Inhibition of Amyloid Aggregation of Bovine Serum Albumin by Sodium Dodecyl Sulfate at Submicellar Concentrations
Xue-Jiao Ma, Yin-Juan Zhang, and Cheng-Ming Zeng*
Shaanxi Normal University, School of Chemistry and Chemical Engineering, Key Laboratory of Analytical Chemistry for Life Science of Shaanxi Province, Xi’an 710119, China; E-mail: chengmingzeng@snnu.edu.cn* To whom correspondence should be addressed.
Received August 27, 2017; Revision received October 6, 2017
Sodium dodecyl sulfate (SDS), as an anionic surfactant, can induce protein conformational changes. Recent investigations demonstrated different effects of SDS on protein amyloid aggregation. In the present study, the effect of SDS on amyloid aggregation of bovine serum albumin (BSA) was evaluated. BSA transformed to β-sheet-rich amyloid aggregates upon incubation at pH 7.4 and 65°C, as demonstrated by thioflavin T fluorescence, circular dichroism, and transmission electron microscopy. SDS at submicellar concentrations inhibited BSA amyloid aggregation with IC50 of 47.5 µM. The inhibitory effects of structural analogs of SDS on amyloid aggregation of BSA were determined to explore the structure–activity relationship, with results suggesting that both anionic and alkyl moieties of SDS were critical, and that an alkyl moiety with chain length ≥10 carbon atoms was essential to amyloid inhibition. We attributed the inhibitory effect of SDS on BSA amyloid aggregation to interactions between the detergent molecule and the fatty acid binding sites on BSA. The bound SDS stabilized BSA, thereby inhibiting protein transformation to amyloid aggregates. This study reports for the first time that the inhibitory effect of SDS on albumin fibrillation is closely related to its alkyl structure. Moreover, the specific binding of SDS to albumin is the main driving force in amyloid inhibition. This study not only provides fresh insight into the role of SDS in amyloid aggregation of serum albumin, but also suggests rational design of novel anti-amyloidogenic reagents based on specific-binding ligands.
KEY WORDS: bovine serum albumin, amyloid aggregation, sodium dodecyl sulfate, fatty acid binding sitesDOI: 10.1134/S000629791801008X
Abbreviations: BSA, bovine serum albumin; CD, circular dichroism; HSA, human serum albumin; SDS, sodium dodecyl sulfate; TEM, transmission electron microscopy; ThT, thioflavin T.
Numerous investigations have demonstrated that amyloid fibrillation of
proteins plays an important role in amyloid-related diseases, including
Alzheimer’s disease, prion diseases, and type 2 diabetes [1-3]. These proteins, despite
their unrelated amino acid sequences and tertiary structures, can
unfold and assemble into fibrils with similar ultrastructures and
identical biochemical properties. Additionally, other proteins, usually
nonpathogenic, also exhibit the ability to form amyloid fibrils. The
ability to form amyloid fibrils is a generic property of the peptide
backbone [4, 5], although the
propensities of different sequences to assemble into amyloid fibrils
differ from one another. Amyloid fibrils are characterized by a common
cross-β-sheet structure, and they can present different isoforms
potentially leading to different biological effects. In addition to
pathogenic isoforms, some amyloids can also exhibit regular biological
functions [6, 7].
Amyloid fibrillation of a protein generally involves a cascade of events that include association/dissociation of the protein oligomers, nucleation, elongation, and maturation of the fibrillar assemblies, corresponding to different kinetic stages of fibril growth [8, 9]. However, amyloid fibrillation of proteins under some conditions does not follow classical nucleation kinetics, showing no seeding reaction and a characteristic absence of a lag phase. These proteins include human serum albumin (HSA) [10], bovine serum albumin (BSA) [11], acylphosphatase [12], β2-microglobulin [13], and transthyretin [14].
During early stage of amyloid formation, native protein monomers undergo conformational changes and unfolding, leading to intermolecular interactions coupled with formation of aggregates with β-sheet-rich structure [15]. Noncovalent interactions between β-sheets give rise to stacking of the peptide chains into fibrillar assemblies. Identification of small molecules capable of disrupting interactions between peptide chains is an essential strategy for screening amyloid inhibitors [16]. Enzymatic inhibitors, antibodies, peptide fragments, synthetic ligands, and some natural molecules have appeared in the list of screened candidates for treating amyloid disorders.
Serum albumins are major soluble-protein constituents of the circulatory system and exhibit a wide spectrum of physiological functions. The most important property of these proteins is their remarkable ability to bind a broad range of hydrophobic molecules, including fatty acids and a variety of drugs [17-19]. Serum albumin generally exhibits a stable structure normally resistant to formation of fibrils due to its high content of disulfide bridge and α-helical structure. However, under some circumstances, serum albumin also exhibits strong propensity to form aggregates, including amyloid fibrils [11, 20-23]. Because amyloid formation appears to reflect generic features of the protein backbones, studying mechanisms of protein fibrillation in the presence of small molecules is useful for gaining better understanding of interactions between amyloid species and ligands.
