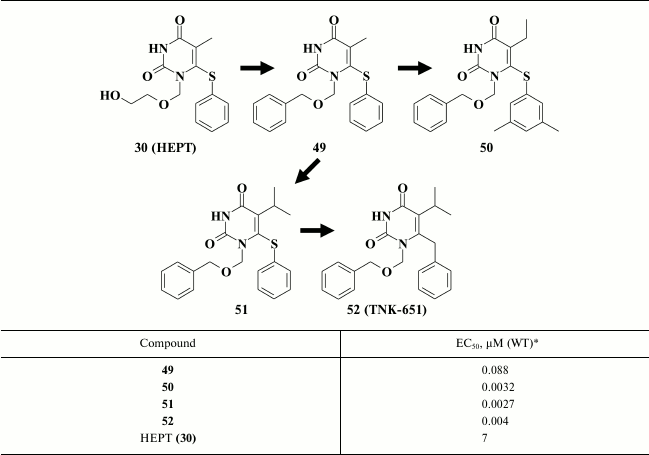
* MT-4 cell line was used in the experiments.
El-Brollosy et al. [132] tested two concepts related to modification of substituents at the N-1 position in HEPT derivatives – replacing the benzene ring for an alkenyl moiety might sustain stacking interactions with aromatic amino acids in the hydrophobic pocket of HIV RT, and incorporation of an alkenyl group into the linker between the pyrimidine moiety and the phenyl substituent at N-1 position. Compounds were found that exhibited high potency against WT HIV, but they abruptly lost their activity in the case of mutations Y181C and K103N+Y181.
X-ray structure analysis data of HIV RT crystalized with TNK-651 (52) [97] and QSAR study [118, 133] explained the high potency of compounds with a phenyl substituent at the N-1 position. An extended substituent at N-1 position is located near the entrance to the hydrophobic pocket composed of amino acids V106, P225, F227, and P236. Oxygen in the linker between the phenyl substituent at N-1 position and the pyrimidine ring makes a hydrogen bond with the side chain of Y318, which is located close to the entrance to the hydrophobic pocket of HIV RT [97, 98]. The phenyl moiety at N-1 position in compound 52 interacts with P236, and the proline residue can change its position in the pocket within 5 Å [97], which allows incorporating bulkier substituents at the N-1 position. Nonetheless, mutation P236L resulted in a sharp loss of inhibitory activity for compounds of the BHAP family [134], which may not be also ruled out for analog 52.
The presence of an extended linker between the pyrimidine ring and the substituent at the N-1 position is not a prerequisite for high potency of inhibitors containing a phenyl substituent. Apart from cycloalkyl substituents, compounds were synthesized with a phenyl substituent (lead compound 53; EC50 = 1.2 nM). Such compounds were inferior to the relevant analogs with a cyclobutyl substituent at N-1 position, but retained inhibitory activity within the submicromolar range as well as the ability to exert the second mode of HIV inhibition related to virus entry into the target cells [135], and they were less toxic. High affinity of the inhibitor to the hydrophobic pocket of HIV RT was reached due to its interaction with Y318 [130].
According to crystallographic data for TNK-651 (52) complexed with HIV RT [97], a bulkier indanyloxymethyl substituent was incorporated at the N-1 position in the pyrimidine moiety [136]. Among a series of synthesized compounds, compound 54 was identified that has subnanomolar inhibitory activity against WT HIV-1 (EC50 = 0.4 nM), but this activity was profoundly lost in case of mutation Y181C commonly found in patients with NNRTI-resistant HIV strains (EC50 Y181C = 1 µM).
In addition, there are HEPT-based NNRTIs having a pyridine ring at the N-1 position in the pyrimidine [103, 137-140], and one of them (55) was identified in a study of 6-phenylamino-derivatives of HEPT [138]. Compound 55 had submicromolar activity against WT HIV-1 (EC50 = 0.08 µM), although its numerous analogs with a pyridine moiety displayed low inhibitory activity. An analog of compound 55 containing a thiophenyl substituent at the C-6 position in the pyrimidine was more potent but also more toxic [141].
High inhibitory activity was found for analogs of compound 55 having a keto group in the linker between the pyrimidine and benzene moieties. Inhibitory activity of the lead compound 56 against HIV-1 WT was as high as for the thiophenyl analog of compound 55, but its cytotoxicity was much lower, thereby resulting in selectivity index higher than 40,000 [141].
Further modification of aromatic groups at the N-1 and C-6 position in compounds containing a keto group in the linker between the pyrimidine and benzene moieties produced compound 57, which acts within the nanomolar range of concentrations and inhibits both WT and NNRTI-resistant HIV-1 mutants (Table 8) [139]. Examining compound 57 crystalized with HIV RT revealed formation of a hydrogen bond between the amino group of the pyridine moiety and the main chain of K103N, underlying its high potency against NNRTI-resistant HIV-1 mutants [140].
Table 8. Inhibitory activity of compounds
57-60 against WT and NNRTI-resistant HIV mutants [139, 140]
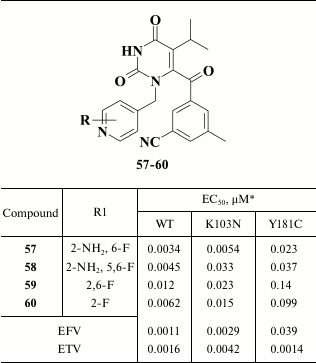
* MT-2 cell line was used in experiments.
Pharmacokinetic study of compound 57 showed that it undergoes microsomal oxidation, defluorination, and hydroxylation at its isopropyl group at the C-5 position in the central pyrimidine ring, which required its administration more than once a day. To level these drawbacks, compounds 58-60 were synthesized (Table 8), but these were inferior to compound 57 in terms of inhibitory activity against NNRTI-resistant HIV strains and poorly matched target pharmacokinetic criteria [140].
HIV-1 NNRTIs BASED ON DIHYDROALKOXYBENZYLOXOPYRIMIDINES
(DABO)
The subclass of dihydroalkoxybenzyloxopyrimidines (DABOs) branched out from HEPT (30) derivatives, and compound 61 considered as a founder of this subclass was synthesized while trying to transfer an extended substituent from the N-1 to the C-2 position in the pyrimidine ring. Compound 61 had weak anti-HIV (EC90 = 86 µM) [142] potency, but it demonstrated a crucial opportunity to generate NNRTIs based on HEPTs with extended substituents at the C-2 position. The DABOs contain an aromatic ring at the C-6 position in the pyrimidine, a prerequisite for its activity [143], thus making it structurally similar to HEPTs. Therefore, many common features used in DABO modifications were taken from the studies on improving inhibitory activity of HEPTs against HIV RT, including:
(i) required presence of an aromatic moiety at the C-6 position in the pyrimidine;
(ii) incorporated alkyl substituents at C-5 position in the pyrimidine [144, 145];
(iii) incorporated methyl groups at the meta-position in the benzene ring into the C-6 position [146], by analogy with compound 39 and GCA-186 (40) [107, 122];
(iv) replaced phenyl moiety at the C-6 position for the naphthyl moiety (DATNO subclass [143]), by analogy with those described in [119];
(v) incorporation of a third aromatic moiety at the C-2 position [147], by analogy with TNK-651 (52) [97];
(vi) substituted phenyl moiety in place of a cyclohexyl one at the C-6 position in the pyrimidine [148], by analogy with HEPT cyclohexyl TNK-6123 (34) derivative [98].
Incorporation of an alkylthio substituent of the alkyloxy group at the C-2 position increased inhibitory activity against WT HIV-1. Compounds with a alkylthio moiety were named S-DABOs (Fig. 7) [146]. While modifying the phenyl moiety at the C-6 position in the pyrimidine, an important difference of DABO from HEPT derivatives was found: it was optimal to incorporate substituents at the ortho-position in the phenyl moiety [149], which goes against HEPT derivatives that optimally contain a phenyl moiety at the meta-position phenyl in the C-6 position of the pyrimidine [96].
Fig. 7. Structural diversity of DABO analogs.
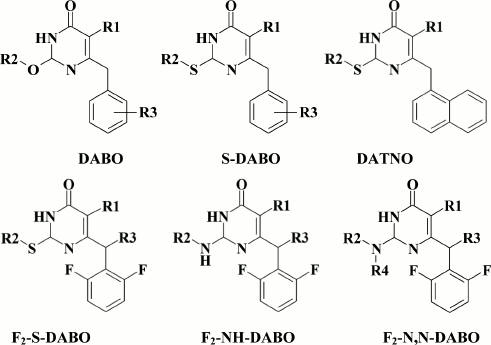
The search for substituents at the meta-position in the phenyl moiety revealed highly potent NNRTIs such as compound 62 with a fluorine at the meta-position in the benzene ring [149], which became a founder for the subclass of 2,6-F2-S-DABOs.
Studying substituents at the C-5 position in the pyrimidine moiety revealed another difference of DABO from HEPT derivatives: despite the fact that incorporation of halogen at the C-5 position in the pyrimidine in both subclasses results in lowering its inhibitory activity, upon incorporating an alkyl substituent at the same C-5 position in the pyrimidine increases inhibitory activity in DABOs in the series Me > Et > i-Pr [150], which contradicts data for HEPT derivatives showing the opposite dependence [106].
Studies on the impact of an aromatic substituent at the C-6 position in the pyrimidine [143] demonstrate that its removal results in complete loss of inhibitory activity, and elongation of the linker between the pyrimidine and phenyl moiety lowers inhibitory activity against HIV-1 as well. Compounds where the phenyl substituent was replaced with a naphthyl moiety (DATNOs) (the most potent compound is 63, with EC50 = 0.33 µM), revealed high inhibitory activity so that α- versus β-naphthyl derivatives were more potent [143], similarly to HEPT derivatives [119]. An important feature of DATNOs was a negative effect triggered by incorporating a substituent at the C-5 position in the pyrimidine moiety, which was not typical of DABOs [150].
