Possible Role of Escherichia coli Protein YbgI
O. V. Sergeeva1,2*, D. O. Bredikhin2, M. V. Nesterchuk1, M. V. Serebryakova2, P. V. Sergiev1,2, and O. A. Dontsova1,2
1Skolkovo Institute of Science and Technology, 143026 Skolkovo, Moscow Region, Russia; E-mail: o.sergeeva@skoltech.ru2Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received September 5, 2017; Revision received December 5, 2017
Proteins containing the NIF3 domain are highly conserved and are found in bacteria, eukaryotes, and archaea. YbgI is an Escherichia coli protein whose gene is conserved among bacteria. The structure of YbgI is known; however, the function of this protein in cells remains obscure. Our studies of E. coli cells with deleted ybgI gene suggest that YbgI is involved in formation of the bacterial cell wall.
KEY WORDS: Escherichia coli, cell stress, bacterial cell wallDOI: 10.1134/S0006297918030070
The genome of one important model organism, the eubacterium Escherichia coli, contains numerous genes whose functions are still unknown. One of them is ybgI, which belongs to an operon that also includes poorly studied genes: ybgJ (predicted hydrolase), ybgK (predicted carboxyl transferase), ybgL (predicted component of lactam metabolism), and nei (endonuclease VIII) genes. Endonuclease VIII is involved in base excision repair (BER) [1]; it can also act as a lyase in the removal of apurinic/apyrimidinic sites from DNA [2].
YbgI protein encoded by the ybgI gene consists of 247 amino acids and has a molecular weight of 27 kDa. It belongs to the DUF34 protein family with unknown function [3]. Proteins of this family contain the highly conserved NIF3 domain that has been found in eukaryotes, bacteria, and archaea [4]. The best-studied representative of this family is the yeast protein NIF3; its amino acid sequence is 22% identical to the sequence of E. coli YbgI [5]. NIF3 interacts with the N-terminal domain of the NGG1p transcription coactivator in yeast [6]. NIF3L1 from Homo sapiens and Nif3l1 from Mus musculus are 22 and 37% identical, respectfully, to yeast NIF3. These proteins are expressed in stem cells, but their role in cells remains obscure. Note that both NIF3L1 and Nif3l1 have highly conserved N- and C-terminal domains connected by a sequence that is absent in yeast NIF3 and most bacterial proteins of the NIF3 family [7].
Studies of the cell stress response revealed that mitomycin C-induced genotoxic stress in E. coli is accompanied by a 3.5-fold upregulation of the expression of ybgI. However, after some time, the level of ybgI expression diminishes despite the presence of mitomycin C [8]. Also, deletion of the ybgI gene decreases by 2 to 3 orders of magnitude the survival of bacterial cells subjected to ionizing radiation (2000 Gy) [9].
According to X-ray crystallography, three dimeric YbgI molecules form a toroid in which each subunit has a metal-binding site at the inner surface. The identity of divalent metal ligands in the YbgI molecule is unknown; it was suggested that they might be iron, zinc, manganese, or magnesium ions [5]. Based on previously obtained data and colocalization of the ybgI and endonuclease VIII genes in the same operon, it was suggested that YbgI is involved in BER under stress conditions. The goal of this work was to investigate the role of YbgI in E. coli cells.
MATERIALS AND METHODS
Analysis of the surface charge of YbgI. To identify and analyze equipotential surfaces in the YbgI molecule, we used the APBS (Adaptive Poisson–Boltzmann Solver) program package [10] for the PyMol molecular graphics system [11], which estimates the electrostatic properties of biomolecules by solving the Poisson–Boltzmann equation.
Affinity chromatography. Escherichia coli cells transformed with the pCA24 plasmid carrying the recombinant His6-YbgI protein gene [12] were grown in Petri dishes on LB agar medium (1% bactotryptone, 0.5% yeast extract, and 171 mM NaCl) containing chloramphenicol. Single colonies were selected for growth in liquid LB medium. The cells were incubated overnight in the presence of chloramphenicol and then used for inoculation of chloramphenicol-containing LB medium. The cells were grown at 37°C on a shaker (180 rpm) to optical density A600 = 0.3. Protein biosynthesis was induced by adding 1 mM IPTG, and the cells were incubated for another 2 h. The cells were cooled, pelleted by centrifugation at 9000 rpm at 4°C for 15 min, and washed with lysis buffer (20 mM HEPES-KOH, pH 7.5, 200 mM NH4OAc, 10 mM imidazole, 0.1% Triton X-100, and 1 mg/ml lysozyme). The pellet was then resuspended in the lysis buffer, and the cells were disintegrated by sonication (10 cycles for 15 s with a 45-s pause between the cycles). The supernatant was mixed with 50% suspension of Ni-NTA-agarose (Qiagen, USA) and incubated on a rotator mixer at 200 rpm for 1 h at 4°C. The resin was separated by centrifugation at 3000 rpm for 1 min, washed with buffer P (20 mM HEPES-KOH, pH 7.5, 200 mM NH4OAc, 40 mM imidazole, and 0.1% Triton X-100) for 15 min, and separated from the buffer by centrifugation at 3000 rpm for 1 min. The protein was eluted with a step-wise gradient of imidazole (40, 60, 80, 100, 150, 200, 250, 300, and 600 mM) in buffer L (20 mM HEPES-KOH, pH 7.5, 200 mM NH4OAc, and 0.1% Triton X-100). At each imidazole concentration, the resin was incubated with the eluent for 15 min with gentle shaking at 4°C and then centrifuged at 3000 rpm for 1 min to collect the supernatant.