Surfactants can be used as denaturants to disrupt the native conformation of a protein. The driving forces in surfactant-induced conformational changes of proteins include electrostatic and hydrophobic interactions, as well as specific binding activity. These interactions can result in protein folding or unfolding depending on the concentrations of both surfactant and protein. Sodium dodecyl sulfate (SDS), as an anionic surfactant, consists of both polar and nonpolar moieties and induces different changes to protein conformation [24, 25] that might promote amyloid formation of the protein [26-28]. However, SDS binding to a protein might also hinder pathways related to protein aggregation, including amyloid assembly [28-30].
BSA is among the most extensively studied serum proteins, particularly due to its structural homology with HSA. Moreover, BSA shares similar biochemical properties with HSA, including ability to bind and transport fatty acids [31, 32]. Furthermore, both BSA and HSA have propensity to transform into amyloid fibrils [10, 11, 22, 23]. In this study, we utilized BSA as an in vitro model to investigate effects of SDS and its structural analogs on protein amyloid fibrillation. Our results indicated that SDS at submicellar concentrations efficiently inhibited amyloid aggregation of BSA, and that its inhibitory role was closely related to its alkyl structure.
MATERIALS AND METHODS
Chemicals. BSA (66.5 kDa), thioflavin T (ThT), SDS, and its structural analogs were purchased from Sigma-Aldrich (USA). Other reagents were of analytical grade.
Preparation and characterization of BSA fibrils. BSA was dissolved in 20 mM Tris-HCl (pH 7.4) in the presence or absence of SDS or its structural analogs to a final concentration of 50 µM. The concentrations of SDS and its structural analogs were between 0 and 150 µM. Aggregation was initiated by incubating the mixture at 65°C for 6 h in a water bath without agitation.
ThT assay. ThT fluorescence was measured in a mixture of 0.5 µM BSA and 10 µM ThT, with excitation at 440 nm and emission at 484 nm, in a Perkin Elmer LS55 spectrofluorimeter (PerkinElmer, USA). We confirmed that SDS and all its structural analogs have no effect on ThT fluorescence under the conditions of the present study.
Transmission electron microscopy (TEM). For TEM measurements, an aliquot of BSA aggregates was diluted 30-fold with water and dropped onto copper-mesh grids. Samples were negatively stained with 5% (w/v) phosphotungstic acid and air-dried at room temperature. Observations were performed using a JEOL JEM-2100 electron microscope (Japan) with an accelerating voltage of 80 kV.
Circular dichroism (CD) measurements. CD measurements were performed on a Chirascan CD spectrometer (Applied Photophysics, UK) at 25°C in a cuvette of 0.1-cm pathlength and at a scan range from 190 to 240 nm. The results were plotted as ellipticity (mdeg) versus wavelength (nm), and the secondary structural contents of BSA were analyzed by the CDNN program (Applied Photophysics).
RESULTS
Inhibitory effects of SDS on BSA fibrillation. Incubation of BSA at an elevated temperature results in formation of amyloid fibrils, with increased temperature [11, 20] or BSA concentration [20] significantly enhancing fibril growth. In this study, we incubated 50 µM BSA in 20 mM Tris-HCl (pH 7.4) at 65°C in the presence or absence of an inhibitor, and the growth of amyloid fibrils was monitored and characterized by ThT fluorescence, CD, and TEM.
ThT is a fluorescent probe used for monitoring formation and growth of amyloid fibrils. The fluorescent intensity of ThT increases significantly when the compound specifically binds to the highly ordered β-sheet structure of amyloid fibrils [33]. As depicted in Fig. 1a, an immediate increase in ThT fluorescence was observed following incubation initiation, with the intensity increasing gradually as amyloid formation progressed until reaching to a plateau after ~3 to 4 h. The lack of a lag phase in fibril growth suggested that nucleation was not involved in fibril assembly [8, 9]. When fresh BSA was incubated with its fibril seeds, ThT fluorescence increased similarly to that observed in the control sample in the absence of seeds (data not shown), indicating that amyloid seeds had no effect on the nucleation-independent fibrillation of BSA and consistent with previous reports [11, 20].
Fig. 1. Inhibitory effects of SDS on BSA amyloid aggregation. a) ThT-related growth curves of BSA fibrils in the absence (filled triangles) or presence of 20 µM (filled squares), 50 µM (open triangles), 100 µM (open squares), and 150 µM (open circles) SDS. b) Inhibition of BSA fibrillation by introduction of SDS at different incubation time points. SDS (150 µM) was introduced into samples at 0.5 h (open circles), 1 h (open triangles), 2 h (filled squares), and 3 h (open squares) after incubation initiation. TEM images of BSA aggregates at 6 h and prepared in the absence (c) or presence (d) of 150 µM SDS. Scale bar represents 200 nm.