Incorporation of a methyl group into the linker between the pyrimidine and phenyl substituent promoted inhibitory activity of the compounds against HIV-1 [151, 152]. The impact of optical isomers on inhibitory activity was examined for the most potent compound 64, showing that the R-isomer versus the S-isomer was more potent by three orders of magnitude [152]. According to computer modeling based on the X-ray structure data of HIV RT crystalized with TNK-651 (52) [97], it was concluded that the methyl group of the R-isomer interacts with the side chain of Y181 [151].
Substitution of sulfur for the amino group within the substituent at the C-2 position in the pyrimidine produced another DABO subclass known as NH-DABO, featured by inhibitory activity similar to the S-DABO inhibitory activity and a high genetic barrier to resistance [153].
Computer modeling of compound 65 inside the hydrophobic pocket of HIV RT suggested that the amino group at the C-2 position in the pyrimidine was able to make a hydrogen bond with K101, and this might compensate for the loss of interaction between the phenyl moiety and Y181 as well as Y188 in case of their mutation, therefore explaining the preserved inhibitory activity of compound 65 [154].
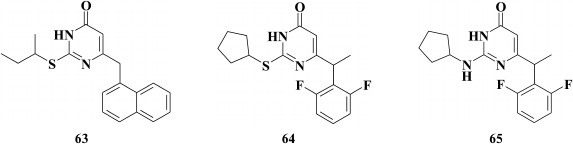
Based on 2,6-F2-NH-DABO, another DABO subclass known as 2,6-F2-N,N-DABOs was synthesized [155] (66). Compounds of this subclass exhibit high inhibitory activity (for 66 EC50 = 0.1 µM) against both WT and resistant HIV RT mutants. It should be noted that by modifying the linker between the phenyl substituent 2,6-F2-N,N-DABO and the pyrimidine ring via incorporating a halogen, hydroxyl, cyano, or keto group into the linker, a generation of compounds with submicromolar activity against WT HIV-1 was formed, which did not extend a genetic barrier to resistance for this subclass of compounds. Even the most potent compounds were found to sharply lose inhibitory activity against the mutation Y181C HIV-1 RT.
In the case of compounds without modifications in the linker at the C-6 position of the pyrimidine, the inhibitory activity against the Y181C mutant decreased to a lesser extent on incorporation of a third aromatic moiety at the amino group of the C-2 position in the pyrimidine (67); however, like the other studied compounds, compound 67 was completely inactive against the double mutation K103N+Y181C [156].
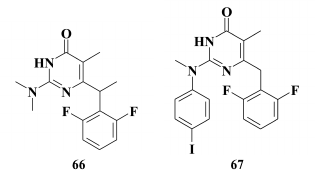
Sharp elevation of the inhibitory activity against WT HIV-1 was achieved by incorporating an aromatic moiety at the C-2 position [157]. Again, this was similar to HEPT derivatives, which were also noted to have a similar impact of the third aromatic ring on inhibitory activity against HIV-1 [97]. It should be noted that incorporating an aromatic moiety at the C-2 position in DABOs cannot compensate for the lack of an aromatic substituent at the C-6 position in the pyrimidine [143]. Using the most potent compound 68 (IC50 = 0.026 µM) against WT HIV RT identified by Manetti et al. [147], the linker between the pyrimidine ring and the phenyl substituent at C-2 position in the pyrimidine was varied. This study showed a positive effect of the extension of the linker length (69) on inhibitory activity against mutation Y188L of HIV RT, whereas incorporation of a double bond into the linker (70) increased the activity against resistant K103N, Y181C, and Y188L HIV mutant strains. Incorporation of a ternary bond into the linker resulted in complete loss of an inhibitory activity. Unfortunately, toxicity of the compounds with unsaturated hydrocarbon moieties was higher than toxicity of unsaturated analogs [157].
Compound with a keto group in the linker between the pyrimidine moiety and the phenyl substituent at the C-2 position in the pyrimidine produced a compound with nano- and subnanomolar activity against WT HIV-1 (for lead compound 71 EC50 = 0.4 nM) [157, 158]. Nonetheless, it remained the major issue for DABOs having three aromatic rings: even the most potent compounds continued to lose activity by several orders of magnitude in the case of mutations K103N, Y181C, and Y188L [159].
According to computer modeling of DABOs having three aromatic rings complexed with HIV RT [157, 159], which were based on HIV RT crystalized with HEPT derivatives containing three aromatic moieties from TNK-651 (52) [97], it was assumed that compounds from both subclasses might be similarly positioned inside the hydrophobic pocket.
In particular, according to computer modeling, substituents at the C-6 position in the pyrimidine of phenylacetyl-DABO and TNK-651 derivatives (52) were found to make a stacking interaction with Y188, and the aromatic substituent at C-2 position interacts with P236, V106, and Y318 (similar to the aromatic substituent at N-1 position in the pyrimidine of TNK-651 (52)), the pyrimidine moiety makes a hydrogen bond with the main chain at K101, whereas the alkyl substituent at C-5 position in the pyrimidine is positioned in a way similar to TNK-651 (52) [157, 159]. The similar position of the alkyl moiety at the C-5 position in the pyrimidine may explain an increased inhibitory activity in the series H < Me < Et < i-Pr for analogs of 71 having various substituents at the C-5 position, which was observed for HEPT analogs (30) [103, 106-109].
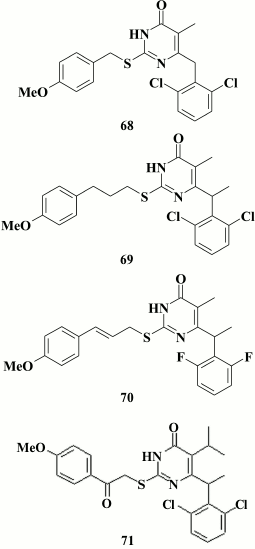
Yu et al. [160] tried to extend the resistance profile for DABOs having three aromatic rings by incorporating an amino group into the linker between the pyrimidine moiety and the phenyl substituent at the C-2 position in the pyrimidine, which might contribute to formation of new hydrogen bonds to amino acids lining the hydrophobic pocket and compensate for the loss of interaction with Y181 and Y188 in case of their mutations. For the same purpose, substituents most distant from Y181 and Y188 in phenyl moiety at C-2 position in the pyrimidine were varied. The most potent compound (72) turned out to be less active against WT HIV RT compared to other DABOs having three aromatic rings, thus indicating lack of prospects for this approach.
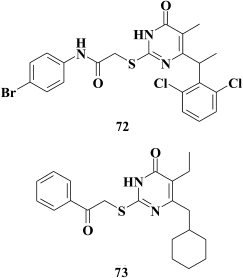
Replacement of the phenyl substituent at the C-6 position in the pyrimidine with a cyclohexyl moiety was a more promising approach [148]. Like HEPT derivatives [98], this yielded highly potent compound 73 displaying inhibitory activity against WT HIV within the nanomolar range (EC50 = 2.4 nM). Computer modeling based on X-ray structure analysis of HIV RT crystalized with TNK-651 (52) [97] suggested that the cyclohexyl moiety of compound 73 is involved in van der Waals interactions with Y181, Y188, W229, and V106, this being confirmed by examining the inhibitory activity of compound 73 in resistant HIV mutants; the activity was sharply decreased in the case of mutations V106A and Y181C in HIV-1 RT [148].
To summarize the data regarding the three classes of pyrimidine-substituted HIV-1 NNRTIs, the following patterns producing novel highly potent compounds with broad resistance profile against drug-resistant mutations in the hydrophobic pocket of HIV-1 RT should be noted.
Compounds should: (i) exhibit conformational flexibility to be able to adapt to mutations within the enzymatic hydrophobic pocket; (ii) interact as much as possible with conserved amino acid W229 in the hydrophobic pocket, and, ideally, with less conserved amino acids F227, L234, P236, and Y318; (iii) make hydrogen bonds with the main chain of the protein, and therefore substantially reduce negative effects on inhibitory activity upon mutations in amino acid residues lining the hydrophobic pocket.
Acknowledgments
This study was performed with financial support from the Russian Science Foundation (project No. 14-50-000-60).
REFERENCES
1.UNAIDS, Fact sheet: 2014 statistics
(2015).
2.HIV-Infection in RF for 2016 (official
statistics); available from
(https://spid-vich-zppp.ru/statistika/ofitsialnaya-statistika-vich-spid-rf-2016.html).
3.Prokofjeva, M. M., Kochetkov, S. N., and Prassolov,
V. S. (2016) Therapy of HIV infection: current approaches and
prospects, Acta Naturae, 8, 23-32.
4.Guidelines for the Use of Antiretroviral Agents
in HIV-1-infected Adults and Adolescents (2017).
5.Resistance to Bactericidal Drugs (2016);
available from
(http://www.who.int/mediacentre/factsheets/fs194/ru/).
6.Touzard, Romo, F., Smeaton, L. M., Campbell, T. B.,
Riviere, C., Mngqibisa, R., Nyirenda, M., Supparatpinyo, K.,
Kumarasamy, N., Hakim, J. G., and Flanigan, T. P. (2014) Renal and
metabolic toxicities following initiation of HIV-1 treatment regimen in
a diverse, multinational setting: a focused safety analysis of ACTG
PEARLS (A5175), HIV Clin. Trials, 15, 246-260.
7.Turner, B. G., and Summers, M. F. (1999) Structural
biology of HIV, J. Mol. Biol., 285, 1-32.
8.Freed, E. O., and Martin, M. A. (2007) HIVs and
their replication, in Fields Virology (Knipe, D. M., and Howley,
P. M., eds.) Lippincott Williams & Wilkins, Philadelphia, pp.
2107-2185.
9.Kuritzkes, D. R., and Walter, B. D. (2007) HIV-1
pathigenesis, clinical manifestations and treatment, in Fields
Virology (Knipe, D. M., and Howley, P. M., eds.) Lippincott
Williams & Wilkins, Philadelphia, pp. 2187-2214.