The resulting protein solutions were dialyzed against buffer D (20 mM HEPES-KOH, pH 7.5, 1 mM Mg(OAc)2, 200 mM NH4OAc, 10% glycerol, and 0.1% Triton X-100) for 2 h at room temperature and analyzed by electrophoresis in denaturing polyacrylamide gel. Protein aliquots were mixed with sample buffer (50 mM Tris-HCl, pH 6.8, 100 mM DTT, 2% SDS, 0.1% Bromophenol Blue, 10% glycerol) and fractionated by 8-12% SDS-PAGE to determine protein purity and concentration. Lysates of wild-type (wt) E. coli cells and cell overexpressing His6-tagged YhhF protein (molecular mass ~22 kDa) were used as controls. The protein was further identified by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) of tryptic hydrolysates of the protein.
Cell growth in the presence of various nitrogen sources. Escherichia coli wt (BW25141) and ybgI-deficient cells (ΔybgI; JW0700 from the Keio collection [13]) were grown in minimal M9 medium without ammonium chloride. The following compounds were used as nitrogen sources (20 mM): adenine, NH4Cl, urea, uridine, and α-amino acids (L-alanine, L-arginine, L-asparagine, L-aspartic acid, L-cysteine, L-glutamine, L-glutamic acid, L-glycine, L-histidine, L-methionine, L-phenylalanine, L-proline, L-serine, L-threonine, L-tryptophan, L-tyrosine, L-valine).
The optical density of the cell suspension was determined every 40 min for 20 h, i.e., before the cells entered the stationary phase. The number of cells after time Δt was calculated using the formula:
N2 = 2Δt/Τ × Ν1.
The doubling time was calculated using the formula:
T = Δt·ln(2)/[ln(N2) – ln(N1)],
where T is the doubling time, N1 and N2 are the values of cell suspension optical density that corresponded to the cell number in a culture, and Δt is the time between measurements N1 and N2.
RT-qPCR for mRNA quantification. Escherichia coli wt and ΔybgI cells were grown in the minimal M9 medium to the logarithmic and stationary phases. Total RNA was isolated with an RNeasy Mini Kit (Qiagen) and used as a template in the reaction of cDNA synthesis with a Maxima Universal First Strand cDNA Synthesis Kit (Thermo Scientific, USA) and random primers.
qRCR was performed using the SYBR Green dye. Based on the results of earlier studies, we amplified the dps, gadA, glmS, pheA, prsA, rsuA, tnaA, ygaU, and ygiW genes from E. coli using primers given in Table 1. 16S rRNA was used as a reference molecule because its concentration in bacterial cells varies much less that the concentrations of other RNAs. Two-fold (or higher) change in the mRNA content was considered significant. All experiments were performed in triplicate.
Table 1. Primers used in the study

Differential two-dimensional (2D) electrophoresis. Cells were centrifuged at 4000 rpm; the pellets were washed twice with TBS (50 mM Tris-HCl, pH 7.6, 150 mM NaCl), centrifuged again at 4000 rpm, resuspended in 40% CHAPS/NP-40, incubated with 0.08 mg/ml RNase A and 1 U/μl DNase I, and lysed by subjecting them to 5 cycles of freezing/thawing (liquid nitrogen/37°C). The lysates were incubated on ice for 30 min, mixed with sample loading buffer (30 mM Tris-HCl, pH 8.5, 7 M urea, 2 M thiourea, 3% CHAPS, 1% Nonidet P-40), and centrifuged at 12,000 rpm. Protein concentration in the supernatants was determined by the Bradford method [14].
Aliquots containing 100 μg protein were mixed with 400 pmol of fluorescent dyes Cy3 (wt cells) or Cy5 (ΔybgI cells) and incubated for 30 min on ice; 10 mM lysine was then added, and the mixtures were incubated on ice for another 15 min.
Gels for isoelectric focusing were cast in glass tubes (diameter, 1 mm). Protein samples from the wt and ΔybgI cells were mixed with each other; 1% ampholytes (BioRad, USA), and 1% dithiothreitol were added. The samples were loaded onto the gels, and isoelectrofocusing was performed in the following regime: linear voltage gradient of 50-600 V for 6 h, 700 V for 10 h, and 900 V for 2 h.
After isofocusing, the gels were extracted from the tubes, incubated for 30 min in the equilibration buffer (150 mM Tris-HCl, pH 6.8, 8 M urea, 2.5% SDS, and 50% glycerol), and placed on 8-16% gradient polyacrylamide gels. Electrophoresis in the second direction was performed at 250 V for 5 h. After the electrophoresis, the gels were fixed in 20% ethanol, 10% acetic acid for 3 h, washed three times with deionized water, and scanned with a Typhoon fluorescence scanner (GE Healthcare Life Sciences, USA) to identify protein zones that differed in the compared samples. The gels were then stained with silver nitrate to cut out the protein spots for further identification. The gels were washed with 0.03% sodium thiosulfate, rinsed three times with water, stained with 0.1% silver nitrate in 0.04% formaldehyde for 15-20 min, washed with water, and incubated with developing solution (4% Na2CO3, 0.0006% Na2S2O3, 0.04% formaldehyde) until full development. The staining was stopped with 10% acetic acid.
