REVIEW: Effect of Environmental Factors on Nuclear Organization and Transformation of Human B Lymphocytes
F. B. Sall1,2,3#, D. Germini1,2#, A. P. Kovina1,2,4, V. Ribrag5,6, J. Wiels1,2, A. O. Toure3, O. V. Iarovaia2,4, M. Lipinski1,2, and Y. Vassetzky1,2,7*
1UMR8126, Université Paris-Sud, CNRS, Institut de Cancérologie Gustave Roussy, 94805 Villejuif, France; E-mail: vassetzky@igr.fr, vassetzky@gmail.com2LIA1066 “Laboratoire Franco–Russe de Recherche en Oncologie”, 94805 Villejuif, France
3Laboratoire d’Hématologie Centre Hospitalier Universitaire Aristide Le Dantec, Université Cheikh Anta Diop, Dakar, Sénégal
4Institute of Gene Biology, Russian Academy of Sciences, 119334 Moscow, Russia
5Institut Gustave Roussy, 94805 Villejuif, France
6Institut National de la Santé et de la Recherche Médicale (INSERM) Unité (U) 1009, Université Paris Sud, Institut Gustave Roussy, 94805 Villejuif, France
7Koltzov Institute of Developmental Biology, Russian Academy of Sciences, 119334 Moscow, Russia
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received November 10, 2017; Revision received December 8, 2017
Chromosomal translocations have long been known for their association with malignant transformation, particularly in hematopoietic disorders such as B-cell lymphomas. In addition to the physiological process of maturation, which creates double strand breaks in immunoglobulin gene loci, environmental factors including the Epstein–Barr and human immunodeficiency viruses, malaria-causing parasites (Plasmodium falciparum), and plant components (Euphorbia tirucalli latex) can trigger a reorganization of the nuclear architecture and DNA damage that together will facilitate the occurrence of deleterious chromosomal rearrangements.
KEY WORDS: Burkitt lymphoma, EBV, HIV, Plasmodium falciparum, Euphorbia tirucalli, oncogenesis, nuclear organizationDOI: 10.1134/S0006297918040119
Abbreviations: AICDA, activation-induced cytosine deaminase; BL, Burkitt lymphoma; CSR, class switch recombination; DSBs, DNA double strand breaks; EBV, Epstein–Barr virus; HIV, human immunodeficiency virus; NHEJ, non-homologous end joining; RAG1(2), recombination activating gene 1(2); ROS, reactive oxygen species; SHM, somatic hypermutation.
In humans, malignant transformation is often associated with specific
chromosomal translocations. Those result from two DNA double strand
breaks (DSBs) erroneously repaired through non-homologous end joining
(NHEJ). Proximity (colocalization) of the two translocation partners in
the 3D nuclear space is necessary for translocations to take place.
DSBs can be generated by a variety of exogenous factors including ionizing radiations, chemotherapeutic drugs, and infectious agents or toxins. The production of reactive oxygen species (ROS) or enzyme-dependent physiological processes such as the chromosomal rearrangements that occur during the normal maturation of B and T lymphocytes also generate DNA breakage.
DYNAMICS OF NUCLEAR ORGANIZATION, DNA DAMAGE, AND
TRANSLOCATIONS
With chromosomes nonrandomly arranged in the nuclear space and occupying so-called chromosome territories [1], genomes are spatially organized in tissue- and cell type-specific ways [2, 3]. In general and regardless of the proliferation status of the cell, gene-rich and smaller chromosomes tend to be located in central areas of the nucleus, while gene-poor and larger chromosomes mostly lie in the nuclear periphery [2, 4-6]. Gene loci also exhibit nonrandom and gene-specific distribution patterns in the interphase nucleus [7]. The interaction between distinct chromosomes can activate genes in trans resulting in colocalization of actively transcribed gene loci on different chromosomes [8]. The precise positioning of genes in the nuclear space, which is important for essential DNA transactions including transcription, replication, and repair ([9, 10], reviewed in [11]), evolves during development, differentiation, and aging, and during cellular responses to stress and various stimuli. The molecular factors that control this 3D (re)positioning remain largely unknown.
Chromosomal movements are known to accompany DNA repair, creating the possibility that two loci normally located away from each other are brought together [12-14]. Two alternative models have been proposed. In the “contact-first” model, both chromosomal partners lie in the vicinity of each other at the time of DNA damage. Live imaging analysis has shown that more than 80% of the translocations are created when DNA breaks are produced on two different chromosomes within a distance less than 2.5 μm [15]. Translocations are less frequent if the breaks are separated by 5 μm or more [15]. Usually, neither damaged nor intact chromatin fibers undergo extensive movements [12, 16]. However, the “break-first” model postulates that chromosomes are first broken away from each other and then move in the nuclear space and find their translocation partner. Indeed, under certain circumstances (e.g. large scale DNA damage or the intervention of the DNA repair factors) an enhanced mobility has been demonstrated for damaged DNA [13, 17]. As often, “break-first” and “contact-first” mechanisms can combine to produce a translocation. In this respect, we reported recently that inducing DSBs in the MYC locus in peripheral B-cells incubated in the presence of an HIV protein results in MYC relocalizing proximal to IGH gene loci (see further below) [18].