10.Greene, W. C., and Peterlin, B. M. (2002)
Charting HIV’s remarkable voyage through the cell: basic science
as a passport to future therapy, Nat. Med., 8,
673-680.
11.Piekna-Przybylska, D. B. R. (2013) Proviral
DNA Synthesis in HIV: Background in Human Immunodeficiency Virus
Reverse Transcriptase. A Bench-to-Bedside Success, Springer, pp.
23-53.
12.Steitz, T. A. (1999) DNA polymerases: structural
diversity and common mechanisms, J. Biol. Chem., 274,
17395-17398.
13.Kohlstaedt, L. A., Wang, J., Friedman, J. M.,
Rice, P. A., and Steitz, T. A. (1992) Crystal structure at 3.5 Å
resolution of HIV-1 reverse transcriptase complexed with an inhibitor,
Science, 256, 1783-1790.
14.Huang, H., Chopra, R., Verdine, G. L., and
Harrison, S. C. (1998) Structure of a covalently trapped catalytic
complex of HIV-1 reverse transcriptase: implications for drug
resistance, Science, 282, 1669-1675.
15.Singh, K., Flores, J. A., Kirby, K. A., Neogi,
U., Sonnerborg, A., Hachiya, A., Das, K., Arnold, E., McArthur, C.,
Parniak, M., and Sarafianos, S. G. (2014) Drug resistance in non-B
subtype HIV-1: impact of HIV-1 reverse transcriptase inhibitors,
Viruses, 6, 3535-3562.
16.Esnouf, R., Ren, J., Ross, C., Jones, Y.,
Stammers, D., and Stuart, D. (1995) Mechanism of inhibition of HIV-1
reverse transcriptase by non-nucleoside inhibitors, Nat. Struct.
Biol., 2, 303-308.
17.Ding, J., Das, K., Tantillo, C., Zhang, W.,
Clark, A. D., Jr., Jessen, S., Lu, X., Hsiou, Y., Jacobo-Molina, A.,
Andries, K., Pauwels, R., Moereels, H., Koymans, L., Janssen, P.,
Smith, R. H., Koepke, M. K., Michejda, C. J., Hughes, S. H., and
Arnold, E. (1995) Structure of HIV-1 reverse transcriptase in a complex
with the non-nucleoside inhibitor alpha-APA R 95845 at 2.8 Å
resolution, Structure, 3, 365-379.
18.Ren, J., Esnouf, R., Garman, E., Somers, D.,
Ross, C., Kirby, I., Keeling, J., Darby, G., Jones, Y., Stuart, D., and
Stammers, D. (1995) High resolution structures of HIV-1 RT from four
RT–inhibitor complexes, Nat. Struct. Biol., 2,
293-302.
19.Ding, J., Das, K., Moereels, H., Koymans, L.,
Andries, K., Janssen, P. A., Hughes, S. H., and Arnold, E. (1995)
Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the
binding of diverse nonnucleoside inhibitors, Nat. Struct. Biol.,
2, 407-415.
20.Tantillo, C., Ding, J., Jacobo-Molina, A., Nanni,
R. G., Boyer, P. L., Hughes, S. H., Pauwels, R., Andries, K., Janssen,
P. A., and Arnold, E. (1994) Locations of anti-AIDS drug binding sites
and resistance mutations in the three-dimensional structure of HIV-1
reverse transcriptase. Implications for mechanisms of drug inhibition
and resistance, J. Mol. Biol., 243, 369-387.
21.Das, K., Martinez, S. E., Bauman, J. D., and
Arnold, E. (2012) HIV-1 reverse transcriptase complex with DNA and
nevirapine reveals non-nucleoside inhibition mechanism, J. Mol.
Biol., 19, 253-259.
22.Schauer, G. D., Huber, K. D., Leuba, S. H., and
Sluis-Cremer, N. (2014) Mechanism of allosteric inhibition of HIV-1
reverse transcriptase revealed by single-molecule and ensemble
fluorescence, Nucleic Acids Res., 42, 11687-11696.
23.Liu, S., Abbondanzieri, E. A., Rausch, J. W., Le
Grice, S. F., and Zhuang, X. (2008) Slide into action: dynamic
shuttling of HIV reverse transcriptase on nucleic acid substrates,
Science, 322, 1092-1097.
24.Wang, J., Smerdon, S. J., Jager, J., Kohlstaedt,
L. A., Rice, P. A., Friedman, J. M., and Steitz, T. A. (1994)
Structural basis of asymmetry in the human immunodeficiency virus type
1 reverse transcriptase heterodimer, Proc. Natl. Acad. Sci. USA,
91, 7242-7246.
25.U.S. Food and Drug Administration.
Antiretroviral Drugs Used in the Treatment of HIV Infection
Table; available from
(http://www.fda.gov/ForPatients/Illness/HIVAIDS/Treatment/ucm118915.htm).
26.Schafer, J. J., and Short, W. R. (2012)
Rilpivirine, a novel non-nucleoside reverse transcriptase inhibitor for
the management of HIV-1 infection: a systematic review, Antivir.
Ther., 17, 1495-1502.
27.Croxtall, J. D. (2012) Etravirine: a review of
its use in the management of treatment-experienced patients with HIV-1
infection, Drugs, 72, 847-869.
28.De Bethune, M. P. (2010) Non-nucleoside reverse
transcriptase inhibitors (NNRTIs), their discovery, development, and
use in the treatment of HIV-1 infection: a review of the last 20 years
(1989-2009), Antiviral Res., 85, 75-90.
29.Li, X., Zhang, L., Tian, Y., Song, Y., Zhan, P.,
and Liu, X. (2014) Novel HIV-1 non-nucleoside reverse transcriptase
inhibitors: a patent review (2011-2014), Expert Opin. Ther.
Pat., 24, 1199-1227.
30.Schuckmann, M. M., Marchand, B., Hachiya, A.,
Kodama, E. N., Kirby, K. A., Singh, K., and Sarafianos, S. G. (2010)
The N348I mutation at the connection subdomain of HIV-1 reverse
transcriptase decreases binding to nevirapine, J. Biol. Chem.,
285, 38700-38709.
31.Betancor, G., Alvarez, M., Marcelli, B., Andres,
C., Martinez, M. A., and Menendez-Arias, L. (2015) Effects of HIV-1
reverse transcriptase connection subdomain mutations on polypurine
tract removal and initiation of (+)-strand DNA synthesis, Nucleic
Acids Res., 43, 2259-2270.
32.Hsiou, Y., Das, K., Ding, J., Clark, A. D., Jr.,
Kleim, J. P., Rosner, M., Winkler, I., Riess, G., Hughes, S. H., and
Arnold, E. (1998) Structures of Tyr188Leu mutant and wild-type HIV-1
reverse transcriptase complexed with the non-nucleoside inhibitor HBY
097: inhibitor flexibility is a useful design feature for reducing drug
resistance, J. Mol. Biol., 284, 313-323.
33.Kertesz, D. J., Brotherton-Pleiss, C., Yang, M.,
Wang, Z., Lin, X., Qiu, Z., Hirschfeld, D. R., Gleason, S., Mirzadegan,
T., Dunten, P. W., Harris, S. F., Villasenor, A. G., Hang, J. Q.,
Heilek, G. M., and Klumpp, K. (2010) Discovery of
piperidin-4-yl-aminopyrimidines as HIV-1 reverse transcriptase
inhibitors. N-benzyl derivatives with broad potency against resistant
mutant viruses, Bioorg. Med. Chem. Lett., 20,
4215-4218.
34.Hsiou, Y., Ding, J., Das, K., Clark, A. D., Jr.,
Boyer, P. L., Lewi, P., Janssen, P. A., Kleim, J. P., Rosner, M.,
Hughes, S. H., and Arnold, E. (2001) The Lys103Asn mutation of HIV-1
RT: a novel mechanism of drug resistance, J. Mol. Biol.,
309, 437-445.
35.Ren, J., Nichols, C. E., Chamberlain, P. P.,
Weaver, K. L., Short, S. A., Chan, J. H., Kleim, J. P., and Stammers,
D. K. (2007) Relationship of potency and resilience to drug resistant
mutations for GW420867X revealed by crystal structures of inhibitor
complexes for wild-type, Leu100Ile, Lys101Glu, and Tyr188Cys mutant
HIV-1 reverse transcriptases, J. Med. Chem., 50,
2301-2309.
36.Antinori, A., Zaccarelli, M., Cingolani, A.,
Forbici, F., Rizzo, M. G., Trotta, M. P., Di Giambenedetto, S.,
Narciso, P., Ammassari, A., Girardi, E., De Luca, A., and Perno, C. F.
(2002) Cross-resistance among nonnucleoside reverse transcriptase
inhibitors limits recycling efavirenz after nevirapine failure, AIDS
Res. Hum. Retroviruses, 18, 835-838.
37.Madruga, J. V., Cahn, P., Grinsztejn, B.,
Haubrich, R., Lalezari, J., Mills, A., Pialoux, G., Wilkin, T.,
Peeters, M., Vingerhoets, J., De Smedt, G., Leopold, L., Trefiglio, R.,
and Woodfall, B. (2007) Efficacy and safety of TMC125 (etravirine) in
treatment-experienced HIV-1-infected patients in DUET-1: 24-week
results from a randomised, double-blind, placebo-controlled trial,
Lancet, 370, 29-38.
38.Lazzarin, A., Campbell, T., Clotet, B., Johnson,
M., Katlama, C., Moll, A., Towner, W., Trottier, B., Peeters, M.,
Vingerhoets, J., De Smedt, G., Baeten, B., Beets, G., Sinha, R., and
Woodfall, B. (2007) Efficacy and safety of TMC125 (etravirine) in
treatment-experienced HIV-1-infected patients in DUET-2: 24-week
results from a randomised, double-blind, placebo-controlled trial,
Lancet, 370, 39-48.