Identification of PAGE-separated proteins. Protein zones from the silver-stained gels were cut out, rinsed twice with washing buffer (50 mM NH4HCO3, 40% acetonitrile) for 20 min, dehydrated in acetonitrile for 5 min, and dried in the open air. Dried pieces of the gel were incubated with 10 ng/µl trypsin solution in 100 mM NH4HCO3 for 4 h at 37°C. After incubation, an equal volume of 0.5% trifluoroacetic acid was added; the samples were loaded on a support, dried in the open air, and analyzed by MALDI-MS using an Ultraflex II mass spectrometer (Bruker, USA). The mass spectra were analyzed with the flexAnalysis 3.2 program; the proteins were identified using the Mascot 2.4.2 program and the NCBI database (see Supplement to this article on the journal website http://protein.bio.msu.ru/biokhimiya and Springer site Link.springer.com).
Intracellular localization of YbgI. Escherichia coli cells expressing chimeric GFP-modified PstB, TktA, CheW, and DnaT proteins from the ASKA E. coli cells’ library [12] were used for preparing competent cells. These proteins were chosen based on information from the MicrobesOnline database and published data on their cell localization. The competent cells were transformed with the plasmid coding for the chimeric mCherry-YbgI protein (obtained in our laboratory). The cells were grown to the logarithmic phase, placed on microscope slides in Mowiol mounting medium (Sigma, USA), covered with coverslips, and analyzed under a Nikon C2 fluorescence microscope (Nikon, Japan) for colocalization of the green and red fluorescence.
Cell response to UV-induced DNA damage. Escherichia coli wt, ΔybgI, and recA-deficient cells were grown in LB medium to the logarithmic phase and the same optical density. Cell aliquots (~10 μl) were placed on a piece of Parafilm, subjected to UV irradiation at 254 nm (dose, 50 mJ/cm2) for different time intervals (from 5 s to 10 min), plated on Petri dishes with LB agar medium, and incubated overnight at 37°C. The grown colonies were counted the next day.
RESULTS
Role of YbgI in cell response to UV-induced DNA damage. Based on the YbgI crystal structure, Ladner et al. [5] suggested that YbgI interacts with DNA. YbgI consists of three domains that form a toroidal structure with an inner diameter of 30 Å. It can be assumed that nucleic acids can bind in the center of the protein complex, because the diameter of the B-DNA double helix is 20 Å [15].
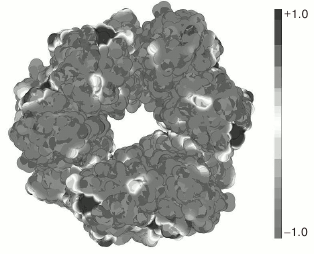
To test this hypothesis, we calculated the distribution of the surface charge on the YbgI molecule using the PyMol program with the APBS plug-in (Fig. 1). As seen in Fig. 1, the protein has positively and negatively charged areas, although most of its surface around the central hole of the toroid is negatively charged, which suggests against nucleic acid binding to YbgI.
Fig. 1. Distribution of electrostatic charge on the surface of YbgI protein (PDB ID: 1NMP). Gray and black colors designate negative and positive surface charges, respectively.
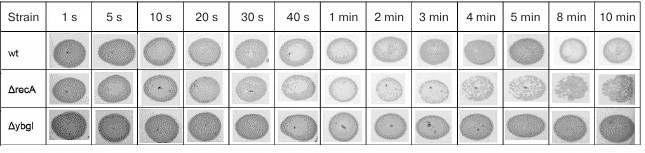
Nevertheless, the location of the ybgI gene in the same operon with the nei gene for endonuclease VIII might indicate a possible role of YbgI in DNA repair. To see if this is the case, we UV-irradiated wt and ΔybgI cells at 254 nm (intensity, 50 mJ) for time intervals of 5 s to 10 min. As a positive control, we used cells deficient in the recA gene required for recombinational DNA repair and SOS response in bacteria [16].
As expected, E. coli recA-deficient cells were more sensitive to DNA damage and started to die already after 4 min of irradiation. At the same time, survival of the ΔybgI cells was not affected even by irradiation for 10 min (Fig. 2). Therefore, it is unlikely that YbgI contributes to the repair of the UV-induced DNA damage to the same extent as RecA.
Fig. 2. Cell survival (number of colonies grown after plating 10 μl of bacterial culture from the exponential growth phase) after UV-irradiation for indicated times.
Cellular localization of YbgI. Cellular localization of YbgI might help in understanding the function of this protein. We expressed the chimeric protein YbgI protein C-terminally fused with the red fluorescent protein mCherry in E. coli AG1 cells. Unexpectedly, YbgI localized to the cell poles (Fig. 3a).