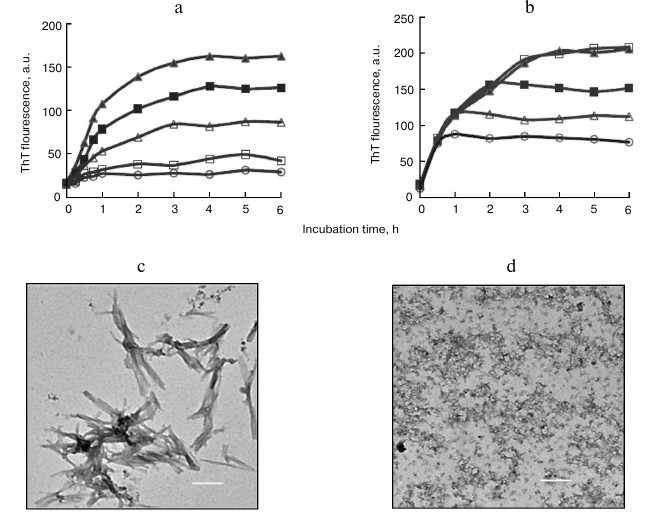
SDS exhibited inhibitory effects on the amyloid formation of BSA in a dose-dependent fashion, with an IC50 value of 47.5 µM (table). Coincubation of BSA with 50 or 150 µM SDS for 6 h resulted in 52.3 and 89.6% reduction in the final intensity of ThT fluorescence, respectively (Fig. 1a).
IC50* values of detergents exhibiting inhibition of BSA
fibrillation

* Half maximal inhibitory concentration.
To explore the role of SDS on amyloid formation after initiation of fibril assembly, SDS was introduced into the samples at later time points. After incubation for 0.5-2 h, the addition of SDS ceased elevations in ThT fluorescence. However, upon the ThT curve reaching a plateau at 3 h, the addition of SDS showed no effect on ThT fluorescence (Fig. 1b). These findings suggested that SDS was capable of interrupting fibril assembly without any effect on existing fibrils.
The inhibitory effects of SDS on BSA fibrillation were supported by TEM analysis of the aggregates. In the absence of SdS, the matured BSA fibrils at 6 h showed amyloid morphology characterized as dense and bundled fibrillar assemblies (Fig. 1c). In the presence of 150 µM SDS, only amorphous aggregates were observed after a 6-h incubation (Fig. 1d), indicating that amyloid fibrillation of BSA was inhibited and consistent with the ThT data.
CD is a widely used spectroscopic technique for study of protein secondary structure. The conversion of α-helix to β-sheet is a typical feature of amyloid formation [34]. The CD curve of BSA obtained before incubation exhibited two minima at 208 and 222 nm, typical of high α-helical content (Fig. 2a). After incubation initiated, the minima at 222 and 208 nm progressively reduced, with a shape change corresponding to a decrease in α-helical structure and an increase in β-sheet structure (Fig. 2a). Secondary structure analysis based on the CD spectra suggests that free BSA has a high α-helix content (63%), with 6% β-sheet, 12% turn, and 19% random coil, which is consistent with a literature report [35]. After incubation at 65°C for 6 h, major reduction of the α-helix from 63 to 28% was recorded, concomitant with a major increase in β-sheet from 6 to 31%.
Fig. 2. Far-UV CD spectra of BSA incubated in the absence or presence of 150 µM SDS. a) CD spectra of native BSA (1) and of native BSA and SDS without incubation (2), and BSA incubated for 1, 3, and 6 h (3-5), respectively. b) CD spectra of native BSA (1) and BSA incubated with 150 µM SDS for 1 or 6 h (2, 3), respectively.
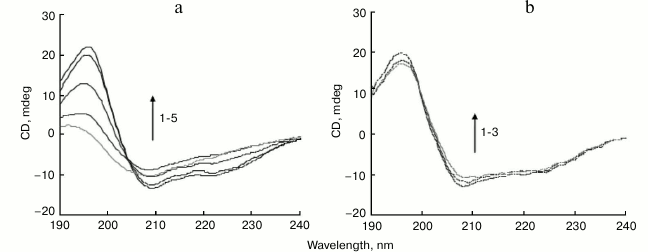
The BSA sample coincubated with 150 µM SDS at 65°C for 1-6 h retained CD spectral features associated with α-helices, showing little change in the peaks at 208 and 222 nm (Fig. 2b). After incubation for 6 h, the secondary structure contents of BSA (α-helix content 62%, with 7% β-sheet) were like those observed in the native protein, suggesting that SDS prevented BSA structural transition from α-helices to β-sheets. SDS hindered the formation of amyloid-prone cross-β-sheet structures in BSA, thereby inhibiting fibril growth.
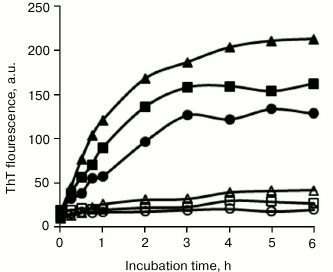
Inhibitory effects of SDS structural analogs on BSA amyloid fibrillation. SDS consists of a polar sulfonic group and a nonpolar dodecyl moiety. To reveal the molecular mechanism associated with amyloid inhibition, the roles of several structural analogs of SDS on BSA fibrillation were determined. As shown in Fig. 3, 150 µM sodium salts of decanesulfonate (C10), tetradecanesulfonate (C14), and hexadecanesulfonate (C16) completely inhibited BSA fibrillation, sharing similar inhibitory characteristics as those observed with SDS. By comparison, 150 µM sodium salts of hexanesulfonate (C6) and octanesulfonate (C8) showed only weak inhibitory activity, implying that the length of the alkyl chain was critical for efficient inhibition of BSA fibrillation by sodium alkyl sulfonate, and that an alkyl moiety with chain length ≥10 carbon atoms was essential. These alkyl sulfonates inhibited BSA fibrillation in a dose-dependent manner, with IC50 values decreasing from decanesulfonate to hexadecanesulfonate (table).
