Expression of Soluble Active Interferon αA in Escherichia coli Periplasm by Fusion with Thermostable Lichenase Using the Domain Insertion Approach
A. A. Tyurin1,2, K. V. Kabardaeva1,2, O. N. Mustafaev3, O. S. Pavlenko1,2, N. S. Sadovskaya1, V. S. Fadeev1, E. A. Zvonova2, and I. V. Goldenkova-Pavlova1*
1Institute of Plant Physiology, Russian Academy of Sciences, 127276 Moscow, Russia; E-mail: irengold58@gmail.com2Russian State Agrarian University – Moscow Timiryazev Agricultural Academy, Department of Genetics and Biotechnology, 127550 Moscow, Russia; E-mail: alexjofar@gmail.com
3Baku State University, Department of Biophysics and Molecular Biology, Baku, AZ 1148, Azerbaijan; E-mail: orkhan@bioset.org
* To whom correspondence should be addressed.
Received August 30, 2017; Revision received November 2, 2017
A recombinant DNA in which the interferon αA (IFN-αA) gene sequence is integrated into a loop region of the gene coding thermostable lichenase was constructed. This approach of insertion fusion with thermostable lichenase is advantageous in terms of increasing the solubility, stability, and production of the fusion partner in soluble form in general and in the periplasm of bacterial cells in particular. Thus, the insertion of IFN-αA into the loop (53 a.a.) of thermostable lichenase from Clostridium thermocellum resulted in effective expression of the soluble form of the recombinant protein in the periplasm of Escherichia coli without any compromise in biological activity of IFN-αA, while the thermostable lichenase retained its ability for functional folding without dramatic loss of its basic activity and thermostability.
KEY WORDS: interferon αA, thermostable lichenase, domain insertion, expression, solubility, biological activityDOI: 10.1134/S0006297918030069
Abbreviations: CPE, cytopathogenic effect; FBS, fetal bovine serum; IFN, interferon; LicB, thermostable lichenase; MBP, maltose-binding protein; P-EP, protein preparation after ethanol precipitation; PP, purified protein; TPL, total protein lysate.
Interferons are a group of cytokines secreted by mammalian cells; they
have antiviral, antiproliferative, and immunomodulating activities.
Based on their structure, functional activity, and receptor
specificity, interferons are divided into three main groups: type I
(IFN-α and IFN-β), type II (IFN-γ), and type III
(IFN-λ) [1]. Recombinant interferon αA
(IFN-αA) is a vivid example of a hydrophobic protein that is used
as the first-line treatment for hepatitis C, and it is also important
in regimes for treatment of hepatitis B and leukemia, but it is
difficult to isolate and purify [2-4]. Considering the great therapeutic potential of
IFN-αA, this protein is of particular interest to researchers.
Although motivation for research on IFN-αA varies, most efforts
of researchers are directed to developing approaches to the production
of this therapeutic protein in soluble and biologically functional form
using Escherichia coli as its effective producer [5]. The reason for this attention to the solution of
the problem of the production of the soluble form of IFN-αA is
because this protein is usually produced in E. coli as inclusion
bodies, which are then refolded, followed by purification in a series
of chromatography steps [6, 7].
Expression of the protein through inclusion bodies is a difficult task,
since the protein with partially incorrect folding increases the
tendency to aggregate. This is considered a risk factor for its
increased immunogenicity. Thus, the production of IFN-αA, as well
as other therapeutic proteins, in soluble and biologically functional
form is desirable both for safety reasons and for significant
simplification of subsequent purification processes [6, 7].
Numerous research efforts have been directed to the development of approaches for the production in E. coli of soluble proteins similar to IFN-αA [8-10]. According to current opinion, some highly soluble proteins can increase the solubility of their fusion partners, thereby preventing the formation of insoluble aggregates, increasing the efficiency of expression of the soluble form of the protein and simplifying its purification. Several fusion strategies have been developed to improve the level of expression and secretion of recombinant proteins in host cells [11]. The direct tandem fusion strategy is the simplest and most common, i.e., the creation of proteins in “head-to-tail” fusion, in which one protein follows the C-terminus of another protein [11]. Widely used direct fusion partners include maltose-binding protein (MBP) [12], glutathione-S-transferase (GST) [13, 14], transcription termination protein (NusA) [15], ubiquitin-like proteins (SUMO) [5, 16, 17], and several others. Experimental studies have shown that direct fusion of the target protein with such proteins can in general provide more stable structure and significantly improve the solubility of the target protein [11].
However, the direct tandem fusion strategy may fail because such fusion proteins, although contributing to the solubility of their in vitro fusion partners, are often more sensitive to proteolytic degradation and are structurally unstable [18-20]. The strategy of creating fusion proteins by insertion into domains may be more useful in such cases [18-20]. Domain insertion is carried out through the integration of a protein domain (“guest” domain) into another (receiving domain), namely in the area of integration sites. The insertion site is carefully selected [18-20]. It should be emphasized that domain insertion is more complex for design than direct fusion, since in most cases it is necessary to search for the optimal site for “insertion” in a protein that accepts an insert that may violate domain stacking if such site is chosen incorrectly [19-21]. Clear successes in using the domain insertion strategy have been demonstrated on only a small list of proteins, because for the successful creation of such fusion proteins it is crucial to predict potential areas of integration in the host protein [21]. The rule is to select a surface loop or turn that does not directly participate in the catalytic regions as the insertion areas, thereby eliminating possible steric collisions between the receiving and integrated domains [20]. It has been convincingly shown that insertion fusion of proteins can be beneficial with regard to the stabilizing effects of recombinant proteins [21]. Thus, both the direct fusion strategy and the domain insertion strategy are useful for improving the expression level of soluble forms of recombinant proteins in many systems [18-23].