Fig. 3. Localization of YbgI in bacterial cells. a) YbgI-mCherry chimeric protein. b) Colocalization of YbgI-mCherry with recombinant PstB-GFP protein. c) Colocalization of YbgI-mCherry with recombinant TktA-GFP protein. Each picture shows a single bacterial cell.

Next, we studied YbgI-mCherry colocalization with some recombinant E. coli proteins modified with GFP at their C-termini. The proteins were chosen based on published data on their cellular localization and analysis of the MicrobesOnline database for potential protein–protein interactions. The plasmid coding for YbgI-mCherry was used for transformation of E. coil AG1 cells expressing these GFP-containing proteins [12]. We found that YbgI colocalized only with two proteins: PstB and TktA (Fig. 3, b and c). TktA is a transketolase and a key enzyme in glycolysis and pentose phosphate pathway [17]; PstB is the ATP-binding subunit of the cell protein complex responsible for phosphate transport across the cell membrane [18].
Search for YbgI-interacting proteins. Recombinant YbgI with the C-terminal His6-tag (pCA24YbgI plasmid) was expressed in E. coli AG1 cells and purified [12] by affinity chromatography with the goal of identifying potential YbgI interaction partners. Bacterial cell lysate containing YbgI-His6 was incubated with Ni-NTA-Sepharose; the bound proteins were eluted with a gradient of imidazole concentration, fractionated in denaturing polyacrylamide gel, and analyzed by MALDI mass spectrometry. We suggested that proteins that copurify with YbgI might interact with it in the cell. As a control, we used N-terminally His6-tagged bacterial methyltransferase YhhF.
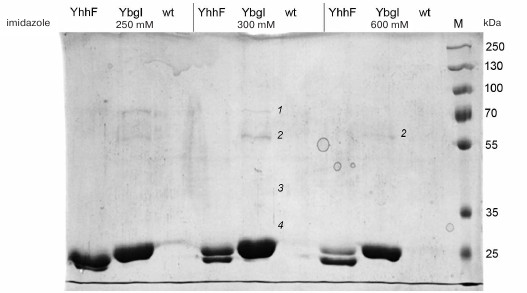
Four proteins copurifying with YbgI were identified by mass spectrometry of the tryptic hydrolysate of the eluted protein (Fig. 4): prephenate dehydratase PheA (1), L-glutamine:D-fructose-6-phosphate aminotransferase GlmS (2), ribose-phosphate pyrophosphokinase PrsA (3), and 16S rRNA pseudouridylate synthase RsuA (4) (see Supplement). GlmS was the only protein found in the fraction with the maximum YbgI content after elution with high imidazole concentration (600 mM); therefore, GlmS might be an interaction partner of YbgI in the cell.
Fig. 4. Affinity purification of YbgI-His6; YhhF and wt are used as controls; 250, 300, and 600, imidazole concentration in the eluent (mM); M, PageRuler Plus Prestained Protein Ladder (ThermoScientific, USA); 1-4) fractions analyzed by MALDI mass spectrometry (see description in the text).
GlmS catalyzes conversion of fructose-6-phosphate into glucosamine-6-phosphate utilizing glutamine as a nitrogen source; one of the reaction products is L-glutamate [19]. This reaction is one of the stages in the synthesis of UDP-N-acetylglucosamine, which is among major components of the cell wall peptidoglycan in bacteria [20].
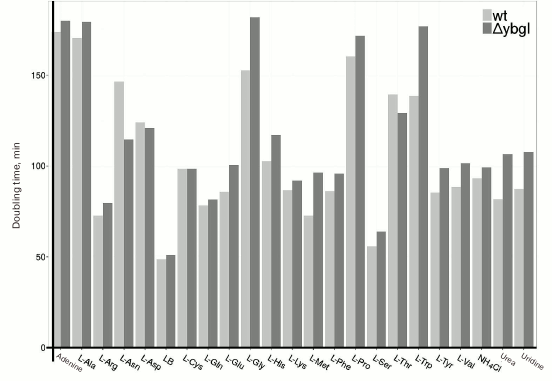
Characterization of ΔybgI cells. GlmS uses glutamine as a nitrogen source and copurifies with YbgI. We compared the growth rates of E. coli ΔybgI [13] and wt cells in minimal medium containing various nitrogen sources (Fig. 5). We found that deletion of the ybgI gene did not affect the growth of cells for any of the nitrogen sources used.
Fig. 5. Doubling times (min) for wt and ΔybgI cells in the presence of different nitrogen sources.
Comparative proteomic analysis of the wt and ΔybgI cells might help to elucidate the role of YbgI protein in the cell. The bacteria were grown in the LB and M9 minimal media. Proteins from the wt cells were labeled with green fluorescent Cy3 dye; proteins from the ΔybgI cells – with red fluorescent Cy5 dye. Both dyes covalently bind to the side chain amino groups in proteins. The samples were fractionated by isoelectric focusing followed by electrophoresis in denaturing polyacrylamide gel. If the content of the protein remained the same, the corresponding zone in the gel was identified by its yellow color; if the level of protein expression changed, the color of the corresponding spot was either green or red (Fig. 6).