Fig. 3. Inhibitory effects of sodium alkyl sulfonates on BSA amyloid aggregation. BSA was incubated in the absence (filled triangles) or presence of 150 µM hexanesulfonate (filled squares), octanesulfonate (filled circles), decanesulfonate (open triangles), tetradecanesulfonate (open squares), and hexadecanesulfonate (open circles).
We also examined the inhibitory effect of sodium laurate (C12) on BSA fibrillation. As shown in Fig. 4a, sodium laurate shared similar inhibitory roles as those observed with SDS on BSA fibrillation, with an IC50 value of 45.1 µM. After incubation for 6 h at 65°C, only amorphous aggregates were observed in the sample containing 50 µM BSA and 150 µM sodium laurate (Fig. 4b). Like the results observed in the presence of alkyl sulfonates, 150 µM sodium salts of hexanoate (C6) and octanoate (C8) exhibited weak inhibitory effects on BSA fibrillation, whereas sodium salts of decanoate (C10), myristate (C14), and palmitate (C16) demonstrated similar levels of inhibition (Fig. 4c) as sodium laurate and SDS. These fatty acids also inhibited BSA fibrillation in a dose-dependent manner, with IC50 values decreasing from decanoate to palmitate (table).
Fig. 4. Inhibitory effects of sodium carboxylates on BSA amyloid aggregation. a) Dose-dependent inhibition of BSA fibrillation by sodium laurate. The concentrations of laurate were 0 µM (filled triangles), 20 µM (filled squares), 50 µM (open triangles), 100 µM (open squares), and 150 µM (open circles). b) TEM images of BSA aggregates at 6 h and prepared in the presence of 150 µM sodium laurate. Scale bar represents 200 nm. c) Inhibitory effects of sodium carboxylates on BSA amyloid formation. BSA was incubated in the absence (filled triangles) or presence of 150 µM sodium salts of hexanoate (filled squares), octanoate (filled circles), decanoate (open squares), myristate (open triangles), and palmitate (open circles). d) The CD spectra of native BSA (1) and BSA incubated for 6 h with 150 µM sodium laurate (2) or without a detergent (3).
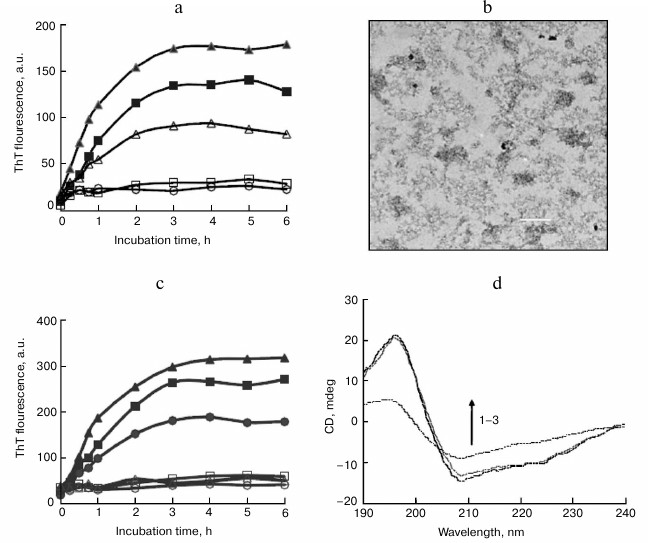
CD measurements were also performed to monitor changes in secondary structure of BSA upon incubation with structural analogs of SDS. As shown in Fig. 4d, after incubation with 150 µM sodium laurate at 65°C for 6 h, the CD spectra of BSA remained almost unchanged, suggesting that sodium laurate prevented the structural transition from α-helix to β-sheet. Other structural analogs, including alkyl sulfonates and carboxylates with chain length ≥10 carbon atoms (150 µM), shared similar effects with SDS and sodium laurate on the CD spectra of BSA (data not shown). These results are consistent with the ThT data.
To determine whether an acidic group is critical to amyloid inhibition, the effects of 150 µM 1-dodecanol (C12) and dodecyl trimethyl ammonium chloride (C12) on BSA fibrillation were determined. The results showed that neither compound influenced BSA fibrillation (data not shown), suggesting that both an alkyl chain and an acidic group are critical for SDS inhibition of BSA fibrillation. Our results indicated that the inhibitory role of SDS on BSA fibrillation could be attributed to the ability of BSA to bind fatty acids.