Recently, epigenetic modifications have been shown to have an impact on the susceptibility of chromatin to undergo DNA DSBs and on the movements of broken chromosomes [19]. Histone posttranslational modifications that regulate gene transcription have such an impact on chromosome breakages and ensuing translocations [20]. It has been reported that the methylation of K4 and K36 on histone H3 facilitates chromatin decondensation and also DSB formation [21]. In addition, chromosomal regions enriched in H3K4me3 constitute preferential targets for two enzymes, Recombination Activating Gene (RAG) 2 and Activation-Induced Cytosine Deaminase (AICDA) involved in V(D)J recombination and Class Switch Recombination (CSR) in normal B cells [22, 23]. That DSBs occur predominantly in transcriptionally active regions [24-26] comes as no surprise, since the recruitment of transcription factors further opens the chromatin structure. In turn, such sites become more susceptible to DNA damage and prone to translocations [27, 28].
Transcription is also regulated by the methylation/demethylation of genomic DNA within CpG islands present in as much as 70% of gene promoters [29]. Interestingly, 40 to 70% of the breakpoints specifically associated with pro-B and/or pre-B leukemias and lymphomas are found in the vicinity of such CpG-rich regions [30]. Moreover, independently of their methylation status, the cytosine residues in the CpG islands are potential targets for AICDA, which can induce their deamination resulting in single-nucleotide mismatches that in turn become targets for nucleases including RAG1/2 [31].
Transcription is not the only process that leads to chromatin decondensation. Local chromatin structures can be modified by many chromatin remodeling complexes involved in different DNA transactions [32, 33]. Along with these changes, large-scale chromatin motions occur due to the increased flexibility of the chromatin fiber [33]. All together, these findings suggest the existence of a functional relationship between mechanisms of epigenetic regulation, the occurrence of DSBs, and the deleterious production of chromosomal rearrangements.
BURKITT LYMPHOMA AND NUCLEAR GENE RELOCALIZATION
Burkitt lymphoma (BL) is a highly aggressive B-cell lymphoma with three clinically distinct presentations [34, 35]. The endemic form is observed in children in Central Africa, particularly in areas with holoendemic malaria where it is virtually always associated with an infection by the Epstein–Barr virus (EBV) [36, 37]. The sporadic form of BL seen all over the world in patients usually in their teens, has an EBV prevalence of ~10-20% [34]. The third form, which occurs in HIV-infected patients, exhibits an intermediary level (~40-50%) of EBV prevalence [34, 36, 38]. What is constant in all three forms, is the existence of one among three possible chromosomal translocations, all involving the MYC oncogene on chromosome 8 rearranged either with the immunoglobulin (Ig) heavy chain gene locus (IGH) on chromosome 14, or with one of the two chromosomes carrying the Ig light chain genes IGK on chromosome 2 or IGL on chromosome 22. The t(8;14) translocation is the most frequent, being found in ~80% of the cases. The resulting juxtaposition of MYC and IGH leads to hyperactivation of MYC [38-40] via its relocalization from the periphery towards a perinucleolar region [41] in a more central position in the nucleus [42]. Similarly to MYC, IGK is often found near the nuclear periphery, but not IGL, which resides closer to the nuclear center while IGH is mostly found in regions located in-between [43]. Thus, the frequency of MYC rearrangement with either one of the three Ig gene loci could be directly related to its relative proximity with each of them [18, 43, 44].
In normal peripheral blood B-cells, the IGH and MYC gene loci are separated by approximately 40% of the nuclear diameter [18, 41, 43]. At the time of Somatic Hypermutation (SHM) and CSR in both human and murine B-cells, MYC relocalizes close to an IGH locus [41, 45] in the vicinity of the so-called “recombination compartment”, which is enriched in AICDA [46], one enzyme absolutely required for Ig gene maturation [47, 48]. Through its enzymatic activity, AICDA deaminates cytidines and produces U:G mismatches [49]. It can also target and modify regions outside IGH, as demonstrated when overexpressed in murine activated B-cells [47, 48, 50]. Indeed, proximity to IGH predisposes to AICDA targeting with 90% of the AICDA hotspots being found in gene loci located in the vicinity of the IGH locus [50]. AICDA can thus induce chromosomal breaks in the MYC region [47, 48], which in turn increases the probability of IGH/MYC translocations. Below, we will briefly review various environmental factors that have been shown to increase the risk of occurrence of a BL-associated chromosomal translocation.