Fig. 6. Proteome of ΔybgI cells grown in LB and M9 media. Protein samples were prepared by mixing cell lysates of wt (Cy3-labeled, green fluorescence) and ΔybgI (Cy5-labeled, red fluorescence) cells and then fractionated by 2D-electrophoresis. Numbers indicate the proteins whose expression changes in ΔybgI cells (see also Table 2).

No differences were found between wt and ΔybgI cells grown in the LB medium under normal conditions. However, in the M9 minimal medium, the expression levels of eight proteins changed in the ΔybgI cells (Table 2). The expression of TnaA and AphA was upregulated, while expression of YgiW, YgaU, Dps, and OsmY was downregulated.
Table 2. Proteins whose expression is
affected by the ybgI gene deletion

Note: Numbers correspond to protein zones on Fig. 6.
To evaluate the role of ybgI in cell signaling pathways, we used several reporter plasmids encoding the fluorescent proteins CER and RFP [27]. In all the plasmid, the RFP gene was under control of the same promoter and had identical ribosome-binding sites, so it was used as an internal control. The CER gene was placed under control of genetic regulatory elements modulated by the cell signaling pathways (Table 3). Escherichia coli wt and ΔybgI cells were transformed with the reporter plasmids (Fig. 7) and grown in LB and M9 media.
Fig. 7. Fluorescence intensity of reporter genes CER/RFP in wt (black) and ΔybgI (grey) cells in LB and M9 media.
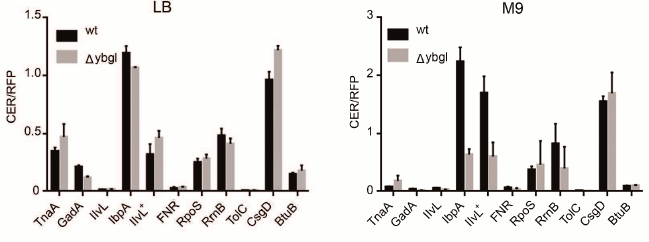
Table 3. Reporter plasmids for expression of
fluorescent proteins CER and RFP

In the ΔybgI cells grown in LB medium, the expression of only two reporter genes was altered; when the same cells were grown in M9 medium, the number of affected genes was higher (Table 4).
Table 4. Reporter constructs whose
expression was affected by ybgI gene deletion
(Коcтя,
берите эту
таблицу из
руccкой
верcии, там
она более
понятно
отформатирована)
To confirm the results of affinity chromatography, proteomic analysis, and differential expression of the reporter constructs, we analyzed the content of certain mRNAs in ΔybgI cells grown to the logarithmic phase in the minimal M9 medium, since these were the conditions under which the most pronounced changes were observed. The data were normalized to the content of 16S rRNA (Fig. 8).
Fig. 8. Ratio between mRNA contents in ΔybgI and wt cells.
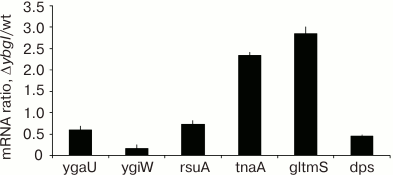
We found that in ΔybgI cells, expression of the tnaA and glmS genes was upregulated, while expression of ygaU, ygiW, and dps was downregulated. The amount of rsuA mRNA did not change. These data correlate well with the results of proteomic analysis and reporter gene expression.
Finally, we analyzed the resistance of ΔybgI cells to various antibiotics: carbenicillin, roxithromycin, levofloxacin, fosfomycin, vancomycin, novobiocin, nalidixic acid, and amoxicillin (Fig. 9, a and b). The choice of the tested antibiotics was based on published data on the sensitivity of ΔybgI cells to various factors [38].
Fig. 9. Zones of inhibition of growth of wt and ΔybgI cells by antibiotics: a) levofloxacin (L); fosfomycin (F); carbenicillin (C); roxithromycin (R); b) amoxicillin (A); vancomycin (V); nalidixic acid (N); novobiocin (No); c) quantification of the zone of antibiotics inhibition.
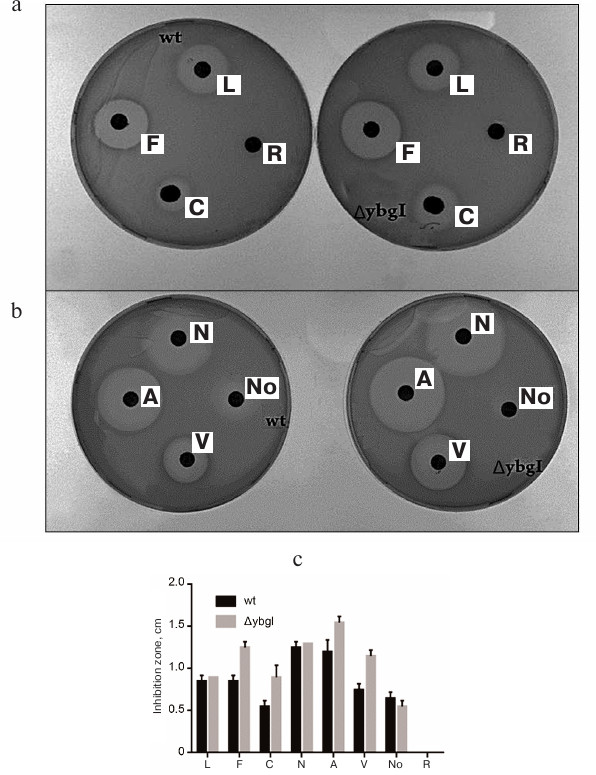
We found that deletion of the ybgI gene resulted in increase in the zone of inhibition for carbenicillin, fosfomycin, vancomycin, and amoxicillin (Fig. 9c). All these antibiotics affect the synthesis of bacterial cell wall [39].