DISCUSSION
BSA is a 583-a.a. protein consisting of three homologous all-α domains and 17 intradomain disulfide bonds. Like apomyoglobin and HSA, which consist of all-α domains, BSA is also capable of transforming into a stable β-sheet fibrillar state under a variety of conditions. In this study, BSA amyloid aggregates were prepared at a neutral pH and 65°C to evaluate the impact of SDS on protein fibrillation. Like previous reports [11, 20], ThT, TEM, and CD data of the present work showed that BSA had an ability to assembly into β-sheet-rich fibrillar structures. In contrast to classical protein amyloid fibrillation, BSA fibrillation showed unique features, including lack of a lag phase and no seeding reaction, with these characteristics also observed in the fibrillation process of other proteins [10, 12-14, 23]. These unique features have been attributed to the existence of different intermediates along the fibrillation pathway associated with serum albumins [20]. Several mechanisms have been proposed to elucidate amyloid formation, including nucleated conformational transition [36] and template-based assembly [37, 38]. The facts that lack of lag phase and no seeding reaction suggest that BSA transform into amyloid aggregates through a different pathway.
Recent reports demonstrate that ring-like oligomers are the basic building units of amyloid fibrils [39, 40]. During early stage of fibril formation, such oligomers associate with each other by a ring-to-ring or ring-on-ring pattern [40]. This mechanism has been shown for several objects, and therefore might be a universal process for amyloid growth [41]. According to this mechanism, we suggest that the assembly of BSA into amyloid species probably involves interactions between monomers and/or dimers leading to formation of ring-like oligomers. The resultant oligomers associate with each other to assemble into larger amyloid structures. The exact molecular mechanism merits further investigation.
SDS can induce protein conformational changes due to its electrostatic and hydrophobic interactions with a protein [31]. There are numerous investigations on the impact of SDS on protein amyloid aggregation, with two contrary effects, inhibition or acceleration of amyloid formation by the detergent reported [26-30, 42]. The role of SDS on protein fibrillation is determined mainly by the concentration of the surfactant, pH value of the medium, and ionic/hydrophobic property of the protein. Rangachari et al. [27] observed that 2 mM SDS promoted Aβ fibril formation, whereas at lower concentration (0.5 mM) SDS showed almost no effect on Aβ fibrillation. Pertinhez et al. [28] reported that SDS at low concentrations accelerated amyloid fibrillation of a 17-residue peptide, whereas at high concentrations, SDS inhibited fibrillation of the peptide. Movaghati et al. [29] demonstrated that SDS at a low concentration (150 µM) promoted HSA fibrillation at pH 2.0, whereas at high concentrations (>400 µM), SDS inhibited fibrillation of the protein. SDS acceleration of BSA fibrillation at pH levels below its isoelectric point (pI 4.7) was also demonstrated by Khan et al. [43, 44]. Here, SDS at submicellar concentrations prevented BSA from amyloid fibrillation at pH 7.4, like a previous report that SDS at low concentrations inhibited protein aggregation [45]. In the present study, the inhibitory roles of SDS structural analogs were determined to explore structure-activity relationships, with results suggesting that alkyl sulfonates shared similar inhibitory effects with carboxylates on BSA fibrillation, and that both the anionic and alkyl moieties of SDS were critical in amyloid inhibition. A previous modeling analysis suggested that SDS can bind the same sites on serum albumins as long-chain fatty acids [46]. Therefore, we attributed SDS inhibition of BSA fibrillation to interactions between the detergent molecule and fatty acid-binding sites on BSA.
An important physiological role of serum albumin involves the transport of fatty acids in the circulatory system. The fatty acid-binding properties of BSA are like those in HSA, and submicellar concentrations of SDS can bind the same sites [31, 47-49]. Like HSA, there are seven main binding pockets for medium-chain and long-chain fatty acids in the three structurally similar domains of BSA [47, 49]. Three of these binding pockets exhibit high affinity for fatty acids, whereas the other four exhibit lower affinity. The high-affinity sites consist of long and narrow hydrophobic pockets that accept the alkyl chain of a fatty acid in its extended conformation while the carboxylate head-group is hydrogen-bonded and coordinated by a charged protein side chain [47, 48]. In principle, up to seven fatty acids can be bound to each albumin molecule; however, under normal physiological conditions, only an average of 0.1 to 2 fatty acid molecules is present in the high-affinity sites [32]. Albumin molecules undergo conformational changes upon binding with a fatty acid, leading to relative rotation of the three protein domains [47, 50]. The bound fatty acid acts as a pin to lock in the altered structural conformation, thereby stabilizing the albumin molecule [50-52]. Additionally, previous reports showed that in the presence of a denaturant or under other denaturing conditions, BSA bound with a fatty acid exhibited higher stability relative to the native protein [53-55]. Our results suggested that SDS and its structural analogs were bound within the binding pockets of BSA normally targeted by fatty acids, and that the bound SDS induced structural changes to stabilize the protein, thereby inhibiting BSA transformation to amyloid aggregates.