We chose a thermostable lichenase – β-1,3-1,4-glucanase (endo-β-1,3;1,4-glucan-D-glycosyl hydrolase, EC 3.2.1.73, P29716) from Clostridium thermocellum – as a new receiving protein. The structure of the protein molecule, as well as the high thermal stability and specific activity of the lichenase, are attractive for the construction of fusion proteins. We have previously shown that the areas of lichenase that allow the introduction of a circular permutation with preservation of the activity and thermostability of the protein also withstand internal integration of small peptides (up to 6 a.a.) [24]. Moreover, the production of this protein in E. coli was shown in soluble form, including in the periplasm [25], suggesting that the lichenase can function as a solubility enhancer.
In this study, the IFN-αA domain was inserted into the thermophilic protein. The insertion of IFN-αA into the loop (53 a.a.) of thermostable lichenase from C. thermocellum resulted in effective expression of the soluble form of the recombinant protein in the periplasm of E. coli without any compromise of the biological activity of IFN-αA, while the thermostable lichenase demonstrated preservation of its activity and thermostability. Thus, these results indicate that the approach of insertion fusion with thermostable lichenase can be beneficial in terms of increasing solubility, stability, and production of fusion partners in soluble form in general and in the periplasm of bacterial cells in particular.
MATERIALS AND METHODS
Gene engineering methods. Standard molecular cloning procedures [26] were used in this work, together with Evrogen (Russia) primers and Promega (USA), Fermentas (Lithuania), QIAexpress (USA), and Novagen (USA) enzymes and reagent kits. The plasmid pQE-NC-L-53 [24] was used as the source of the licB gene. The gene sequence encoding interferon αA (IFN-αA; X01628.1) was produced by chemical enzymatic synthesis by Evrogen and was cloned in two variants. In the first variant, the nucleotide sequence contains the start and stop codons and is flanked by the unique restriction sites BamHI (in the 5′-region) and ApaI-XhoI (in the 3′-region). The second variant is flanked by the same restriction sites, but it does not contain the start and stop codons. Both sequence variants were cloned into the pET system vector, and the resulting vectors are designated as pET-IFN-αA and pET-IFN-P2, respectively.
Construction of expression plasmids carrying native and hybrid genes encoding thermostable lichenase and interferon. The plasmid pET-LicB-53 was constructed in two stages. Initially, the sequence of licB-53 was obtained by PCR using the pQE-NC-L-53 [24] plasmid as a template and forward AGATCTATGGTAAATACGCCT and reverse CTCGAGACCGTTAGGAGTATT primers with introduced BglII and XhoI restriction sites. The amplicon was cloned as a BglII-XhoI fragment into the pET-32 plasmid hydrolyzed with the BamHI and XhoI restriction endonucleases. The plasmid pET-32 carries the His-Tag coding sequence and the sequence encoding the leader peptide for periplasmic expression. The plasmid pET-LicB-53-IFN-αA was constructed by cloning the BamHI-ApaI fragment of plasmid pET-IFN-P2 into plasmid pET-LicB-53 hydrolyzed with the corresponding restriction endonucleases. The reading frame of the hybrid genes was verified by sequencing (Evrogen).
Recombinant protein expression, purification, and fractionation procedures. Escherichia coli Rosetta™(DE3) (Novagen) strains harboring pET-LicB-53, pET-IFN-αA, and pET-LicB-53-IFN-αA were grown to OD600 = 0.6 at 37°C in LB medium and then induced with 0.5 mM isopropyl β-D-1-thiogalactoside at 37°C for 16 h.
The cells were lysed using a French pressure cell press (Aminco, USA) and cleared by centrifugation (total protein lysate, TPL). The cell lysate was loaded on a Ni2+-NTA-agarose column (Qiagen, Germany). The eluted enzyme was gradually dialyzed against 50 mM Tris-HCl (pH 8.0) (purified protein, PP). A part of the TPL was treated with three volumes of 96% ethanol at 4°C for 8-10 h, followed by centrifugation. The precipitate was dried and then dissolved in 50 mM Tris-HCl (pH 8.0). The insoluble fraction was separated by centrifugation, and the supernatant was used as protein preparations after ethanol precipitation (P-EP).
Protein samples were isolated from periplasm fractions, cytoplasm, and inclusion bodies in several stages. The cell pellet was resuspended in sucrose extraction buffer (50 mM Tris-HCl, pH 8.0, 25% sucrose, 100 µg/ml lysozyme, 25 µg/ml RNase), incubated 20 min on ice, and then centrifuged. The supernatant was collected and used as the periplasmic fraction. The pellet was resuspended in lysis buffer (20 mM Tris-HCl, pH 7.4, 2 mM EDTA, 5 mM CaCl2, 5 mM MgCl2), supplemented with 0.1% deoxycholate and deoxyribonuclease (Sigma, USA), incubated for 15 min at 37°C, and centrifuged. The supernatant was used as the soluble cytoplasmic fraction, and the precipitate was used as the fraction of inclusion bodies. The purified proteins (PP and P-EP samples) were concentrated by ultrafiltration to the concentration of 1 mg/ml. Endotoxin content was measured with the Endosafe-PTS system using cartridges with sensitivity of 0.03 EU/ml (Charles River Laboratories, USA).
Analytical characterization of proteins. Plate assay was performed as described previously [24]. Briefly, Petri dishes with agar medium containing 0.05% lichenan in 50 mM Tris-HCl (pH 8.0) were used. Samples containing an equal amount of total protein were applied to preformed wells and incubated at 70°C for 1 h. The activity of lichenase in the samples was determined by staining with 0.5% Congo red solution (Sigma) followed by washing in 1 M NaCl. In this case, an enlightened area is found on the site of active lichenase, since the dye binds only to non-hydrolyzed lichenan. The activity of the lichenase was determined using lichenan (Megazyme, Ireland) as a substrate (incubation time 10 min). The reducing sugars released from the substrate were determined according to the method of Wood and Bhat [27]. The content of reducing sugars was determined from a calibration schedule constructed for glucose. The unit of activity was taken as the amount of enzyme forming 1 µmol of reducing sugars (as the equivalent of glucose) per minute (in terms of mg of protein). The amount of protein in the preparations was determined by the Bradford method [28].