DISCUSSION
There is very little published information on the function of YbgI protein. It was found that YbgI molecules have a toroidal structure, which led to the suggestion that the protein interacts with nucleic acids [5]. To test this hypothesis, we modeled the electrostatic charge distribution on the YbgI protein molecule surface and found that the surface is mostly negatively charged, which would prevent binding of nucleic acids. Another suggested role of YbgI is its involvement in cell response to DNA damage. However, we found that deletion of the ybgI gene did not affect cell survival after UV-induced DNA damage. Nevertheless, data were published on cell response to radiation-induced damage that demonstrated that YbgI might be involved in this process [8].
To elucidate the role of YbgI in cells, we used different approaches. First, we identified proteins that copurify with YbgI and found that the protein that most reliably interacts with YbgI is GlmS. Second, we analyzed the proteome of ΔybgI cells and showed that the amount of YgiW, OsmY, YgaU, and Dps proteins decreased, while amount of TnaA and AphA proteins increased compared to the wt cells. Analysis of the gene expression by RT-PCR confirmed upregulation of tnaA and glmS gene expression and downregulation of ygaU, ygiW, and dps gene expression in ΔybgI cells. Let us consider these changes in more detail. GlmS is an enzyme that catalyzes the first stage of biosynthesis of hexosamine, which is an essential precursor of cell wall peptidoglycan [40]. TnaA is a component of the system for tryptophan decomposition into indole, an important signaling molecule in bacteria [41]. GadA is a glutamate decarboxylase involved in the glutamate-dependent mechanism of bacterial cell resistance to acidification of the medium [42]. YgiW is a component of the cell response to peroxide stress [22, 43]. A homolog of this protein binds to the cell wall peptidoglycan [44]. YgaU is a potassium-binding protein; its deletion affects the structure of the cell wall peptidoglycans [45]. OsmY and Dps are involved in cell stress response; AphA and PheA participate in the biosynthesis of amino acids involving L-glutamate. We found that ΔybgI cells are especially sensitive to the antibiotics that affect the cell wall biosynthesis: carbenicillin (interferes with peptidoglycan binding), fosfomycin (suppresses the first stage of cell wall biosynthesis), amoxicillin (inhibits transpeptidase needed for the suppression of peptidoglycan biosynthesis), and vancomycin (changes permeability of the cell wall) [39].
Peptidoglycans of bacterial cell wall are composed of N-acetylglucosamine and N-acetylmuramic acid connected with short peptide chains via N-acetylmuramic acid lactate residues [46]. The peptides are composed mainly of alanine, glutamine, and lysine residues. This three-dimensional structure makes the bacterial cell wall rigid and protects the cell from osmotic stress. Our results demonstrate that the loss of the ΔybgI gene leads to the changes in the translation and transcription of genes involved in the biosynthesis and function of bacterial cell wall peptidoglycans. The observed effects of various antibiotics on ΔybgI cells confirmed the suggestion of the involvement of YbgI in the formation of bacterial cell wall in E. coli. It is also possible that YbgI is an additional, rather than essential protein, since its deletion is not lethal, but it causes very slight changes in the cell functioning. It is possible that YbgI has several functions that are manifested under normal and stress conditions [5].
Acknowledgments
This work was supported by the Russian Science Foundation (project 14-14-00072) and the Russian Foundation for Fundamental Research (projects 16-04-01100 and 17-00-00366).
REFERENCES
1.Jiang, D., Hatahet, Z., Blaisdell, J. O., Melamede,
R. J., and Wallace, S. S. (1997) Escherichia coli endonuclease
VIII: cloning, sequencing, and overexpression of the nei
structural gene and characterization of nei and nei nth
mutants, J. Bacteriol., 179, 3773-3782.
2.Jiang, D., Hatahet, Z., Melamede, R. J., Kow, Y.
W., and Wallace, S. S. (1997) Characterization of Escherichia
coli endonuclease VIII, J. Biol. Chem., 272,
32230-32239.
3.Pfam: Protein families database of alignments and
HMMs, https://pfam.xfam.org/.
4.Galperin, M. Y., and Koonin, E. V. (2004) Conserved
hypothetical proteins: prioritization of targets for experimental
study, Nucleic Acids Res., 32, 5452-5463.
5.Ladner, J. E., Obmolova, G., Teplyakov, A., Howard,
A. J., Khil, P. P., Camerini-Otero, R. D., and Gilliland, G. L. (2003)
Crystal structure of Escherichia coli protein ybgI, a toroidal
structure with a dinuclear metal site, BMC Struct. Biol.,
3, 7.
6.Martens, J. A., Genereaux, J., Saleh, A., and
Brandl, C. J. (1996) Transcriptional activation by yeast PDR1p is
inhibited by its association with NGG1p/ADA3p, J. Biol. Chem.,
271, 15884-15890.