The binding affinity of albumins for fatty acids is dependent on chain length, as affinity increases from hexanoate (103 M) to palmitate (107 M) [32, 56]. In this study, fatty acids, whether carboxyl or sulfonic, with shorter alkyl chains (C6 and C8) showed only weak inhibitory effects on BSA fibrillation, possibly because the protein failed to be stabilized by the weak binding of these short-chain fatty acids. By contrast, fatty acids with longer alkyl chains (from C10 to C16) were capable of inhibiting BSA fibrillation, with their inhibitory activity proportional to their binding affinity for the protein.
In conclusion, this study demonstrated that SDS at submicellar concentrations is capable of inhibiting BSA amyloid fibrillation. Compared with the inhibition exerted by its structural analogs, SDS acts through interactions between the detergent molecule and the fatty acid-binding sites on BSA. Bound SDS stabilized BSA, thereby inhibiting transformation of the protein to amyloid aggregates. This study reports for the first time that the alkyl chain length of SDS are critical to amyloid inhibition. Moreover, the specific binding of SDS to albumin is the main driving force in amyloid inhibition. The information of the present study not only provide a fresh insight into the roles of SDS on amyloid aggregation of serum albumin, but also suggest rational design of novel anti-amyloidogenic reagents based on specific-binding ligands.
Acknowledgments
We are grateful to Gai-Tao Li and Feng-Qun He for their preliminary measurements.
REFERENCES
1.Dobson, C. M. (2003) Protein folding and
misfolding, Nature, 426, 884-890.
2.Nizhnikov, A. A., Antonets K. S., and
Inge-Vechtomov, S. G. (2015) Amyloids: from pathogenesis to function,
Biochemistry (Moscow), 80, 1127-1144.
3.Stefani, M. (2004) Protein misfolding and
aggregation: new examples in medicine and biology of the dark side of
the protein world, Biochim. Biophys. Acta, 1739,
5-25.
4.Dobson, C. M. (1999) Protein misfolding, evolution
and disease, Trends Biochem. Sci., 24, 329-332.
5.Gazit, E. (2002) The correctly folded state of
proteins: is it a metastable state? Angew. Chem. Int. Ed.,
41, 257-259.
6.Shewmaker, F., McGlinchey, R. P., and Wickner, R.
B. (2011) Structural insights into functional and pathological amyloid,
J. Biol. Chem., 286, 16533-16540.
7.Martino, P. D. (2016) Bap: a new type of functional
amyloid, Trends Microbiol., 24, 682-684.
8.Huang, B., He, J., Ren, J., Yan, X. Y., and Zeng,
C. M. (2009) Cellular membrane disruption by amyloid fibrils involved
intermolecular disulfide cross-linking, Biochemistry, 48,
5794-5800.
9.Fandrich, M. (2007) Absolute correlation between
lag time and growth rate in the spontaneous formation of several
amyloid-like aggregates and fibrils, J. Mol. Biol., 365,
1266-1270.
10.Juarez, J., Taboada, P., and Mosquera, V. (2009)
Existence of different structural intermediates on the fibrillation
pathway of human serum albumin, Biophys. J., 96,
2353-2370.
11.Holm, N. K., Jespersen, S. K., Thomassen, L. V.,
Wolff, T. Y., Sehgal, P., Thomsen, L. A., Christiansen, G., Andersen,
C. B., Knudsen, A. D., and Otzen, D. E. (2007) Aggregation and
fibrillation of bovine serum albumin, Biochim. Biophys. Acta,
1774, 1128-1138.
12.Chiti, F., Webster, P., Taddei, N., Clark, A.,
Stefani, M., Ramponi, G., and Dobson, C. M. (1999) Designing conditions
for in vitro formation of amyloid protofilaments and fibrils,
Proc. Natl. Acad. Sci. USA, 96, 3590-3594.
13.Sasahara, K., Yagi, H., Sakai, M., Naiki, H., and
Goto, Y. (2008) Amyloid nucleation triggered by agitation of
β2-microglobulin under acidic and neutral pH conditions,
Biochemistry, 47, 2650-2660.
14.Hurshman, A. R., White, J. T., Powers, E. T., and
Kelly, J. W. (2004) Transthyretin aggregation under partially
denaturing conditions is a downhill polymerization,
Biochemistry, 43, 7365-7381.
15.Ahmad, A., Uversky, V. N., Hong, D., and Fink, A.
L. (2005) Early events in the fibrillation of monomeric insulin, J.
Biol. Chem., 280, 42669-42675.
16.Doig, A. J., and Derreumaux, P. (2015) Inhibition
of protein aggregation and amyloid formation by small molecules,
Curr. Opin. Struct. Biol., 30, 50-56.
17.Peters, T., Jr. (1985) Serum albumin, Adv.
Protein Chem., 37, 161-245.
18.Olson, R. E., and Christ, D. D. (1996) Plasma
protein binding of drugs, Annu. Rep. Med. Chem., 31,
327-336.
19.Simard, J. R., Zunszain, P. A., Ha, C. E., Yang,
J. S., Bhagavan, N. V., Petitpas, I., Curry, S., and Hamilton, J. A.
(2005) Locating high-affinity fatty acid-binding sites on albumin by
X-ray crystallography and NMR spectroscopy, Proc. Natl. Acad. Sci.