The protein preparations were subjected to electrophoresis in polyacrylamide gel under denaturing conditions according to the Laemmli method [29]. Zymograms were obtained by staining the gel after separation of the proteins containing the lichenan, as previously described [24]. Briefly, electrophoresis was performed in 12% SDS-polyacrylamide gel. Lichenan (0.1%) was added to the separation gel prior to polymerization. After electrophoresis, the gel was incubated at 70°C for 1 h. Enzymatic activity was determined by staining the gel with a 0.5% solution of Congo red, followed by washing in 1 M NaCl. The molecular masses of proteins were determined using Prestained Protein Ladder (Fermentas, Lithuania).
The thermal stability of proteins incubated in 50 mM Tris-HCl (pH 8.0) at 70°C for 240 min was determined by the activity of lichenase as described above.
Western blot analysis was performed with anti-humIFN-α goat antiserum (ab193823; Abcam, UK) as primary antibodies and rabbit anti-goat HRP (ab6741; Abcam) as secondary antibodies. After washing, each PVDF membrane (Bio-Rad, USA) was stained by electro-chemiluminescent method (ECL) (GE Healthcare, USA). Chemiluminescence was detected using a Fusion FX5 device (VilberLourmat, France).
The antiviral activity of the native (IFN-αA) and hybrid (LicB-53-IFN-αA) interferon, as well as the thermostable lichenase LicB-53 (control), was determined by suppressing the cytopathogenic effect (CPE) of the vesicular stomatitis Indiana virus (VSV) (at a dose of 100 CPE50) in a culture of L929 inoculated cells grown in 96-well culture plates (Linbro, USA). Briefly, L929 cells (Sigma) were diluted with medium containing 10% fetal bovine serum (FBS) to (2.5-3.5)·105 cells/ml, and then 100 µl of diluted cells were plated in each well of a 96-well plate. Diluted in a medium containing 10% FBS, protein samples at the required concentrations (0.21, 0.39, 0.78, 1.56, 3.13, 6.25, and 12.5 ng/ml) were incubated with L929 cells for 16-18 h at 37°C. Then the medium was replaced with DMEM containing 7% FBS and 100 CPE50 VSV. After 24-h incubation, the viability of the cells was measured using the MTT assay [30]. Optical density (OD) was measured using a PHERAstar FSX multimodal reader (BMG LABTECH, Germany). The results were analyzed using Graph Pad Prism 5 software using a four-parameter logistic analysis model that corresponds to the sigmoid dose–response curve. A plot of cell survival (in %) versus drug concentration (log C(ng/ml)) was used to calculate the drug concentration at which 50% protection of cells from the cytopathogenic action of the test virus (CPE50) is observed. Note that, due to different experimental conditions, the values of the CPE50 may differ in different laboratories. The experiments were carried out in triplicate.
Computational analysis. The tertiary structures of the hybrid proteins were predicted using the I-TASSER server [31]. The 3D-structures of the proteins were drawn with YASARA (www.yasara.org) and POVRay (www.povray.org). The solubility of the protein was predicted using the following tools: Espresso (Estimation of protein expression and solution) [32] and PROSOII [33]. The protein expression in the periplasm of E. coli was predicted by Periscope [34]. Software packages used for computational analysis are freely available online.
Statistical analysis. All experiments were performed independently at least three times. The statistical processing of the results was carried out using the Statistica for Windows v. 9.0 and Microsoft Office Excel 2007 software unless otherwise noted. The figures and tables show the arithmetic mean values and their standard errors unless otherwise indicated.
RESULTS AND DISCUSSION
Design of thermostable lichenase with insertion of interferon αA and in silico sequence analysis. For the successful construction of proteins with domain insertion, it is extremely important that the receiving domain (in our case, thermostable lichenase) tolerate a discontinuity caused by the insertion domain (in our case, IFN-αA) while maintaining its functionality. In this approach, selecting the appropriate integration site in the receiving domain is a critical step [21, 22], and this domain of the “host” domain should not be critical to its stability [35]. We chose the insertion position in lichenase on the general criterion of localization in the loop section [36]. Moreover, the choice of the loop region (53 a.a.) of lichenase was made based on previous results on the construction of cyclic permutated versions of lichenase and assessment of their activity and thermal stability. We found that new ends in the region of 53 a.a. do not affect the activity or thermo-tolerance of the thermostable lichenase, as well as the integration of a small peptide into this 53 a.a. region of the lichenase [24].
Lichenase is a monomeric protein (215 a.a.). It has a compact structure, and it is characterized by high thermal stability and specific activity [25, 36, 37]. For integration, a modified version of the gene was used. This variant encoding thermostable lichenase with insertion fusion of a small peptide in the catalytic domain region of 53 a.a., which we designated earlier as NC-L-53 (in this work it is designated as LicB-53) (Fig. 1) [24]. Since the codons encoding the integrated peptide at the level of the nucleotide sequence form unique restriction sites, namely, BamHI and ApaI [24], we used them for insertion fusion with the gene sequence coding for IFN-αA (Fig. 1).
Fig. 1. Sequences of LicB-53 (initial version of lichenase) and LicB-53-IFN-αA (variants of lichenase with insertion of the IFN-αA domain into the 53 a.a. loop). Light gray blocks designate sequences of lichenase; dark gray blocks – an integrated small peptide or its parts; gray-white block – IFN-αA. The numbers of the amino acid residues denote their ordinal numbers in the protein, the numbers in parentheses indicate the ordinal numbers of the amino acid residues from the catalytic domain of thermostable lichenase.