BL-ASSOCIATED ENVIRONMENTAL FACTORS AND NUCLEAR
ORGANIZATION
Viruses (EBV and HIV), the malaria parasite Plasmodium falciparum, and the latex of the plant Euphorbia tirucalli are some of the etiological factors that have been associated with the endemic form of BL, but the mechanism whereby these factors contribute to lymphomagenesis remain largely unknown. Their ability to affect the nuclear organization could play a role in BL oncogenesis. Here we will describe and summarize their impact on B-cells nuclear organization and the consequences in terms of lymphomagenesis.
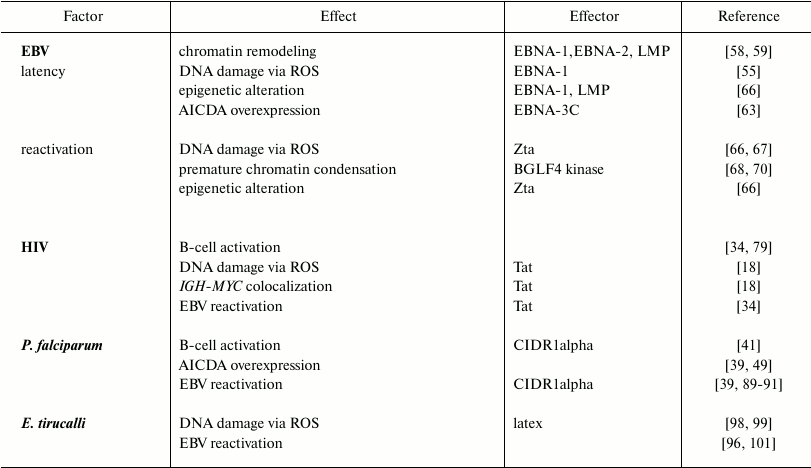
EBV. BL was the first human tumor associated with a virus [51]. The Epstein–Barr virus (EBV) is a human gamma-herpesvirus with a tropism for B lymphocytes and an ability to establish long term persistence in memory B-cells. The EBV life cycle includes two phases: viral particles are produced in the lytic phase; in the latent phase, extrachromosomal episome persists in the nucleus [52].
EBV can remodel the nuclear organization, changing the positioning of chromosomes in infected cells [53]. EBV-positive cells also exhibit additional chromosomal aberrations such as dicentric chromosomes, chromosome fragments, and chromatid gaps, suggesting that chromosome breaks are produced along with defects in DNA repair [54, 55].
EBV latency. The latent phase of EBV infection is characterized by expression of several nuclear antigens (Epstein–Barr Nuclear Antigen [EBNA]-1, -2, -3A, -3B, -3C, and -LP), membrane proteins (Latent Membrane Protein [LMP]-1, -2A, and -2B), and noncoding RNA (EBER-1 and -2) [56]. The EBV Nuclear Antigen-1 (EBNA-1), expressed in both the latent and lytic phases of EBV infection [57], was found to induce DSBs via production of ROS with consequent chromosomal instability [55]. Its expression leads to the deregulation of several genes involved in the maintenance of chromatin organization [58]. EBNA-1 can also induce chromatin decondensation [59]. EBNA-2, a transcriptional activator of both viral and cellular genes, is expressed during latent infection of B lymphocytes and is necessary for B-cell immortalization [60]. It can interact with histone acetyl transferases (HATs) and the SWI/SNF chromatin remodeler complex, thus disturbing the chromatin organization including in the upstream region of MYC [61]. EBNA-3 proteins also play an important role in B-cell immortalization [62]. EBNA-3C can activate the expression of AICDA by directly binding to upstream regulatory elements, where it provokes increased H3K4 trimethylation [63]. The latent protein LMP-1 participates in epigenetic regulation of the host genome by modulating the expression of three DNA methyltransferases – DNMT1, DNMT3A, and DNMT3B [64].
EBV reactivation from latency. The activation of BZLF1 gene transcription leads to the switch from the latent to the lytic phases. The BZLF1 protein (Zebra or Zta) is a transcriptional transactivator that promotes transcription of viral genes inducing the viral lytic cycle [65]. The Zta protein also induces oxidative stress-mediated DNA damage by interacting with p53 [66, 67]. Inducing the EBV lytic cycle distorts the nuclear architecture, resulting in chromatin condensation at the nuclear periphery and formation of transcription factories in the nuclear center [68]. BGLF4 kinase, another key protein for EBV reactivation, phosphorylates and activates BZLF [69]. BGLF4 may play a role in marginal chromatin condensation by activating DNA topoisomerase II and phosphorylating condensin [70]. This could increase the risk of DNA breaks, as premature chromatin condensation is associated with a high risk of chromosomal breaks at common fragile sites [71].