USA, 102, 17958-17963.
20.Vetri, V., D’Amico M., Fodera, V., Leone,
M., Ponzoni, A., Sberveglieri, G., and Militello, V. (2011) Bovine
serum albumin protofibril-like aggregates formation: solo but
not simple mechanism, Arch. Biochem. Biophys., 508,
13-24.
21.Veerman, C., Sagis, L. M. C., Heck, J., and Van
der Linden, E. (2003) Mesostructure of fibrillar bovine serum albumin
gels, Int. J. Biol. Macromol., 31, 139-146.
22.Bhattacharya, M., Jain, N., and Mukhopadhyay, S.
(2011) Insights into the mechanism of aggregation and fibril formation
from bovine serum albumin, J. Phys. Chem. B, 115,
4195-4205.
23.Taboada, P., Barbosa, S., Castro, E., and
Mosquera, V. (2006) Amyloid fibril formation and other aggregate
species formed by human serum albumin association, J. Phys. Chem.
B, 110, 20733-20736.
24.Rizo, J., Blanco, F. J., Kobe, B., Bruch, M. D.,
and Gierasch, L. M. (1993) Conformational behavior of Escherichia
coli OmpA signal peptides in membrane mimetic environments,
Biochemistry, 32, 4881-4894.
25.Waterhous, D. V. (1994) Importance of environment
in determining secondary structure in proteins, Biochemistry,
33, 2121-2128.
26.Giehm, L., Oliveira, C. L. P., Christiansen, G.,
Pedersen, J. S., and Otzen, D. E. (2010) SDS-induced fibrillation of
α-synuclein: an alternative fibrillation pathway, J. Mol.
Biol., 401, 115-133.
27.Rangachari, V., Reed, D. K., Moore, B. D., and
Rosenberry, T. L. (2006) Secondary structure and interfacial
aggregation of amyloid-β(1-40) on sodium dodecyl sulfate micelles,
Biochemistry, 45, 8639-8648.
28.Pertinhez, T. A., Bouchard, M., Smith, R. A. G.,
Dobson, C. M., and Smith, L. J. (2002) Stimulation and inhibition of
fibril formation by a peptide in the presence of different
concentrations of SDS, FEBS Lett., 529, 193-197.
29.Movaghati, S., Moosavi-Movahedi, A. A.,
Khodagholi, F., Digaleh, H., Kachooei, E., and Sheibani, N. (2014)
Sodium dodecyl sulphate modulates the fibrillation of human serum
albumin in a dose-dependent manner and impacts the PC12 cells
retraction, Colloids Surf. B Biointerfaces, 122,
341-349.
30.Ahmad, M. F., Ramakrishna, T., Raman, B., and
Rao, C. M. (2006) Fibrillogenic and non-fibrillogenic ensembles of
SDS-bound human alpha-synuclein, J. Mol. Biol., 364,
1061-1072.
31.Gelamo, E. L., Silva, C. H. T. P., Imasato, H.,
and Tabak, M. (2002) Interaction of bovine (BSA) and human (HSA) serum
albumins with ionic surfactants: spectroscopy and modeling, Biochim.
Biophys. Acta, 1594, 84-99.
32.Van der Vusse, G. J. (2009) Albumin as fatty acid
transporter, Drug Metab. Pharmacokinet., 24, 300-307.
33.An, T. T., Feng, S., and Zeng, C. M. (2017)
Oxidized epigallocatechin gallate inhibited lysozyme fibrillation more
strongly than the native form, Redox Biol., 11,
315-321.
34.Fandrich, M., Fletcher M. A., and Dobson, C. M.
(2001) Amyloid fibrils from muscle myoglobin, Nature,
410, 165-166.
35.Charbonneau, D. M., and Tajmir-Riahi, H. A.
(2010) Study on the interaction of cationic lipids with bovine serum
albumin, J. Phys. Chem. B, 114, 1148-1155.
36.Serio, T. R., Cashikar, A. G., Kowal, A. S.,
Sawicki, G. J., Moslehi, J. J., Serpell, L., Arnsdorf, M. F., and
Lindquist, S. L. (2000) Nucleated conformational conversion and the
replication of conformational information by a prion determinant,
Science, 289, 1317-1321.
37.Esler, W. P., Stimson, E. R., Jennings, J. M.,
Vinters, H. V., Ghilardi, J. R., Lee, J. P., Mantyh, P. W., and Maggio,
J. E. (2000) Alzheimer’s disease amyloid propagation by a
template-dependent dock-lock mechanism, Biochemistry, 39,
6288-6295.
38.Nguyen, P. H., Li, M. S., Stock, G., Straub, J.
E., and Thirumalai, D. (2007) Monomer adds to preformed structured
oligomers of Aβ-peptides by a two-stage dock-lock mechanism,
Proc. Natl. Acad. Sci. USA, 104, 111-116.