39.Janssen, P. A., Lewi, P. J., Arnold, E.,
Daeyaert, F., De Jonge, M., Heeres, J., Koymans, L., Vinkers, M.,
Guillemont, J., Pasquier, E., Kukla, M., Ludovici, D., Andries, K., De
Bethune, M. P., Pauwels, R., Das, K., Clark, A. D., Jr., Frenkel, Y.
V., Hughes, S. H., Medaer, B., De Knaep, F., Bohets, H., De Clerck, F.,
Lampo, A., Williams, P., and Stoffels, P. (2005) In search of a novel
anti-HIV drug: multidisciplinary coordination in the discovery of
4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile
(R278474, rilpivirine), J. Med. Chem., 48, 1901-1909.
40.Maiga, A. I., Descamps, D., Morand-Joubert, L.,
Malet, I., Derache, A., Cisse, M., Koita, V., Akonde, A., Diarra, B.,
Wirden, M., Tounkara, A., Verlinden, Y., Katlama, C., Costagliola, D.,
Masquelier, B., Calvez, V., and Marcelin, A. G. (2010)
Resistance-associated mutations to etravirine (TMC-125) in
antiretroviral-naive patients infected with non-B HIV-1 subtypes,
Antimicrob. Agents Chemother., 54, 728-733.
41.Bunupuradah, T., Ananworanich, J., Chetchotisakd,
P., Kantipong, P., Jirajariyavej, S., Sirivichayakul, S., Munsakul, W.,
Prasithsirikul, W., Sungkanuparph, S., Bowonwattanuwong, C.,
Klinbuayaem, V., Petoumenos, K., Hirschel, B., Bhakeecheep, S., and
Ruxrungtham, K. (2011) Etravirine and rilpivirine resistance in HIV-1
subtype CRF01AE-infected adults failing non-nucleoside reverse
transcriptase inhibitor-based regimens, Antivir. Ther.,
16, 1113-1121.
42.Brehm, J. H., Koontz, D. L., Wallis, C. L.,
Shutt, K. A., Sanne, I., Wood, R., McIntyre, J. A., Stevens, W. S.,
Sluis-Cremer, N., and Mellors, J. W. (2012) Frequent emergence of N348I
in HIV-1 subtype C reverse transcriptase with failure of initial
therapy reduces susceptibility to reverse-transcriptase inhibitors,
Clin. Infect. Dis., 55, 737-745.
43.Johnson, V. A., Calvez, V., Gunthard, H. F.,
Paredes, R., Pillay, D., Shafer, R. W., Wensing, A. M., and Richman, D.
D. (2013) Update of the drug resistance mutations in HIV-1: March 2013,
Top. Antivir. Med., 21, 6-14.
44.Wensing, A. M., Calvez, V., Gunthard, H. F.,
Johnson, V. A., Paredes, R., Pillay, D., Shafer, R. W., and Richman, D.
D. (2014) 2014 update of the drug resistance mutations in HIV-1,
Top. Antivir. Med., 22, 642-650.
45.Lambert-Niclot, S., Charpentier, C., Storto, A.,
Fofana, D., Soulie, C., Fourati, S., Wirden, M., Morand-Joubert, L.,
Masquelier, B., Flandre, P., Calvez, V., Descamps, D., and Marcelin, A.
G. (2014) Rilpivirine, emtricitabine and tenofovir resistance in
HIV-1-infected rilpivirine-naive patients failing antiretroviral
therapy, J. Antimicrob. Chemother., 69, 1086-1089.
46.Gatell, J. M., Morales-Ramirez, J. O., Hagins, D.
P., Thompson, M., Keikawus, A., Hoffmann, C., Rugina, S., Osiyemi, O.,
Escoriu, S., Dretler, R., Harvey, C., Xu, X., and Teppler, H. (2014)
Forty-eight-week efficacy and safety and early CNS tolerability of
doravirine (MK-1439), a novel NNRTIs, with TDF/FTC in ART-naive
HIV-positive patients, J. Int. AIDS Soc., 17, 19532.
47.Jackson, A., and McGowan, I. (2015) Long-acting
rilpivirine for HIV prevention, Curr. Opin. HIV AIDS, 10,
253-257.
48.Margolis, D. A., Brinson, C. C., Smith, G. H., De
Vente, J., Hagins, D. P., Eron, J. J., Griffith, S. K., St. Clair, M.
H., Stevens, M. C., Williams, P. E., Ford, S. L., Stancil, B. S.,
Bomar, M. M., Hudson, K. J., Smith, K. Y., and Spreen, W. R. (2015)
Cabotegravir plus rilpivirine, once a day, after induction with
cabotegravir plus nucleoside reverse transcriptase inhibitors in
antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised,
phase 2b, dose-ranging trial, Lancet Infect. Dis., 15,
1145-1155.
49.Kovarova, M., Council, Date, A. A., Long, J. M.,
Nochi, T., Belshan, M., Shibata, A., Vincent, H., Baker, C. E., Thayer,
W. O., Kraus, G., Lachaud-Durand, S., Williams, P., Destache, C. J.,
and Garcia, J. V. (2015) Nanoformulations of rilpivirine for topical
pericoital and systemic coitus-independent administration efficiently
prevent HIV transmission, PLoS Pathog., 11, e1005075.
50.Miller, C. D., Crain, J., Tran, B., and Patel, N.
(2011) Rilpivirine: a new addition to the anti-HIV-1 armamentarium,
Drugs Today (Barc.), 47, 5-15.
51.Chen, X., Zhan, P., Li, D., De Clercq, E., and
Liu, X. (2011) Recent advances in DAPYs and related analogues as HIV-1
NNRTIss, Curr. Med. Chem., 18, 359-376.
52.Pauwels, R., Andries, K., Debyser, Z., Van Daele,
P., Schols, D., Stoffels, P., De Vreese, K., Woestenborghs, R.,
Vandamme, A. M., Janssen, C. G., Anne, J., Cauwenbergh, G., Desmyter,
J., Heykants, J., Janssen, Mac., De Clercq, E., and Janssen, C. G.
(1993) Potent and highly selective human immunodeficiency virus type 1
(HIV-1) inhibition by a series of alpha-anilinophenylacetamide
derivatives targeted at HIV-1 reverse transcriptase, Proc. Natl.
Acad. Sci. USA, 90, 1711-1715.
53.Ludovici, D. W., Kukla, M. J., Grous, P. G.,
Krishnan, S., Andries, K., De Bethune, M. P., Azijn, H., Pauwels, R.,
De Clercq, E., Arnold, E., and Janssen, P. A. (2001) Evolution of
anti-HIV drug candidates. Part 1: From alpha-anilinophenylacetamide
(alpha-APA) to imidoyl thiourea (ITU), Bioorg. Med. Chem. Lett.,
11, 2225-2228.
54.Ludovici, D. W., Kavash, R. W., Kukla, M. J., Ho,
C. Y., Ye, H., De Corte, B. L., Andries, K., De Bethune, M. P., Azijn,
H., Pauwels, R., Moereels, H. E., Heeres, J., Koymans, L. M., De Jonge,
M. R., Van Aken, K. J., Daeyaert, F. F., Lewi, P. J., Das, K., Arnold,
E., and Janssen, P. A. (2001) Evolution of anti-HIV drug candidates.
Part 2: Diaryltriazine (DATA) analogues, Bioorg. Med. Chem.
Lett., 11, 2229-2234.
55.De Corte, B. L. (2005) From
4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk](1,4)benzodiazepin-2(1H)-one
(TIBO) to etravirine (TMC125): fifteen years of research on
non-nucleoside inhibitors of HIV-1 reverse transcriptase, J. Med.
Chem., 48, 1689-1696.
56.Ludovici, D. W., De Corte, B. L., Kukla, M. J.,
Ye, H., Ho, C. Y., Lichtenstein, M. A., Kavash, R. W., Andries, K., De
Bethune, M. P., Azijn, H., Pauwels, R., Lewi, P. J., Heeres, J.,
Koymans, L. M., De Jonge, M. R., Van Aken, K. J., Daeyaert, F. F., Das,
K., Arnold, E., and Janssen, P. A. (2001) Evolution of anti-HIV drug
candidates. Part 3: Diarylpyrimidine (DAPY) analogues, Bioorg. Med.
Chem. Lett., 11, 2235-2239.
57.Heeres, J., and Lewi, L. P. (2007) The medicinal
chemistry of the DATA and DAPY series of HIV-1 non-nucleoside reverse
transcriptase inhibitors (NNRTIss), in Advances in Antiviral Drug
Design, Elsevier Science, pp. 213-242.
58.Lansdon, E. B., Brendza, K. M., Hung, M., Wang,
R., Mukund, S., Jin, D., Birkus, G., Kutty, N., and Liu, X. (2010)
Crystal structures of HIV-1 reverse transcriptase with etravirine
(TMC125) and rilpivirine (TMC278): implications for drug design, J.
Med. Chem., 53, 4295-4299.
59.Das, K., Bauman, J. D., Clark, A. D., Jr.,
Frenkel, Y. V., Lewi, P. J., Shatkin, A. J., Hughes, S. H., and Arnold,
E. (2008) High-resolution structures of HIV-1 reverse
transcriptase/TMC278 complexes: strategic flexibility explains potency
against resistance mutations, Proc. Natl. Acad. Sci. USA,
105, 1466-1471.
60.Di Fabio, S., Van Roey, J., Giannini, G., Van den
Mooter, G., Spada, M., Binelli, A., Pirillo, M. F., Germinario, E.,
Belardelli, F., de Bethune, M. P., and Vella, S. (2003) Inhibition of
vaginal transmission of HIV-1 in hu-SCID mice by the non-nucleoside
reverse transcriptase inhibitor TMC120 in a gel formulation,
AIDS, 17, 1597-1604.
61.Van Herrewege, Y., Vanham, G., Michiels, J.,
Fransen, K., Kestens, L., Andries, K., Janssen, P., and Lewi, P. (2004)
A series of diaryltriazines and diarylpyrimidines are highly potent
nonnucleoside reverse transcriptase inhibitors with possible
applications as microbicides, Antimicrob. Agents Chemother.,
48, 3684-3689.