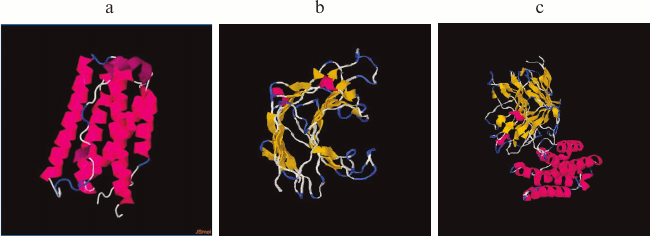
To avoid mutual destabilization of the two domains when a recombinant protein is constructed using the domain insertion approach, it is important not only to use strictly defined domains of the receiving domain, but also the relative proximity of the N- or C-terminal regions is extremely important for the insertion protein. This increases the chance of successful construction of proteins with domain insertion [35, 38]. It is known that in a case where the N or C ends of the insertion protein are sufficiently far apart, the number of degrees of freedom in the dynamics of the proteins is reduced. This can lead to undesirable consequences such as incorrect folding of the proteins, low protein production, and the loss of biological activity due to steric hindrance [21, 22]. IFN-αA is a monomeric hydrophobic protein consisting of 166 a.a., and according to its 3D structure the relative proximity of the N- and C-terminal regions is characteristic (Fig. 2a). To determine whether the thermostable lichenase and IFN-αA retain their inherent 3D structures after integrating IFN-αA into the 53 a.a. loop of lichenase, we modeled the spatial structure of the recombinant protein [31]. According to the predicted 3D models of native (LicB-53 and IFN-αA) and fusion (LicB-53-IFN-αA) proteins, integrated IFN-αA is represented in the corresponding region of the thermostable lichenase (Fig. 2b), and the 3D structures inherent in the native proteins are retained (Fig. 2).
Fig. 2. Spatial structures of proteins IFN-αA (a), LicB-53 (b), and LicB-53-IFN-αA (c).
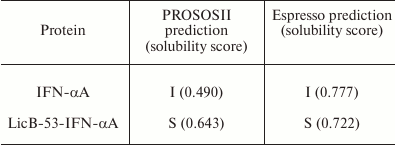
Comparative in silico analysis of the amino acid sequences of native (IFN-α) and fusion (LicB-53-IFN-αA) proteins using two servers [32, 33] predicted the form (soluble or insoluble) of native IFN-αA and recombinant LicB-53-IFN-αA proteins in E. coli cells (Table 1). The prediction indicated that the recombinant LicB-53-IFN-αA protein can be expressed in soluble form, in contrast to native IFN-αA. In addition, theoretical calculations of the efficiency of expression of the soluble form of native IFN-αA and the fused LicB-53-IFN-αA proteins in the periplasm of E. coli were made using the Periscope server [34]. Thus, according to the classification matrix, the expression level of native IFN-αA was predicted to be low, and LicB-53-IFN-αA as average, with the calculated expression level of the protein in the periplasm of E. coli for IFN-αA to be 0.0664 mg/liter, and for LicB-53-IFN-αA 4.8705 mg/liter.
Table 1. Results of prediction of expression
form – insoluble (I) and soluble (S) – for IFN-αA and
LicB-53-IFN-αA
Thus, the results of modeling the 3D protein structures suggest the preservation of IFN-αA and thermostable lichenase functionality in the recombinant protein LicB-53-IFN-αA. The results of the prediction of the expression of native IFN-αA and fused LicB-53-IFN-αA proteins in E. coli indicate the possibility of a significant increase in the expression of the recombinant LicB-53-IFN-αA protein in the periplasm of bacterium in soluble form compared to native IFN-αA. To verify the predictions, a recombinant DNA was constructed in which the sequence of the IFN-αA gene was integrated into the LicB-53 gene (Fig. 1). The recombinant genes (licB-53, IFN-αA and licB-53-IFN-αA) were transferred to expression plasmids based on the pET plasmid system and bacterial transformants designated by us according to the name of the proteins – LicB-53, IFN-αA, and LicB-53-IFN-αA.
Analytical characterization of recombinant proteins LicB-53, IFN-αA, and LicB-53-IFN-αA. During the study, we tried to determine (i) whether the thermostable lichenase can undergo stable and functional folding when integrating IFN-αA as an internal domain; (ii) whether the recombinant LicB-53-IFN-αA protein will be more efficiently expressed in the E. coli periplasm in soluble form compared to native IFN-αA; (iii) whether IFN-αA as an inserted domain in the thermostable lichenase retain its function.
To answer these questions, recombinant proteins were produced in E. coli, and protein lysates (TPL) including affinity-chromatography purified (PP) and ethanol-precipitated (P-EP) were obtained. Analysis of the TPL by the cup test showed that the enzymatic activity inherent in thermostable lichenase of C. thermocellum was found for both LicB-53 and LicB-53-IFN-αA (Fig. 3a). This indicates that the thermostable lichenase assumes functionally active folding when IFN-αA is integrated as an internal domain, and LicB-53-IFN-αA retains the functional activity of the lichenase.