7.Tascou, S., Uedelhoven, J., Dixkens, C., Nayernia,
K., Engel, W., and Burfeind, P. (2000) Isolation and characterization
of a novel human gene, NIF3L1, and its mouse ortholog,
Nif3l1, highly conserved from bacteria to mammals, Cytogen.
Genome Res., 90, 330-336.
8.Khil, P. P., and Camerini-Otero, R. D. (2002) Over
1000 genes are involved in the DNA damage response of Escherichia
coli, Mol. Microbiol., 44, 89-105.
9.Byrne, R. T., Chen, S. H., Wood, E. A., Cabot, E.
L., and Cox, M. M. (2014) Escherichia coli genes and
pathways involved in surviving extreme exposure to ionizing radiation,
J. Bacteriol., 196, 3534-3545.
10.Baker, N. A., Sept, D., Simpson, J., Holst, M.
J., and McCammon, A. J. (2001) Electrostatics of nanosystems:
application to microtubules and the ribosome, Proc. Natl. Acad. Sci.
USA, 98, 10037-10041.
11.DeLano, W. L. (2002) The PyMOL Molecular
Graphics System, DeLano Scientific, San Carlos, USA.
12.Kitagawa, M., Ara, T., Arifuzzaman, M.,
Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H., and Mori, H. (2005)
Complete set of ORF clones of Escherichia coli ASKA library (a
complete set of E. coli K-12 ORF archive): unique resources for
biological research, DNA Res., 12, 291-299.
13.Baba, T., Ara, T., Hasegawa, M., Takai, Y.,
Okumura, Y., Baba, M., Datsenko, K. A., Tomita, M., Wanner, B. L., and
Mori, H. (2006) Construction of Escherichia coli K-12
in-frame, single-gene knockout mutants: the Keio collection, Mol.
Syst. Biol., 2, 2006.0008; Epub 2006 Feb 21.
14.Bradford, M. M. (1976) A rapid and sensitive
method for the quantitation of microgram quantities of protein
utilizing the principle of protein–dye binding, Anal.
Biochem., 72, 248-254.
15.Ghosh, A., and Bansal, M. (2003) A glossary of
DNA structures from A to Z, Acta Crystallogr. D Biol.
Crystallogr., 59, 620-626.
16.Gascon, J., Oubina, A., Perez-Lezaun, A., and
Urmeneta, J. (1995) Sensitivity of selected bacterial species to UV
radiation, Curr. Microbiol., 30, 177-182.
17.Iida, A., Teshiba, S., and Mizobuchi, K. (1993)
Identification and characterization of the tktB gene encoding a
second transketolase in Escherichia coli K-12, J.
Bacteriol., 175, 5375-5783.
18.Gardner, S. G., Johns, K. D., Tanner, R., and
McCleary, W. R. (2012) The PhoU protein from Escherichia
coli interacts with PhoR, PstB, and metals to form a
phosphate-signaling complex at the membrane, J. Bacteriol.,
196, 1741-1752.
19.Brooks, K. M., and Hampel, K. J. (2011) Rapid
steps in the glmS ribozyme catalytic pathway: cation and ligand
requirements, Biochemistry, 50, 2424-2433.
20.Dutka-Malen, S., Mazodier, P., and Badet,
B. (1988) Molecular cloning and overexpression of the
glucosamine synthetase gene from Escherichia coli,
Biochimie, 70, 287-290.
21.Watanabe, T., and Snell, E. E. (1977) The
interaction of Escherichia coli tryptophanase with
various amino and their analogs. Active site mapping, J.
Biochem., 82, 733-745.
22.Lee, J., Hiibel, S. R., Reardon, K. F., and Wood,
T. K. (2010) Identification of stress-related proteins in
Escherichia coli using the pollutant
cis-dichloroethylene, J. Appl. Microbiol., 108,
2088-2102.
23.Ashraf, K. U., Josts, I., Mosbahi, K., Kelly, S.
M., Byron, O., Smith, B. O., and Walker, D. (2016) The potassium
binding protein Kbp is a cytoplasmic potassium sensor,
Structure, 24, 741-749.
24.Almiron, M., Link, A. J., Furlong, D., and
Kolter, R. (1992) A novel DNA-binding protein with regulatory and
protective roles in starved Escherichia coli, Genes Dev.,
6, 2646-2654.
25.Yim, H. H., and Villarejo, M. (1992) OsmY,
a new hyperosmotically inducible gene, encodes a periplasmic protein in
Escherichia coli, J. Bacteriol., 174,
3637-3644.
26.Thaller, M. C., Schippa, S., Bonci, A., Cresti,
S., and Rossolini, G. M. (1997) Identification of the gene
(aphA) encoding the class B acid phosphatase/phosphotransferase
of Escherichia coli MG1655 and characterization of its product,
FEMS Microbiol. Lett., 146, 191-198.
27.Osterman, I. A., Prokhorova, I. V., Sysoev, V.