62.Devlin, B., Nuttall, J., Wilder, S., Woodsong,
C., and Rosenberg, Z. (2013) Development of dapivirine vaginal ring for
HIV prevention, Antiviral Res., 100 (Suppl.), S3-8.
63.Lewi, P. J., De Jonge, M., Daeyaert, F., Koymans,
L., Vinkers, M., Heeres, J., Janssen, P. A., Arnold, E., Das, K.,
Clark, A. D., Jr., Hughes, S. H., Boyer, P. L., De Bethune, M. P.,
Pauwels, R., Andries, K., Kukla, M., Ludovici, D., De Corte, B.,
Kavash, R., and Ho, C. (2003) On the detection of multiple-binding
modes of ligands to proteins, from biological, structural, and modeling
data, J. Comput. Aided Mol. Des., 17, 129-134.
64.Daeyaert, F., De Jonge, M., Heeres, J., Koymans,
L., Lewi, P., Vinkers, M. H., and Janssen, P. A. (2004) A pharmacophore
docking algorithm and its application to the cross-docking of 18
HIV-NNRTIs’s in their binding pockets, Proteins,
54, 526-533.
65.Guillemont, J., Pasquier, E., Palandjian, P.,
Vernier, D., Gaurrand, S., Lewi, P. J., Heeres, J., De Jonge, M. R.,
Koymans, L. M., Daeyaert, F. F., Vinkers, M. H., Arnold, E., Das, K.,
Pauwels, R., Andries, K., De Bethune, M. P., Bettens, E., Hertogs, K.,
Wigerinck, P., Timmerman, P., and Janssen, P. A. (2005) Synthesis of
novel diarylpyrimidine analogues and their antiviral activity against
human immunodeficiency virus type 1, J. Med. Chem., 48,
2072-2079.
66.Mordant, C., Schmitt, B., Pasquier, Demestre, C.,
Queguiner, L., Masungi, C., Peeters, A., Smeulders, L., Bettens, E.,
Hertogs, K., Heeres, J., Lewi, P., and Guillemont, J. (2007) Synthesis
of novel diarylpyrimidine analogues of TMC278 and their antiviral
activity against HIV-1 wild-type and mutant strains, Eur. J. Med.
Chem., 42, 567-579.
67.Lewi, P., Heeres, J., and Baert L. E. C. (2006)
Prevention of HIV-Infection with TMC278, Patent WO 2006106103
A3.
68.Ripamonti, D., Bombana, E., and Rizzi, M. (2014)
Rilpivirine: drug profile of a second-generation non-nucleoside reverse
transcriptase HIV-inhibitor, Expert. Rev. Anti. Infect. Ther.,
12, 13-29.
69.Feng, X. Q., Zeng, Z. S., Liang, Y. H., Chen, F.
E., Pannecouque, C., Balzarini, J., and De Clercq, E. (2010) Synthesis
and biological evaluation of 4-(hydroxyimino)arylmethyl
diarylpyrimidine analogues as potential non-nucleoside reverse
transcriptase inhibitors against HIV, Bioorg. Med. Chem.,
18, 2370-2374.
70.Zeng, Z. S., Liang, Y. H., Feng, X. Q., Chen, F.
E., Pannecouque, C., Balzarini, J., and De Clercq, E. (2010) Lead
optimization of diarylpyrimidines as non-nucleoside inhibitors of HIV-1
reverse transcriptase, ChemMedChem, 5, 837-840.
71.Gu, S. X., He, Q. Q., Yang, S. Q., Ma, X. D.,
Chen, F. E., De Clercq, E., Balzarini, J., and Pannecouque, C. (2011)
Synthesis and structure–activity relationship of novel
diarylpyrimidines with hydromethyl linker (CH(OH)-DAPYs) as HIV-1
NNRTIss, Bioorg. Med. Chem., 19, 5117-5124.
72.Das, K., Clark, A. D., Lewi, P. J., Heeres, J.,
De Jonge, M. R., Koymans, L. M., Vinkers, H. M., Daeyaert, F.,
Ludovici, D. W., Kukla, M. J., De Corte, B., Kavash, R. W., Ho, C. Y.,
Ye, H., Lichtenstein, M. A., Andries, K., Pauwels, R., De Bethune, M.
P., Boyer, P. L., Clark, P., Hughes, S. H., Janssen, P. A., and Arnold,
E. (2004) Roles of conformational and positional adaptability in
structure-based design of TMC125-R165335 (etravirine) and related
non-nucleoside reverse transcriptase inhibitors that are highly potent
and effective against wild-type and drug-resistant HIV-1 variants,
J. Med. Chem., 47, 2550-2560.
73.Gu, S. X., Li, Z. M., Ma, X. D., Yang, S. Q., He,
Q. Q., Chen, F. E., De Clercq, E., Balzarini, J., and Pannecouque, C.
(2012) Chiral resolution, absolute configuration assignment and
biological activity of racemic diarylpyrimidine CH(OH)-DAPY as
potent nonnucleoside HIV-1 reverse transcriptase inhibitors, Eur. J.
Med. Chem., 53, 229-234.
74.Feng, X. Q., Liang, Y. H., Zeng, Z. S., Chen, F.
E., Balzarini, J., Pannecouque, C., and De Clercq, E. (2009) Structural
modifications of DAPY analogues with potent anti-HIV-1 activity,
ChemMedChem, 4, 219-224.
75.Liang, Y. H., Feng, X. Q., Zeng, Z. S., Chen, F.
E., Balzarini, J., Pannecouque, C., and De Clercq, E. (2009) Design,
synthesis, and SAR of naphthyl-substituted diarylpyrimidines as
non-nucleoside inhibitors of HIV-1 reverse transcriptase,
ChemMedChem, 4, 1537-1545.
76.Liang, Y. H., He, Q. Q., Zeng, Z. S., Liu, Z. Q.,
Feng, X. Q., Chen, F. E., Balzarini, J., Pannecouque, C., and Clercq,
E. D. (2010) Synthesis and anti-HIV activity of 2-naphthyl substituted
DAPY analogues as non-nucleoside reverse transcriptase inhibitors,
Bioorg. Med. Chem., 18, 4601-4605.
77.Lee, W. G., Frey, K. M., Gallardo-Macias, R.,
Spasov, K. A., Chan, A. H., Anderson, K. S., and Jorgensen, W. L.
(2015) Discovery and crystallography of bicyclic arylaminoazines as
potent inhibitors of HIV-1 reverse transcriptase, Bioorg. Med. Chem.
Lett., 25, 4824-4827.
78.Gu, S. X., Yang, S. Q., He, Q. Q., Ma, X. D.,
Chen, F. E., Dai, H. F., Clercq, E. D., Balzarini, J., and Pannecouque,
C. (2011) Design, synthesis and biological evaluation of cycloalkyl
arylpyrimidines (CAPYs) as HIV-1 NNRTIss, Bioorg. Med. Chem.,
19, 7093-7099.
79.Huang, B., Kang, D., Yang, J., Zhan, P., and Liu,
X. (2016) Novel diarylpyrimidines and diaryltriazines as potent HIV-1
NNRTIss with dramatically improved solubility: a patent evaluation of
US20140378443A1, Expert. Opin. Ther. Pat., 26,
281-289.
80.Sergeyev, S., Yadav, A. K., Franck, P., Michiels,
J., Lewi, P., Heeres, J., Vanham, G., Arien, K. K., Vande Velde, C. M.,
De Winter, H., and Maes, B. U. (2016)
2,6-Di(arylamino)-3-fluoropyridine derivatives as HIV non-nucleoside
reverse transcriptase inhibitors, J. Med. Chem., 59,
1854-1868.
81.Andries, K., Azijn, H., Thielemans, T., Ludovici,
D., Kukla, M., Heeres, J., Janssen, P., De Corte, B., Vingerhoets, J.,
Pauwels, R., and De Bethune, M. P. (2004) TMC125, a novel
next-generation nonnucleoside reverse transcriptase inhibitor active
against nonnucleoside reverse transcriptase inhibitor-resistant human
immunodeficiency virus type 1, Antimicrob. Agents Chemother.,
48, 4680-4686.
82.Usach, I., Melis, V., and Peris, J. E. (2013)
Non-nucleoside reverse transcriptase inhibitors: a review on
pharmacokinetics, pharmacodynamics, safety and tolerability, J. Int.
AIDS Soc., 16, 1-14.
83.Sun, L. Q., Qin, B., Huang, L., Qian, K., Chen,
C. H., Lee, K. H., and Xie, L. (2012) Optimization of
2,4-diarylanilines as non-nucleoside HIV-1 reverse transcriptase
inhibitors, Bioorg. Med. Chem. Lett., 22, 2376-2379.
84.Weuts, I., Van Dycke, F., Voorspoels, J., De
Cort, S., Stokbroekx, S., Leemans, R., Brewster, M. E., Xu, D.,
Segmuller, B., Turner, Y. T., Roberts, C. J., Davies, M. C., Qi, S.,
Craig, D. Q., and Reading, M. (2011) Physicochemical properties of the
amorphous drug, cast films, and spray dried powders to predict
formulation probability of success for solid dispersions: etravirine,
J. Pharm. Sci., 100, 260-274.
85.Zeng, Z. S., He, Q. Q., Liang, Y. H., Feng, X.
Q., Chen, F. E., De Clercq, E., Balzarini, J., and Pannecouque, C.
(2010) Hybrid diarylbenzopyrimidine non-nucleoside reverse
transcriptase inhibitors as promising new leads for improved anti-HIV-1
chemotherapy, Bioorg. Med. Chem., 18, 5039-5047.
86.Smith, S. J., Pauly, G. T., Akram, A., Melody,
K., Rai, G., Maloney, D. J., Ambrose, Z., Thomas, C. J., Schneider, J.