Then we analyzed the purified protein (PP) by the zymogram method. Single enlightened bands of activity are detected on the zymogram, which may indicate that LicB-53-IFN-αA, as well as LicB-53, in the samples are present in the form of monomers (Fig. 3b), and that the molecular mass of the recombinant protein LicB-53-IFN-αA corresponds to the theoretically calculated value of 45 kDa. It should also be noted that the recombinant LicB-53-IFN-αA, like LicB-53, retains the ability to renature to the active form of the enzyme in the gel after electrophoresis under denaturing conditions (Fig. 3b). These results indicate that the catalytic domain of the lichenase in the composition of both LicB-53 and LicB-53-IFN-αA regains functional conformation after denaturation. Comparative analysis of the specific activity of the purified protein (PP) estimated at 70°C showed that the recombinant protein LicB-53-IFN-αA retains a high level of specific activity, which in relation to LicB-53 is more than 80% (Table 2). Further, we comparatively analyzed the specific activity of lichenases of TPL and samples after ethanol precipitation (P-EP). It was found that for both LicB-53 and LicB-53-IFN-αA, the enzymatic activity of the lichenase not only persists, but also significantly increases (more than 10-fold) (Table 3). Such an increase in the level of enzymatic activity can be explained by a significant decrease in the proportion of other total soluble proteins (from 10-20-fold) in TPL after ethanol precipitation (Table 3). These results also suggest that the recombinant LicB-53-IFN-αA protein, like the thermostable LicB-53 lichenase, is capable of in vitro refolding after denaturation with ethanol.
Fig. 3. Properties of the initial version of thermostable lichenase (LicB-53) and a variant of the lichenase with the insertion of IFN-αA (LicB-53-IFN-αA). a) A plate test of the total protein lysates obtained from bacterial transformants: LicB-53 (1), IFN-αA (2), LicB-53-IFN-αA (3), and control (bacterial transformant transformed with the empty plasmid pET-32) (4). b) Zymogram of purified proteins: LicB-53 (1), LicB-53-IFN-αA (2); M, molecular weight marker. c) Thermostability of LicB-53 and LicB-53-IFN-αA.
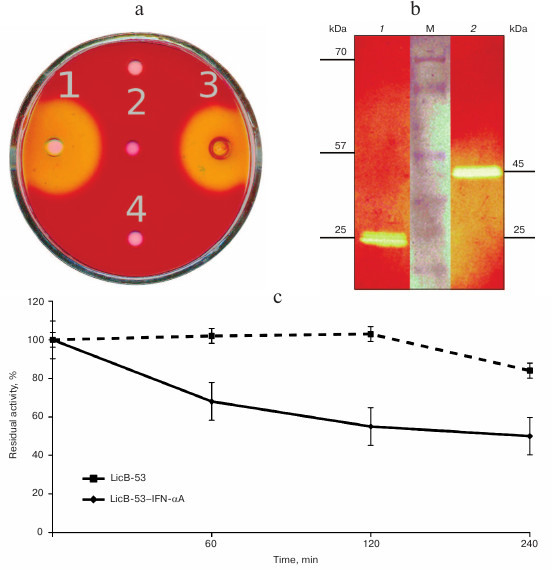
Table 2. Specific activity of the
lichenase
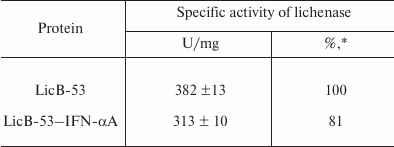
* Protein activity relative to activity of LicB-53.
Table 3. Specific activity of the lichenase
and level of total soluble proteins (TSP) in total protein lysates
(TPL) and in samples after ethanol precipitation (P-EP)
Comparative analysis of the thermal stability of the proteins established that LicB-53-IFN-αA showed no dramatic reduction in thermal stability – its activity was not less than 50% after 240-min incubation at 70°C (Fig. 3c). These results are consistent with the results with a cyclic permutated version of lichenase (53 a.a.), which retains about 40% of activity after 240-min incubation at 70°C [24]. Thus, it is convincingly shown that the thermostable lichenase is capable of functional folding when integrating IFN-αA as its internal domain, without dramatically losing its basic properties, activity, and thermal stability (Fig. 3 and Tables 2 and 3).
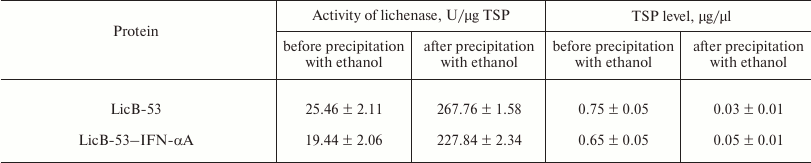
We then evaluated the production of LicB-53-IFN-αA and IFN-αA proteins in different cell fractions – E. coli periplasm, soluble (cytoplasmic), and insoluble (inclusion bodies). To do this, the corresponding fractions were examined by SDS-PAGE followed by Ponceau staining (Fig. 4a) and by Western blot hybridization (Fig. 4b). As shown in Fig. 4b, IFN-αA was detected exclusively in the insoluble fraction (inclusion body), in contrast to LicB-53-IFN-αA, 38 and 11% of which are detected in the periplasmic and soluble (cytoplasmic) fractions, respectively (Fig. 4c). These data indicate an increase in the level of LicB-53-IFN-αA expression in soluble form compared to IFN-αA and agree with the theoretical predictions presented earlier (Table 1). According to these data, about half of the recombinant protein LicB-53-IFN-αA is expressed in E. coli cells in soluble form (cytoplasmic and periplasmic fraction) (Fig. 4c). Moreover, the calculations made on the basis of experimental data convincingly show a significant increase in the level of the recombinant protein LicB-53-IFN-αA in E. coli cells (Fig. 4c), which can be due to peculiarities of folding of the thermostable lichenase, whose catalytic domain is folded into a classical β-jellyroll architecture [36], which is generally remarkably conformationally stable, resisting changes in both the amino acid sequence and the topology of the chains [39]. This allows making a well-founded assumption that the conformational stability of the recombinant protein LicB-53-IFN-αA, in comparison with IFN-αA, can result in more efficient expression of this protein in E. coli cells. According to current opinion, proteins with increased stability are tolerant to mutational events, including terminal fusions and insertions of the domain(s), and retain their functionality [40, 41], which we showed in relation to thermostable lichenase earlier [24, 25].