O., Boykova, Y. V., Efremenkova, O. V., Svetlov, M. S., Kolb, V. A.,
Bogdanov, A. A., Sergiev, P. V., and Dontsova, O. A. (2012)
Attenuation-based dual-fluorescent-protein reporter for screening
translation inhibitors, Antimicrob. Agents Chemother.,
56, 1774-1783.
28.Li, G., and Young, K. D. (2015) A new suite of
tnaA mutants suggests that Escherichia coli tryptophanase
is regulated by intracellular sequestration and by occlusion of its
active site, BMC Microbiol., 15, 14.
29.Moreau, P. L. (2007) The lysine decarboxylase
CadA protects Escherichia coli starved of phosphate
against fermentation acids, J. Bacteriol., 189,
2249-2261.
30.Vitreschak, A. G., Lyubetskaya, E. V., Shirshin,
M. A., Gelfand, M. S., and Lyubetsky, V. A. (2004) Attenuation
regulation of amino acid biosynthetic operons in proteobacteria:
comparative genomics analysis, FEMS Microbiol. Lett.,
234, 357-370.
31.Laskowska, E., Wawrzynow, A., and Taylor, A.
(1996) IbpA and IbpB, the new heat-shock proteins, bind to endogenous
Escherichia coli proteins aggregated intracellularly by
heat shock, Biochimie, 78, 117-122.
32.Unden, G., and Schirawski, J. (1997) The
oxygen-responsive transcriptional regulator FNR of Escherichia
coli: the search for signals and reactions, Mol. Microbiol.,
25, 205-210.
33.Maciag, A., Peano, C., Pietrelli, A., Egli, T.,
De Bellis, G., and Landini, P. (2011) In vitro transcription
profiling of the σS subunit of bacterial RNA polymerase:
re-definition of the σS regulon and identification of
σS-specific promoter sequence elements, Nucleic Acids
Res., 39, 5338-5355.
34.Gralla, J. D. (2005) Escherichia coli
ribosomal RNA transcription: regulatory roles for ppGpp, NTPs,
architectural proteins and a polymerase-binding protein, Mol.
Microbiol., 55, 973-977.
35.Wiriyathanawudhiwong, N., Ohtsu, I., Li, Z. D.,
Mori, H., and Takagi, H. (2009) The outer membrane TolC is involved in
cysteine tolerance and overproduction in Escherichia coli,
Appl. Microbiol. Biotech., 81, 903-913.
36.Hammar, M., Arnqvist, A., Bian, Z., Olsen, A.,
and Normark, S. (1995) Expression of two csg operons is
required for production of fibronectin- and Congo Red-binding curli
polymers in Escherichia coli K-12, Mol. Microbiol.,
18, 661-670.
37.Roth, J. R., Lawrence, J. G., and Bobik, T. A.
(1996) Cobalamin (coenzyme B12): synthesis and biological significance,
Annu. Rev. Microbiol., 50, 137-181.
38.Liu, A., Tran, L., Becket, E., Lee, K., Chinn,
L., Park, E., Tran, K., and Miller, J. H. (2010) Antibiotic sensitivity
profiles determined with an Escherichia coli gene knockout
collection: generating an antibiotic bar code, Antimicrob. Agents
Chemother., 54, 1393-1403.
39.Bush, K. (2012) Antimicrobial agents targeting
bacterial cell walls and cell membranes, Rev. Sci. Tech.,
31, 43-56.
40.Barreteau, H., Kovac, A., Boniface, A., Sova, M.,
Gobec, S., and Blanot, D. (2008) Cytoplasmic steps of peptidoglycan
biosynthesis, FEMS Microbiol. Rev., 32, 168-207.
41.Awano, N., Wada, M., Kohdoh, A., Oikawa, T.,
Takagi, H., and Nakamori, S. (2005) Effect of cysteine desulfhydrase
gene disruption on L-cysteine overproduction in Escherichia
coli, Appl. Microbiol. Biotech., 62, 239-243.
42.De Biase, D., Tramonti, A., John, R. A., and
Bossa, F. (1996) Isolation, overexpression, and biochemical
characterization of the two isoforms of glutamic acid decarboxylase
from Escherichia coli, Protein Expr. Purif.,
8, 430-438.
43.Juarez-Rodriguez, M. D., Torres-Escobar, A., and
Demuth, D. R. (2013) ygiW and qseBC are co-expressed in Aggregatibacter
actinomycetemcomitans and regulate biofilm growth, Microbiology, 159,
989-1001.
44.Moreira, C. G., Herrera, C. M., Needham, B. D.,
Parker, C. T., Libby, S. J., Fang, F. C., Trent, M. S., and Sperandio,
V. (2013) Virulence and stress-related periplasmic protein (VisP) in
bacterial/host associations, Proc. Natl. Acad. Sci. USA,
110, 1470-1475.
45.Bernal-Cabas, M., Ayala, J. A., and Raivio, T. L.
(2015) The Cpx envelope stress response modifies peptidoglycan
cross-linking via the L,D-transpeptidase LdtD and the novel protein
YgaU, J. Bacteriol., 197, 603-614.
46.Vollmer, W., and Bertsche, U. (2008) Murein
(peptidoglycan) structure, architecture and biosynthesis in
Escherichia coli, Biochim. Biophys. Acta, 1778,
1714-1734.
Supplemental Data (PDF)