T., and Hughes, S. H. (2016) Rilpivirine analogs potently inhibit
drug-resistant HIV-1 mutants, Retrovirology, 13, 11.
87.Corbett, J. W., Ko, S. S., Rodgers, J. D.,
Jeffrey, S., Bacheler, L. T., Klabe, R. M., Diamond, S., Lai, C. M.,
Rabel, S. R., Saye, J. A., Adams, S. P., Trainor, G. L., Anderson, P.
S., and Erickson-Viitanen, S. K. (1999) Expanded-spectrum nonnucleoside
reverse transcriptase inhibitors inhibit clinically relevant mutant
variants of human immunodeficiency virus type 1, Antimicrob. Agents
Chemother., 43, 2893-2897.
88.Corbett, J. W., and Rodgers, J. D. (2002)
Discovery of second generation quinazolinone non-nucleoside reverse
transcriptase inhibitors of HIV-1, Prog. Med. Chem., 40,
63-105.
89.Girardet, J. L., Koh, Y. H., Shaw, S., and Kin,
H. W. (2006) Diaryl-purine, Azapurines and Deazapurines as
Non-nucleoside Reverse Transcriptase Inhibitors for Treatment of
HIV, Patent WO 2006122003 A2.
90.Lai, M. T., Feng, M., Falgueyret, J. P., Tawa,
P., Witmer, M., DiStefano, D., Li, Y., Burch, J., Sachs, N., Lu, M.,
Cauchon, E., Campeau, L. C., Grobler, J., Yan, Y., Ducharme, Y., Cote,
B., Asante-Appiah, E., Hazuda, D. J., and Miller, M. D. (2014) In
vitro characterization of MK-1439, a novel HIV-1 nonnucleoside
reverse transcriptase inhibitor, Antimicrob. Agents
Chemother., 58, 1652-1663.
91.Ren, J., Chamberlain, P. P., Stamp, A., Short, S.
A., Weaver, K. L., Romines, K. R., Hazen, R., Freeman, A., Ferris, R.
G., Andrews, C. W., Boone, L., Chan, J. H., and Stammers, D. K. (2008)
Structural basis for the improved drug resistance profile of new
generation benzophenone non-nucleoside HIV-1 reverse transcriptase
inhibitors, J. Med. Chem., 51, 5000-5008.
92.Tang, G., Kertesz, D. J., Yang, M., Lin, X.,
Wang, Z., Li, W., Qiu, Z., Chen, J., Mei, J., Chen, L., Mirzadegan, T.,
Harris, S. F., Villasenor, A. G., Fretland, J., Fitch, W. L., Hang, J.
Q., Heilek, G., and Klumpp, K. (2010) Exploration of
piperidine-4-yl-aminopyrimidines as HIV-1 reverse transcriptase
inhibitors. N-Phenyl derivatives with broad potency against resistant
mutant viruses, Bioorg. Med. Chem. Lett., 20,
6020-6023.
93.Chen, X., Liu, X., Meng, Q., Wang, D., Liu, H.,
De Clercq, E., Pannecouque, C., and Balzarini, J. (2013) Novel
piperidinylamino-diaryl-pyrimidine derivatives with dual structural
conformations as potent HIV-1 non-nucleoside reverse transcriptase
inhibitors, Bioorg. Med. Chem. Lett., 23, 6593-6597.
94.Wan, Z. Y., Yao, J., Tao, Y., Mao, T. Q., Wang,
X. L., Lu, Y. P., Wang, H. F., Yin, H., Wu, Y., Chen, F. E., De Clercq,
E., Daelemans, D., and Pannecouque, C. (2015) Discovery of
piperidin-4-yl-aminopyrimidine derivatives as potent non-nucleoside
HIV-1 reverse transcriptase inhibitors, Eur. J. Med. Chem.,
97, 1-9.
95.Miyasaka, T., Tanaka, H., Baba, M., Hayakawa, H.,
Walker, R. T., Balzarini, J., and De Clercq, E. (1989) A novel lead for
specific anti-HIV-1 agents:
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine, J. Med.
Chem., 32, 2507-2509.
96.Chen, W., Zhan, P., Wu, J., Li, Z., and Liu, X.
(2012) The development of HEPT-type HIV non-nucleoside reverse
transcriptase inhibitors and its implications for DABO family, Curr.
Pharm. Des., 18, 4165-4186.
97.Hopkins, A. L., Ren, J., Esnouf, R. M., Willcox,
B. E., Jones, E. Y., Ross, C., Miyasaka, T., Walker, R. T., Tanaka, H.,
Stammers, D. K., and Stuart, D. I. (1996) Complexes of HIV-1 reverse
transcriptase with inhibitors of the HEPT series reveal conformational
changes relevant to the design of potent non-nucleoside inhibitors,
J. Med. Chem., 39, 1589-1600.
98.Hopkins, A. L., Ren, J., Tanaka, H., Baba, M.,
Okamato, M., Stuart, D. I., and Stammers, D. K. (1999) Design of
MKC-442 (emivirine) analogues with improved activity against
drug-resistant HIV mutants, J. Med. Chem., 42,
4500-4505.
99.Tanaka, H., Baba, M., Ubasawa, M., Takashima, H.,
Sekiya, K., Nitta, I., Shigeta, S., Walker, R. T., De Clercq, E., and
Miyasaka, T. (1991) Synthesis and anti-HIV activity of 2-, 3-, and
4-substituted analogues of
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT), J. Med.
Chem., 34, 1394-1399.
100.Tang, J., Maddali, K., Dreis, C. D., Sham, Y.
Y., Vince, R., Pommier, Y., and Wang, Z. (2011) N-3 hydroxylation of
pyrimidine-2,4-diones yields dual inhibitors of HIV reverse
transcriptase and integrase, ACS Med. Chem. Lett., 2,
63-67.
101.Tang, J., Maddali, K., Dreis, C. D., Sham, Y.
Y., Vince, R., Pommier, Y., and Wang, Z. (2011)
6-Benzoyl-3-hydroxypyrimidine-2,4-diones as dual inhibitors of HIV
reverse transcriptase and integrase, Bioorg. Med. Chem. Lett.,
21, 2400-2402.
102.Tang, J., Maddali, K., Metifiot, M., Sham, Y.
Y., Vince, R., Pommier, Y., and Wang, Z. (2011)
3-Hydroxypyrimidine-2,4-diones as an inhibitor scaffold of HIV
integrase, J. Med. Chem., 54, 2282-2292.
103.Balzarini, J., Baba, M., and De Clercq, E.
(1995) Differential activities of
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives against
different human immunodeficiency virus type 1 mutant strains,
Antimicrob. Agents Chemother., 39, 998-1002.
104.Tanaka, H., Takashima, H., Ubasawa, M., Sekiya,
K., Nitta, I., Baba, M., Shigeta, S., Walker, R. T., De Clercq, E., and
Miyasaka, T. (1992) Synthesis and antiviral activity of deoxy analogs
of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) as potent
and selective anti-HIV-1 agents, J. Med. Chem., 35,
4713-4719.
105.Baba, M., Shigeta, S., Tanaka, H., Miyasaka,
T., Ubasawa, M., Umezu, K., Walker, R. T., Pauwels, R., and De Clercq,
E. (1992) Highly potent and selective inhibition of HIV-1 replication
by 6-phenylthiouracil derivatives, Antiviral. Res., 17,
245-264.
106.Kim, D. K., Gam, J., Kim, Y. W., Lim, J., Kim,
H. T., and Kim, K. H. (1997) Synthesis and anti-HIV-1 activity of a
series of 1-alkoxy-5-alkyl-6-(arylthio)uracils, J. Med. Chem.,
40, 2363-2373.
107.Kim, D. K., Gam, J., Kim, G., Lim, J., Lee, N.,
Kim, H. T., and Kim, K. H. (1996) Synthesis and anti-HIV-1 activity of
a series of 1-(alkoxymethyl)-5-alkyl-6-(arylselenenyl)uracil and
-2-thiouracils, J. Heterocycl. Chem., 33, 1275-1283.
108.Tanaka, H., Takashima, H., Ubasawa, M., Sekiya,
K., Inouye, N., Baba, M., Shigeta, S., Walker, R. T., De Clercq, E.,
and Miyasaka, T. (1995) Synthesis and antiviral activity of 6-benzyl
analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) as
potent and selective anti-HIV-1 agents, J. Med. Chem.,
38, 2860-2865.
109.Mitchell, M. L., Son, J. C., Guo, H., Im, Y.
A., Cho, E. J., Wang, J., Hayes, J., Wang, M., Paul, A., Lansdon, E.
B., Chen, J. M., Graupe, D., Rhodes, G., He, G. X., Geleziunas, R., Xu,
L., and Kim, C. U. (2010) N1-Alkyl pyrimidinediones as non-nucleoside
inhibitors of HIV-1 reverse transcriptase, Bioorg. Med. Chem.
Lett., 20, 1589-1592.
110.Baba, M., Shigeta, S., Yuasa, S., Takashima,
H., Sekiya, K., Ubasawa, M., Tanaka, H., Miyasaka, T., Walker, R. T.,
and De Clercq, E. (1994) Preclinical evaluation of MKC-442, a highly
potent and specific inhibitor of human immunodeficiency virus type 1
in vitro, Antimicrob. Agents Chemother., 38,
688-692.
111.Yuasa, S., Sadakata, Y., Takashima, H., Sekiya,
K., Inouye, N., Ubasawa, M., and Baba, M. (1993) Selective and
synergistic inhibition of human immunodeficiency virus type 1 reverse
transcriptase by a non-nucleoside inhibitor, MKC-442, Mol.
Pharmacol., 44, 895-900.
112.Szczech, G. M., Furman, P., Painter, G. R.,
Barry, D. W., Borroto-Esoda, K., Grizzle, T. B., Blum, M. R.,
Sommadossi, J., Endoh, R., Niwa, T., Yamamoto, M., and Moxham, C.