Fig. 4. Production of LicB-53-IFN-αA and IFN-αA in different E. coli cell fractions. a) Electrophoregram of total protein lysates, Ponceau staining. b) Western blot hybridization of total protein lysates. For panels (a) and (b): soluble (cytoplasmic) fraction – LicB-53-IFN-αA (1) and IFN-αA (4); insoluble fraction (inclusion body) – LicB-53-IFN-αA (2) and IFN-αA (5); periplasm – LicB-53-IFN-αA (3) and IFN-αA (6); M, molecular mass markers. Marker molecular masses are shown on the left, molecular masses of target proteins on the right. c) Level of production of IFN-αA and LicB-53-IFN-αA proteins in different cell fractions (determined by densitometry).
Bacterial expression systems are effective for production of many recombinant proteins (including IFN-αA), which are usually expressed as inclusion bodies [42, 43]. Although insoluble and with improper folding, the proteins can be purified and functionally folded, but these manipulations reduce the yield of the target protein [5, 23, 43]. It should be noted that the expression of soluble proteins in E. coli periplasm can have several advantages; it not only simplifies the purification process of the target protein, but it also increases the probability of obtaining proteins with proper folding and biologically activity. To increase the solubility of recombinant proteins, N- or C-terminal fusions of the target protein with a variety of proteins that increase the solubility of the target protein as a fusion partner can be successful [5, 23, 42]. Such direct fusion proteins have been successfully used for IFN-αA [23, 42], but none of those cases provided the production of IFN-αA in the periplasm of the bacterial cells. Our results indicate that the integration of IFN-αA as an internal domain of thermostable lichenase is a useful approach both for increasing the solubility of the recombinant IFN-αA and production of the recombinant protein LicB-53-IFN-αA in the periplasm of E. coli (Fig. 4).
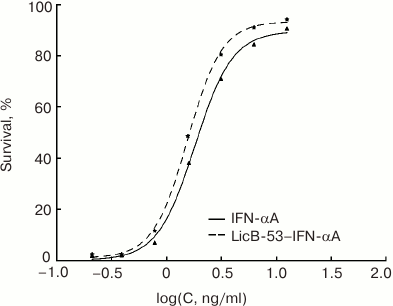
The biological activity of the purified LicB-53-IFN-αA was determined using an antiviral assay (L929/VSV). LicB-53-IFN-αA was tested in parallel with IFN-αA and LicB-53. Note that all test proteins were obtained by an identical expression and purification method (see “Materials and Methods”) to exclude any potential differences in activity because of the use of different expression systems or purification methods. Samples of purified proteins (LicB-53-IFN-αA, IFN-αA, and LicB-53) were free of endotoxins: the endotoxin content was 4-7 EU/mg protein as determined by the Endosafe-PTS system. As expected, the thermostable lichenase LicB-53 shows no antiviral activity (data not shown) in comparison with the biological activity of LicB-53-IFN-αA, which demonstrates antiviral activity with a CPE50 of 1.85 ± 0.24 ng/ml, similar to that for IFN-αA (1.99 ± 0.42 ng/ml) (Fig. 5). Thus, we have shown that IFN-αA as a domain integrated in a thermostable lichenase preserves the capacity for functional folding, which is confirmed by the preservation of antiviral activity similar to native interferon (Fig. 5).
Fig. 5. Comparison of the antiviral activity of recombinant protein LicB-53-IFN-αA (insertion of IFN-αA into thermostable lichenase) and native IFN-αA obtained and purified by the same protocol.
In this study, we used an approach based on the insertion of the IFN-αA domain into a thermophilic protein. Insertion of IFN-αA into the loop (53 a.a.) of the thermostable lichenase enzyme of C. thermocellum (LicB-53) led to effective expression of the soluble form of the recombinant protein in the periplasm of E. coli without any compromises in the biological activity of IFN-αA (Fig. 5), as well as in the activity and thermal stability of the lichenase (Fig. 3). Although insertion of long sequences is believed to be destabilizing of the recombinant protein [44, 45], presumably due to an entropy-unfavorable condition for contact between amino acid residues leading to rapid folding of the protein [46] and the formation of nonfunctional protein complexes such as inclusion bodies [18], our results show that the insertion fusion of interferon (IFN-αA) with the thermostable lichenase probably stabilizes IFN-αA, and it contributes to the prevention of structural changes and the formation of aggregate species (Fig. 4). It is known that, for example, maltose-binding proteins (MBPs) stabilize partner proteins fused to it by increasing the solubility of the fusion proteins [45]. Thus, it can be assumed that the improved stabilization of IFN-αA by domain insertion into the thermostable lichenase may also be due to an increase in the solubility of the recombinant LicB-53-IFN-αA protein. In addition, we have demonstrated that fusion with the MBP also increases the level of expression of recombinant proteins, and it is suggested that this is also due to an increase in their solubility [45]. It is likely that an increase in the solubility of the LicB-53-IFN-αA protein, along with its conformational stability due to the surprisingly stable jellyroll tertiary structure of the thermostable lichenase, can provide more efficient expression of this protein in E. coli cells (Fig. 4). Effective expression of a soluble form IFN-αA in the periplasm of E. coli achieved by the described method can potentially be applied to other proteins. As noted above, for the successful construction of proteins with domain insertion, there is a requirement for the N and C termini of the inserted protein – they must be close to each other in space. Because ~50% of proteins in Protein Data Bank (PDB) are proximal in this respect, many proteins can be open to this strategy [38].