39.Grigorashvili, E. I., Selivanova, O. M.,
Dovidchenko, N. V., Dzhus, U. F., Mikhailina, A. O., Suvorina, M. Y.,
Marchenkov, V. V., Surin, A. K., and Galzitskaya, O. V. (2016)
Determination of size of folding nuclei of fibrils formed from
recombinant Aβ(1-40) peptide, Biochemistry (Moscow),
81, 538-547.
40.Selivanova, O. M., Glyakina, A. V., Gorbunova, E.
Y., Mustaeva, L. G., Suvorina, M. Y., Grigorashvili, E. I., Nikulin, A.
D., Dovidchenko, N. V., Rekstina, V. V., Kalebina, T. S., Surin, A. K.,
and Galzitskaya, O. V. (2016) Structural model of amyloid fibrils for
amyloidogenic peptide from Bgl2p glucantransferase of S.
cerevisiae cell wall and its modifying analog. New morphology of
amyloid fibrils, Biochim. Biophys. Acta, 1864,
1489-1499.
41.Selivanova, O. M., Suvorina, M. Y., Surin, A. K.,
Dovidchenko, N. V., and Galzitskaya, O. V. (2017) Insulin and Lispro
insulin: what is common and different in their behavior? Curr.
Protein Pept. Sci., 18, 57-64.
42.Hoover, C. E., Davenport, K. A., Henderson, D.
M., Zabel, M. D., and Hoover, E. A. (2017) Endogenous brain lipids
inhibit prion amyloid formation in vitro, J. Virol.,
91, e02162-02166.
43.Khan, J. M., Qadeer, A., Chaturvedi, S. K.,
Ahmad, E., Rehman, S. A. A., Gourinath, S., and Khan, R. H. (2012) SDS
can be utilized as an amyloid inducer: a case study on diverse
proteins, PLoS One, 7, e29694.
44.Khan, J. M., Abdulrehman, S. A., Zaidi, F. K.,
Gourinath, S., and Khan, R. H. (2014) Hydrophobicity alone cannot
trigger aggregation in protonated mammalian serum albumins, Phys.
Chem. Chem. Phys., 16, 5150-5161.
45.Rafikova, E. R., Panyukov, Y. V., Arutyunyan, A.
M., Yaguzhinsky, L. S., Drachev, V. A., and Dobrov, E. N. (2004) Low
sodium dodecyl sulfate concentrations inhibit tobacco mosaic virus coat
protein amorphous aggregation and change the protein stability,
Biochemistry (Moscow), 69, 1372-1378.
46.Santos, S. F., Zanette, D., Fischer, H., and
Itri, R. (2003) A systematic study of bovine serum albumin (BSA) and
sodium dodecyl sulfate (SDS) interactions by surface tension and small
angle X-ray scattering, J. Colloid Interf. Sci., 262,
400-408.
47.Bujacz, A. (2012) Structures of bovine, equine
and leporine serum albumin, Acta Crystallogr. D, 68,
1278-1289.
48.Hamilton, J. A., Era, S., Bhamidipati, S. P., and
Reed, R. G. (1991) Locations of the three primary binding sites for
long-chain fatty acids on bovine serum albumin, Proc. Natl. Acad.
Sci. USA, 88, 2051-2054.
49.De Sousa Neto, D., Salmon, C. E., Alonso, A., and
Tabak, M. (2009) Interaction of bovine serum albumin (BSA) with ionic
surfactants evaluated by electron paramagnetic resonance (EPR)
spectroscopy, Colloids Surf. B Biointerfaces, 70,
147-156.
50.Bhattacharya, A. A., Grune T., and Curry, S.
(2000) Crystallographic analysis reveals common modes of binding of
medium and long-chain fatty acids to human serum albumin, J. Mol.
Biol., 303, 721-732.
51.Curry, S., Mandelkow, H., Brick, P., and Franks,
N. (1998) Crystal structure of human serum albumin complexed with fatty
acid reveals an asymmetric distribution of binding sites, Nat.
Struct. Biol., 5, 827-835.
52.Curry, S., Brick, P., and Franks, N. P. (1999)
Fatty acid binding to human serum albumin: new insights from
crystallographic studies, Biochim. Biophys. Acta, 1441,
131-140.
53.Ahmad, N., and Qasim, M. A. (1995) Fatty acid
binding to bovine serum albumin prevents formation of intermediate
during denaturation, Eur. J. Biochem., 227, 563-565.
54.Takeda, K., and Moriyama, Y. (2015) Kinetic
aspects of surfactant-induced structural changes of proteins –
unsolved problems of two-state model for protein denaturation, J.
Oleo Sci., 64, 1143-1158.
55.Matei, I., Ariciu, A. M., Neacsu, M. V.,
Collauto, A., Salifoglou, A., and Ionita, G. (2014) Cationic spin probe
reporting on thermal denaturation and complexation–decomplexation
of BSA with SDS. Potential applications in protein purification
processes, J. Phys. Chem. B, 118, 11238-11252.
56.Ashbrook, J. D., Spector, A. A., and Fletche, J.
E. (1972) Medium chain fatty acid binding to human plasma albumin,
J. Biol. Chem., 247, 7038-7042.