(2000) Safety assessment, in vitro and in vivo, and
pharmacokinetics of emivirine, a potent and selective nonnucleoside
reverse transcriptase inhibitor of human immunodeficiency virus type 1,
Antimicrob. Agents Chemother., 44, 123-130.
113.Tanaka, H., Baba, M., Hayakawa, H., Sakamaki,
T., Miyasaka, T., Ubasawa, M., Takashima, H., Sekiya, K., Nitta, I.,
Shigeta, S., Walker, R. T., Balzarini, J., and Declercq, E. (1991) A
new class of HIV-1-specific 6-substituted acyclouridine derivatives:
synthesis and anti-HIV-1 activity of 5- or 6-substituted analogues of
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT), J. Med.
Chem., 34, 349-357.
114.Goudgaon, N. M., and Schinazi, R. F. (1991)
Activity of acyclic 6-(phenylselenenyl)pyrimidine nucleosides against
human immunodeficiency viruses in primary lymphocytes, J. Med.
Chem., 34, 3305-3309.
115.Petersen, L., Jessen, C. H., Pedersen, E. B.,
and Nielsen, C. (2003) Synthesis and evaluation of new potential HIV-1
non-nucleoside reverse transcriptase inhibitors. New analogues of
MKC-442 containing Michael acceptors in the C-6 position, Org.
Biomol. Chem., 1, 3541-3545.
116.Pan, H. C., Chen, C. Z., Piras, G., Dutschman,
G. E., Rowe, E. C., Chen, Y. C., and Chu, S. H. (1994) Synthesis and
anti-HIV-1 activities of 6-arylthio- and 6-arylselenoacyclonucleosides,
J. Heterocycl. Chem., 31, 177-185.
117.Buckheit, R. W., Fliakas-Boltz, V., Decker, W.
D., Roberson, J. L., Stup, T. L., Pyle, C. A., White, E. L., McMahon,
J. B., Currens, M. J., Boyd, M. R., and Bader, J. P. (1995) Comparative
anti-HIV evaluation of diverse HIV-1-specific reverse transcriptase
inhibitor-resistant virus isolates demonstrates the existence of
distinct phenotypic subgroups, Antiviral. Res., 26,
117-132.
118.Meng, G., De Clercq, E., and Dai, H. F. (2003)
Interactive study between two types of 6-naphthyl-methyl HEPT analogs
and HIV-1 reverse transcriptase inhibitors, Process Engineer.
Chem., 1, 23-27.
119.Meng, G., Chen, F. E., De Clercq, E.,
Balzarini, J., and Pannecouque, C. (2003) Nonnucleoside HIV-1 reverse
transcriptase inhibitors: Part I. Synthesis and
structure–activity relationship of
1-alkoxymethyl-5-alkyl-6-naphthylmethyl uracils as HEPT analogues,
Chem. Pharm. Bull. (Tokyo), 51, 779-789.
120.Sun, G. F., Chen, X. X., Chen, F. E., Wang, Y.
P., De Clercq, E., Balzarini, J., and Pannecouque, C. (2005)
Nonnucleoside HIV-1 reverse-transcriptase inhibitors: Part 5. Synthesis
and anti-HIV-1 activity of novel 6-naphthylthio HEPT analogues,
Chem. Pharm. Bull. (Tokyo), 53, 886-892.
121.Ji, L., Chen, F. E., Xie, B., De Clercq, E.,
Balzarini, J., and Pannecouque, C. (2007) Synthesis and anti-HIV
activity evaluation of 1-[(alkenyl or alkynyl or
alkyloxy)methyl]-5-alkyl-6-(1-naphthoyl)-2,4-pyrimidinediones as novel
non-nucleoside HIV-1 reverse transcriptase inhibitors, Eur. J. Med.
Chem., 42, 198-204.
122.Tanaka, H., Takashima, H., Ubasawa, M., Sekiya,
K., Nitta, I., Baba, M., Shigeta, S., Walker, R. T., De Clercq, E., and
Miyasaka, T. (1992) Structure–activity relationships of
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine analogues: effect of
substitutions at the C-6 phenyl ring and at the C-5 position on
anti-HIV-1 activity, J. Med. Chem., 35, 337-345.
123.Gaudio, A. C., and Montanari, C. A. (2002) HEPT
derivatives as non-nucleoside inhibitors of HIV-1 reverse
transcriptase: QSAR studies agree with the crystal structures, J.
Comput. Aided. Mol. Des., 16, 287-295.
124.Goudgaon, N., McMillan, A., and Schinazi, R.
(1992) 1-(Ethoxymethyl)-6-(phenyl-selenenyl)pyrimidines with
activity against human immunodeficiency virus types 1 and 2,
Antivir. Chem. Chemother., 3, 263-266.
125.Chiu, T. L., and So, S. S. (2004) Development
of neural network QSPR models for Hansch substituent constants. 2.
Applications in QSAR studies of HIV-1 reverse transcriptase and
dihydrofolate reductase inhibitors, J. Chem. Inf. Comput. Sci.,
44, 154-160.
126.Sorensen, E. R., El-Brollosy, N. R., Jorgensen,
P. T., Pedersen, E. B., and Nielsen, C. (2005) Synthesis of
6-(3,5-dichlorobenzyl) derivatives as isosteric analogues of the HIV
drug 6-(3,5-dimethylbenzyl)-1-(ethoxymethyl)-5-isopropyluracil
(GCA-186), Arch. Pharm. (Weinheim), 338, 299-304.
127.El-Brollosy, N. R., Sorensen, E. R., Pedersen,
E. B., Sanna, G., La Colla, P., and Loddo, R. (2008) Synthesis and
antiviral evaluation of 6-(trifluoromethylbenzyl) and 6-(fluorobenzyl)
analogues of HIV drugs emivirine and GCA-186, Arch. Pharm.
(Weinheim), 341, 9-19.
128.Danel, K., Larsen, E., Pedersen, E. B.,
Vestergaard, B. F., and Nielsen, C. (1996) Synthesis and potent
anti-HIV-1 activity of novel 6-benzyluracil analogues of
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine, J. Med.
Chem., 39, 2427-2431.
129.Buckheit, R. W., Jr., Watson, K.,
Fliakas-Boltz, V., Russell, J., Loftus, T. L., Osterling, M. C.,
Turpin, J. A., Pallansch, L. A., White, E. L., Lee, J. W., Lee, S. H.,
Oh, J. W., Kwon, H. S., Chung, S. G., and Cho, E. H. (2001) SJ-3366, a
unique and highly potent nonnucleoside reverse transcriptase inhibitor
of human immunodeficiency virus type 1 (HIV-1) that also inhibits
HIV-2, Antimicrob. Agents Chemother., 45, 393-400.
130.Buckheit, R. W., Jr., Hartman, T. L., Watson,
K. M., Kwon, H. S., Lee, S. H., Lee, J. W., Kang, D. W., Chung, S. G.,
and Cho, E. H. (2007) The structure–activity relationships of
2,4(1H,3H)-pyrimidinedione derivatives as potent HIV type 1 and type 2
inhibitors, Antivir. Chem. Chemother., 18, 259-275.
131.Tanaka, H., Baba, M., Saito, S., Miyasaka, T.,
Takashima, H., Sekiya, K., Ubasawa, M., Nitta, I., Walker, R. T.,
Nakashima, H., and De Clercq, E. (1991) Specific anti-HIV-1
“acyclonucleosides” which cannot be phosphorylated:
synthesis of some deoxy analogues of
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine, J. Med.
Chem., 34, 1508-1511.
132.El-Brollosy, N. R., Jorgensen, P. T., Dahan,
B., Boel, A. M., Pedersen, E. B., and Nielsen, C. (2002) Synthesis of
novel N-1 (allyloxymethyl) analogues of
6-benzyl-1-(ethoxymethyl)-5-isopropyluracil (MKC-442, emivirine)
with improved activity against HIV-1 and its mutants, J. Med.
Chem., 45, 5721-5726.
133.Hannongbua, S., Nivesanond, K., Lawtrakul, L.,
Pungpo, P., and Wolschann, P. (2001) 3D-quantitative
structure–activity relationships of HEPT derivatives as HIV-1
reverse transcriptase inhibitors, based on ab initio
calculations, J. Chem. Inf. Comput. Sci., 41,
848-855.
134.Dueweke, T. J., Pushkarskaya, T., Poppe, S. M.,
Swaney, S. M., Zhao, J. Q., Chen, I. S., Stevenson, M., and Tarpley, W.
G. (1993) A mutation in reverse transcriptase of
bis(heteroaryl)piperazine-resistant human immunodeficiency virus type 1
that confers increased sensitivity to other nonnucleoside inhibitors,
Proc. Natl. Acad. Sci. USA, 90, 4713-4717.
135.Buckheit, R. W., Jr., Watson Buckheit, K.,
Sturdevant, C. B., and Buckheit, R. W., 3rd (2013) Selection and
characterization of viruses resistant to the dual acting
pyrimidinedione entry and non-nucleoside reverse transcriptase
inhibitor IQP-0410, Antiviral. Res., 100, 382-391.
136.El-Brollosy, N. R., Nielsen, C., and Pedersen,
E. (2005) Synthesis of N-1-(indanyloxymethyl) and
N-1-(4-hydroxybut-2-enyloxymethyl) analogues of the HIV drugs emivirine
and GCA-186, Monatsh. Chem., 136, 1247-1254.
137.Pontikis, R., Benhida, R., Aubertin, A. M.,
Grierson, D. S., and Monneret, C. (1997) Synthesis and anti-HIV
activity of novel N-1 side chain-modified analogs of
1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT), J. Med.
Chem., 40, 1845-1854.
138.Cho, E. H., Chung, Kim, J. Y., Lee, S. H.,
Kwon, H. S., Lee, J. E., Joo, J. H., Kim, B. C., and Kang, D. W. (1999)
Antiviral Substituted Pyrimidinedione Homocarbocyclic Nucleoside
Derivatives and Methods for the Preparation thereof and Compositions
Containing the Same as Active Ingredients, Patent W097/30979.