We note several properties of the thermostable lichenase as a host protein when using the domain insertion approach. First, the thermostable lichenase preserves functionality with significant dimensions of the embedded domain (Fig. 2 and Table 3). Second, the persistence of lichenase activity and thermal stability in the precipitation of non-target proteins with ethanol can be used for rapid and economical purification of fusion proteins (Table 3). Thus, it was shown earlier that by the incubation of protein lysates at 65°C, up to 50% of the polluting plant proteins are removed, leaving the proteins fused with the lichenase preserved [47]. Third, the zymogram method can be used to accurately determine the molecular weights of fusion proteins that include the lichenase. The zymogram method is characterized by high sensitivity and does not require specific antibodies to the protein of interest (Fig. 2b). Thus, our results support the use of the approach based on the insertion of the protein domain into the thermostable lichenase to improve the expression level of soluble forms of recombinant proteins, including those in the periplasm of E. coli.
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (project No. 16-34-00249; AAT).
REFERENCES
1.Pestka, S., and Baron, S. (1981) Definition and
classification of the interferons, Methods Enzymol., 78,
3-14.
2.Pestka, S., Langer, J. A., Zoon, K. C., and Samuel,
C. E. (1987) Interferons and their actions, Annu. Rev. Biochem.,
56, 727-777.
3.Borden, E. C., Sen, G. C., Uze, G., Silverman, R.
H., Ransohoff, R. M., and Foster, G. R. (2007) Interferons at age 50:
past, current and future impact on biomedicine, Nat. Rev.
Drug Discov., 6, 975-990.
4.Gutterman, J. U. (1994) Cytokine therapeutics:
lessons from interferon alpha, Proc. Natl. Acad. Sci. USA,
91, 1198-1205.
5.Peciak, K., Tommasi, R., Choi, J., Brocchini, S.,
and Laurine, E. (2014) Expression of soluble and active interferon
consensus in SUMO fusion expression system in E. coli,
Protein Expr. Purif., 99, 18-26.
6.Clark, E. D. (2001) Protein refolding for
industrial processes, Curr. Opin. Biotechnol., 12,
202-207.
7.Middelberg, A. P. J. (2002) Preparative protein
refolding, Trends Biotechnol., 20, 437-443.
8.Cabrita, L. D., Dai, W., and Bottomley, S. P.
(2006) A family of E. coli expression vectors for laboratory
scale and high throughput soluble protein production, BMC
Biotechnol., 6, 12.
9.Weickert, M. J., Doherty, D. H., Best, E. A., and
Olins, P. O. (1996) Optimization of heterologous protein production in
Escherichia coli, Curr. Opin. Biotechnol., 7,
494-499.
10.Burgess-Brown, N. A., Sharma, S., Sobott, F.,
Loenarz, C., Oppermann, U., and Gileadi, O. (2008) Codon optimization
can improve expression of human genes in Escherichia coli: a
multi-gene study, Protein Expr. Purif., 59,
94-102.
11.Yang, H., Liu, L., and Xu, F. (2016) The promises
and challenges of fusion constructs in protein biochemistry and
enzymology, Appl. Microbiol. Biotechnol., 100,
8273-8281.
12.Kapust, R. B., and Waugh, D. S. (1999)
Escherichia coli maltose-binding protein is uncommonly effective
at promoting the solubility of polypeptides to which it is fused,
Protein Sci., 8, 1668-1674.
13.Rabhi-Essafi, I., Sadok, A., Khalaf, N., and
Fathallah, D. M. (2007) A strategy for high-level expression of soluble
and functional human interferon alpha as a GST-fusion protein in E.
coli, Protein Eng. Des. Sel., 20, 201-209.
14.Smith, D. B., and Johnson, K. S. (1988)
Single-step purification of polypeptides expressed in Escherichia
coli as fusions with glutathione S-transferase, Gene,
67, 31-40.
15.De Marco, V., Stier, G., Blandin, S., and de
Marco, A. (2004) The solubility and stability of recombinant proteins
are increased by their fusion to NusA, Biochem. Biophys. Res.
Commun., 322, 766-771.
16.Butt, T. R., Edavettal, S. C., Hall, J. P., and
Mattern, M. R. (2005) SUMO fusion technology for difficult-to-express
proteins, Protein Expr. Purif., 43, 1-9.
17.Zhu, F., Wang, Q., Pu, H., Gu, S., Luo, L., and
Yin, Z. (2013) Optimization of soluble human interferon-c production in
Escherichia coli using SUMO fusion partner, World J.
Microbiol. Biotechnol., 29, 319-325.
18.Betton, J. M., Jacob, J. P., Hofnung, M., and
Broome-Smith, J. K. (1997) Creating a bifunctional protein by insertion
of beta-lactamase into the maltodextrin-binding protein, Nat.
Biotechnol., 15, 1276-1279.
19.Doi, N., and Yanagawa, H. (1999) Insertional gene
fusion technology, FEBS Lett., 457, 1-4.
20.Ay, J., Gotz, F., Borriss, R., and Heinemann, U.
(1998) Structure and function of the Bacillus hybrid enzyme
GluXyn-1: native-like jellyroll fold preserved after insertion of
autonomous globular domain, Proc. Natl. Acad. Sci. USA,
95, 6613-6618.
21.Collinet, B., Herve, M., Pecorari, F., Minard,
P., Eder, O., and Desmadril, M. (2000) Functionally accepted insertions
of proteins within protein domains, J. Biol. Chem., 275,
17428-17433.
22.Yu, K., Liu, C., Kim, B. G., and Lee, D. Y.
(2015) Synthetic fusion protein design and applications, Biotechnol.
Adv., 33, 155-164.
23.Elleuche, S. (2015) Bringing functions together
with fusion enzymes from Nature’s inventions to biotechnological
applications, Appl. Microbiol. Biotechnol., 99,
1545-1556.
24.Tyurin, A. A., Sadovskaya, N. S., Nikiforova, K.
R., Mustafaev, O. N., Komakhin, R. A., Fadeev, V. S., and
Goldenkova-Pavlova, I. V. (2015) Clostridium thermocellum
thermostable lichenase with circular permutations and modifications in
the N-terminal region retains its activity and thermostability,
Biochim. Biophys. Acta, 1854, 10-19.
25.Musiychuk, K. A., Goldenkova, I. V., Abdeev, R.
M., Kobets, N. S., and Piruzian, E. S. (2000) Preparation and
properties of Clostridium thermocellum lichenase deletion
variants and their use for construction of bifunctional hybrid
proteins, Biochemistry (Moscow), 65, 1397-1402.
26.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor, Cold Spring Harbor Laboratory Press, N. Y.
27.Wood, T. M., and Bhat, K. M. (1988) Methods for
measuring cellulase activities, Methods Enzymol., 160,
87-112.
28.Bradford, M. A. (1976) Rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing the
principle of protein–dye binding, Anal. Biochem.,
72, 248-254.
29.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
30.Mosmann, T. (1983) Rapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assays, J. Immunol. Methods, 65, 55-63.
31.Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J.,
and Zhang, Y. (2015) The I-TASSER Suite: protein structure and function
prediction, Nat. Methods, 12, 7-8.
32.Hirose, S., Kawamura, Y., Yokota, K., Kuroita,
T., Natsume, T., Komiya, K., Tsutsumi, T., Suwa, Y., Isogai, T.,
Goshima, N., and Noguchi, T. (2011) Statistical analysis of features
associated with protein expression/solubility in an in vivo
Escherichia coli expression system and a wheat germ cell-free
expression system, J. Biochem., 150, 73-81.
33.Smialowski, P., Doose, G., Torkler, P., Kaufmann,
S., and Frishman, D. (2012) PROSO II – a new method for protein
solubility prediction, FEBS J., 279, 2192-2200.
34.Chang, C. C., Li, C., Webb, G. I., Tey, B., Song,
J., and Ramanan, R. N. (2016) Periscope: quantitative prediction of
soluble protein expression in the periplasm of Escherichia coli,
Sci. Rep., 6.
35.Zoldak, G., Carstensen, L., Scholz, C., and
Schmid, F. X. (2009) Consequences of domain insertion on the stability
and folding mechanism of a protein, J. Mol. Biol., 386,
1138-1152.
36.Chen, C. C., Huang, J. W., Zhao, P., Ko, T. P.,
Huang, C. H., Chan, H. C., Huang, Z., Liu, W., Cheng, Y. S., Liu, J.
R., and Guo, R. T. (2015) Structural analyses and yeast production of
the beta-1,3-1,4-glucanase catalytic module encoded by the licB
gene of Clostridium thermocellum, Enzyme Microb.
Technol., 71, 1-7.
37.Zverlov, V. V., and Schwarz, W. H. (2008)
Bacterial cellulose hydrolysis in anaerobic environmental subsystems
– Clostridium thermocellum and Clostridium
stercorarium, thermophilic plant-fiber degraders, Ann.
N.Y. Acad. Sci., 1125, 298-307.
38.Edwards, W. R., Williams, A. J., Morris, J. L.,
Baldwin, A. J., Allemann, R. K., and Jones, D. D. (2010) Regulation of
beta-lactamase activity by remote binding of heme: functional coupling
of unrelated proteins through domain insertion, Biochemistry,
49, 6541-6549.
39.Heinemann, U., Ay, J., Gaiser, O., Muller, J. J.,
and Ponnuswamy, M. N. (1996) Enzymology and folding of natural and
engineered bacterial beta-glucanases studied by X-ray crystallography,
Biol. Chem., 377, 447-454.
40.Bloom, J. D., Labthavikul, S. T., Otey, C. R.,
and Arnold, F. H. (2006) Protein stability promotes evolvability,
Proc. Natl. Acad. Sci. USA, 103, 5869-5874.
41.Segall-Shapiro, T. H., Nguyen, P. Q., Dos Santos,
E. D., Subedi, S., Judd, J., Suh, J., and Silberg, J. J. (2011)
Mesophilic and hyperthermophilic adenylate kinases differ in their
tolerance to random fragmentation, J. Mol. Biol., 406,
135-148.
42.Bis, R. L., Stauffer, T. M., Singh, S. M.,
Lavoie, T. B., and Mallela, K. M. (2014) High yield soluble bacterial
expression and streamlined purification of recombinant human interferon
α-2a, Protein Expr. Purif., 99, 138-146.
43.EL-Baky, N. A., Linjawi, M. H., and Redwan, E. M.
(2015) Auto-induction expression of human consensus interferon-alpha in
Escherichia coli, BMC Biotechnol., 15, 14.
44.Viguera, A. R., and Serrano, L. (1997) Loop
length, intramolecular diffusion and protein folding, Nat. Struct.
Biol., 4, 939-946.
45.Kim, C. S., Pierre, B., Ostermeier, M., Looger,
L. L., and Kim, J. R. (2009) Enzyme stabilization by domain insertion
into a thermophilic protein, Protein Eng., 22,
615-623.
46.Zhou, H. X. (2004) Loops, linkages, rings,
catenanes, cages, and crowders: entropy-based strategies for
stabilizing proteins, Acc. Chem. Res., 37, 123-130.
47.Musiychuk, K., Stephenson, N., Bi, H., Farrance,
C. E., Orozovic, G., Brodelius, M., Brodelius, P., Horsey, A., Ugulava,
N., Shamloul, A. M., Mett, V., Rabindran, S., Streatfield, S. J., and
Yusibov, V. (2007) A launch vector for the production of vaccine
antigens in plants, Influenza Other Respir. Viruses, 1,
19-25.