139.Guo, H. K., Lee, Lee, I. Y., Mitchell, M. L.,
Son, J. C., and Xu, L. (2008) The Novel HIV Reverse Transcriptase
Inhibitors, Patent WO 2009005674 A3.
140.Mitchell, M. L., Son, J. C., Lee, I.
Y., Lee, C. K., Kim, H. S., Guo, H., Wang,
J., Hayes, J., Wang, M., Paul, A., Lansdon, E.
B., Chen, J. M., Eisenberg, G., Geleziunas, R., Xu,
L., and Kim, C. U. (2010) N1-heterocyclic pyrimidinediones as
non-nucleoside inhibitors of HIV-1 reverse transcriptase, Bioorg.
Med. Chem. Lett., 20, 1585-1588.
141.Son, J. C., Kim, S. K., Lee, C. K., and Kim, H.
S. (2004) Antiviral 2,4-Pyrimidinedione Derivatives and Process for
the Preparation thereof, Patent WO 1997043266 A1.
142.De Clercq, E. (1994) Non-nucleoside reverse
transcriptase inhibitors (NNRTIss), Exp. Opin. Invest. Drugs,
3, 253-271.
143.Mai, A., Artico, M., Sbardella, G., Quartarone,
S., Massa, S., Loi, A. G., De Montis, A., Scintu, F., Putzolu, M., and
La Colla, P. (1997) Dihydro(alkylthio)(naphthylmethyl)oxopyrimidines:
novel non-nucleoside reverse transcriptase inhibitors of the S-DABO
series, J. Med. Chem., 40, 1447-1454.
144.Tramontano, E., Marongiu, M. E., De Montis, A.,
Loi, A. G., Artico, M., Massa, S., Mai, A., and La Colla, P. (1994)
Characterization of the anti-HIV-1 activity of
3,4-dihydro-2-alkoxy-6-benzyl-4-oxopyrimidines (DABOs), new
non-nucleoside reverse transcriptase inhibitors, New Microbiol.,
17, 269-279.
145.Artico, M, Massa, S., Mai, A., Marongiu, M. E.,
Piras, G., Tramontano, E., and Lacolla, P. (1993)
3,4-Dihydro-2-alkoxy-6-benzyl-4-oxopyrimidines (DABOs): a new
class of specific inhibitors of human immunodeficiency virus type 1,
Antiviral Chem. Chemother., 4, 361-368.
146.Massa, S. M. A., Artico, M., Sbardella, G.,
Tramontano, E., Loi, A. G., Scano, P., and La Colla, P. (1995)
Synthesis and antiviral activity of new
3,4-dihydro-2-alkoxy-6-benzyl-4-oxopyrimidines (DABOs), specific
inhibitors of human immunodeficiency virus type-1, Antiviral Chem.
Chemother., 6, 1-8.
147.Manetti, F., Este, J. A., Clotet-Codina, I.,
Armand-Ugon, M., Maga, G., Crespan, E., Cancio, R., Mugnaini, C.,
Bernardini, C., Togninelli, A., Carmi, C., Alongi, M., Petricci, E.,
Massa, S., Corelli, F., and Botta, M. (2005) Parallel solution-phase
and microwave-assisted synthesis of new S-DABO derivatives endowed with
subnanomolar anti-HIV-1 activity, J. Med. Chem., 48,
8000-8008.
148.Zhang, X. J., Lu, L. H., Wang, R. R., Wang, Y.
P., Luo, R. H., Cong Lai, C., Yang, L. M., He, Y. P., and Zheng, Y. T.
(2013) DB-02, a C-6-cyclohexylmethyl substituted pyrimidinone HIV-1
reverse transcriptase inhibitor with nanomolar activity, displays an
improved sensitivity against K103N or Y181C than S-DABOs, PLoS
One, 8, e81489.
149.Mai, A., Artico, M., Sbardella, G., Massa, S.,
Novellino, E., Greco, G., Loi, A. G., Tramontano, E., Marongiu, M. E.,
and La Colla, P. (1999)
5-Alkyl-2-(alkylthio)-6-(2,6-dihalophenylmethyl)-3,4-dihydropyrimidin-4(3H)-ones:
novel potent and selective dihydro-alkoxy-benzyl-oxopyrimidine
derivatives, J. Med. Chem., 42, 619-627.
150.Sbardella, G., Mai, A., Artico, M., Chimenti,
P., Massa, S., Loddo, R., Marongiu, M. E., La Collat, P., and Pani, A.
(2001) Structure–activity relationship studies on new DABOS:
effect of substitutions at pyrimidine C-5 and C-6 positions on
anti-HIV-1 activity, Antivir. Chem. Chemother., 12,
37-50.
151.Quaglia, M., Mai, A., Sbardella, G., Artico,
M., Ragno, R., Massa, S., Del Piano, D., Setzu, G., Doratiotto, S., and
Cotichini, V. (2001) Chiral resolution and molecular modeling
investigation of
rac-2-cyclopentylthio-6-[1-(2,6-difluorophenyl)ethyl]-3,4-dihydro-5-methylpyrimid
in-4(3H)-one (MC-1047), a potent anti-HIV-1 reverse transcriptase agent
of the DABO class, Chirality, 13, 75-80.
152.Mai, A., Sbardella, G., Artico, M., Ragno, R.,
Massa, S., Novellino, E., Greco, G., Lavecchia, A., Musiu, C., La
Colla, M., Murgioni, C., La Colla, P., and Loddo, R. (2001)
Structure-based design, synthesis, and biological evaluation of
conformationally restricted novel
2-alkylthio-6-[1-(2,6-difluorophenyl)alkyl]-3,4-dihydro-5-alkylpyrimidin-4(3H)-ones
as non-nucleoside inhibitors of HIV-1 reverse transcriptase, J. Med.
Chem., 44, 2544-2554.
153.Ragno, R., Mai, A., Sbardella, G., Artico, M.,
Massa, S., Musiu, C., Mura, M., Marturana, F., Cadeddu, A., and La
Colla, P. (2004) Computer-aided design, synthesis, and anti-HIV-1
activity in vitro of
2-alkylamino-6-[1-(2,6-difluorophenyl)alkyl]-3,4-dihydro-5-alkylpyrimidin-4(3H)-ones
as novel potent non-nucleoside reverse transcriptase inhibitors, also
active against the Y181C variant, J. Med. Chem., 47,
928-934.
154.Mai, A., Artico, M., Ragno, R., Sbardella, G.,
Massa, S., Musiu, C., Mura, M., Marturana, F., Cadeddu, A., Maga, G.,
and La Colla, P. (2005)
5-Alkyl-2-alkylamino-6-(2,6-difluorophenylalkyl)-3,4-dihydropyrimidin-4(3H)-ones,
a new series of potent, broad-spectrum non-nucleoside reverse
transcriptase inhibitors belonging to the DABO family, Bioorg. Med.
Chem., 13, 2065-2077.
155.Mai, A., Artico, M., Rotili, D., Tarantino, D.,
Clotet-Codina, I., Armand-Ugon, M., Ragno, R., Simeoni, S., Sbardella,
G., Nawrozkij, M. B., Samuele, A., Maga, G., and Este, J. A. (2007)
Synthesis and biological properties of novel
2-aminopyrimidin-4(3H)-ones highly potent against HIV-1 mutant strains,
J. Med. Chem., 50, 5412-5424.
156.Loksha, Y. M., Pedersen, E. B., Loddo, R., and
La Colla, P. (2016) Synthesis and anti-HIV-1 evaluation of some novel
MC-1220 analogs as non-nucleoside reverse transcriptase inhibitors,
Arch. Pharm. (Weinheim), 349, 363-372.
157.Mugnaini, C., Alongi, M., Togninelli, A.,
Gevariya, H., Brizzi, A., Manetti, F., Bernardini, C., Angeli, L.,
Tafi, A., Bellucci, L., Corelli, F., Massa, S., Maga, G., Samuele, A.,
Facchini, M., Clotet-Codina, I., Armand-Ugon, M., Este, J. A., and
Botta, M. (2007) Dihydro-alkylthio-benzyl-oxopyrimidines as inhibitors
of reverse transcriptase: synthesis and rationalization of the
biological data on both wild-type enzyme and relevant clinical mutants,
J. Med. Chem., 50, 6580-6595.
158.He, Y., Chen, F., Sun, G., Wang, Y., De Clercq,
E., Balzarini, J., and Pannecouque, C. (2004) 5-Alkyl-2-[(aryl and
alkyloxylcarbonylmethyl)thio]-6-(1-naphthylmethyl)pyrimidin-4(3H)-ones
as an unique HIV reverse transcriptase inhibitors of S-DABO series,
Bioorg. Med. Chem. Lett., 14, 3173-3176.
159.Nawrozkij, M. B., Rotili, D., Tarantino, D.,
Botta, G., Eremiychuk, A. S., Musmuca, I., Ragno, I., Samuele, A.,
Zanoli, S., Armand-Ugon, M., Clotet-Codina, I., Novakov, I. A.,
Orlinson, B. S., Maga, G., Este, J. A., Artico, M., and Mai, A. (2008)
5-Alkyl-6-benzyl-2-(2-oxo-2-phenylethylsulfanyl)pyrimidin-4(3H)-ones, a
series of anti-HIV-1 agents of the dihydro-alkoxy-benzyl-oxopyrimidine
family with peculiar structure–activity relationship profile,
J. Med. Chem., 51, 4641-4652.
160.Yu, M., Liu, X., Li, Z., Liu, S., Pannecouque,
C., and Clercq, E. D. (2009) Synthesis and biological evaluation of
novel 2-(substituted
phenylaminocarbonylmethylthio)-6-(2,6-dichlorobenzyl)-pyrimidin-4(3H)-ones
as potent HIV-1 NNRTIss, Bioorg. Med. Chem., 17,
7749-7754.