The genome of the EBV resembles a small human chromosome in many ways. During latency, the EBV genome interacts with repressive heterochromatin, but upon spontaneous reactivation when EBV transcription drastically increases, the episome leaves the repressive environment and surrounds itself with active chromatin, leading to rearrangement of neighboring gene loci [72]. EBV can also integrate into the host genome [73] in sites located inside or in the vicinity of regions that harbor potential genome structural variations [74]. Thus, EBV integration can induce genome instability.
HIV-1. Infection by HIV-1 is associated with a significantly elevated incidence of cancer, particularly B-cell lymphomas [75, 76]. In Europe and in the US, the BL incidence is significantly higher in HIV-infected individuals compared with healthy subjects [18, 77]. This is also true for BL endemic zones where BL patients are 10-12 times more prone to be HIV-positive than healthy people [78]. Intriguingly, HIV cannot infect B-cells. Thus, the elevated risk of BL in HIV patients can be caused either by immunodeficiency induced by AIDS or by HIV having a lymphomagenic role by itself.
According to one hypothesis, HIV induces chronic B-cell activation through an immune dysfunction leading to a deregulated clonal expansion of B lymphocytes [79]. Uncontrolled persistent stimulation of B lymphocytes may favor the uncontrolled monoclonal proliferation with as a consequence an increased risk of acquiring critical genetic alterations, ultimately leading to lymphoma development [79]. As already mentioned, AICDA is overexpressed in hyperactivated B-cells [48, 80]. Moreover, in EBV-infected patients, HIV infection can lead to EBV lytic cycle reactivation [34]. In these conditions of immunodepression induced by HIV, EBV-induced cell proliferation fails to be fully controlled by the immune system, resulting in an increased number of latently infected B-cells [34, 81].
Alternatively, since the risk of BL remains elevated in patients under combined antiretroviral therapy (cART) and with normal CD4 counts [82, 83], a direct role of HIV in BL oncogenesis can be postulated. Our recent studies have demonstrated that the HIV-1 transactivator protein Tat, a small protein that is secreted into blood by infected cells and capable of penetrating B-cells, can modify their nuclear organization. It induces the displacement of one MYC allele from the periphery of the nucleus to the center, in close proximity with IGH, as a consequence of aberrant activation of RAG1 and MYC [18]. This proximity between MYC and IGH could predispose to BL development. Tat can also significantly increase oxidative stress-derived DNA damage in B-cells leading to a general chromosomal instability, which could potentially induce diffuse large B-cells lymphoma or Hodgkin lymphoma [84].
Plasmodium falciparum. The association of malaria with endemic BL, the most common tumor in young children in tropical Africa, was observed for the first time almost 50 years ago [39, 85]. Malarial infection is caused by a protozoan parasite of the genus Plasmodium. It is transmitted by a bite of a female mosquito of the genus Anopheles, mostly present in hot areas. Even though several species of Plasmodium exist, only five can cause the disease in humans [86]. Among them, the P. falciparum species is associated with a high morbidity and mortality in Africa [87]. Such infections can profoundly affect B-cell metabolism, promoting polyclonal activation and an abnormal production of antibodies. Plasmodium falciparum-infected erythrocytes express erythrocyte membrane protein 1 (PfEMP1) [88], whose cystein-rich inter-domain region 1alpha can activate B-cells [89]. The MYC and IGH loci move close to each other in activated B-cells [41, 45], thus the probability of the t(8;14) translocation is increased. Moreover, P. falciparum and P. chabaudi can induce an aberrant expression of AICDA in germinal center B-cells in both humans and mice [39, 49]. As a consequence, mice chronically infected with P. chabaudi exhibit a widespread genomic instability and develop mature B-cell lymphomas [49].
Also, the memory B-cells that are most susceptible to the CIDR1alpha domain-mediated activation often harbor a latent EBV. It has been demonstrated that CIDR1alpha can also induce EBV reactivation and its lytic cycle [89]. Indeed, chronic and repeated exposure to P. falciparum leads to virus reactivation from latency, an increased number of latently infected B cells in germinal centers, and elevated EBV viral load [36, 39, 90, 91]. In addition, P. falciparum alters the immune surveillance against EBV since exposure to holoendemic malaria induces suppression of EBV-specific T cell immunosurveillance [92, 93] and defects in EBV-specific CD8+ T-cell differentiation [94].
Euphorbia tirucalli. Euphorbia tirucalli, a plant belonging to the Euphorbiaceae family, is commonly used as an ornamental or hedge plant. It is endemic in African countries including Angola, Erythrea, Ethiopia, Kenya, Malawi, Mauritius, Rwanda, Senegal, Sudan, Tanzania, Uganda, and Zanzibar [95]. Interestingly, its geographical distribution is very similar to that of the endemic BL [96]. The plant has been frequently observed in houses of BL patients in the Eastern Africa [97], suggesting a possible link with BL pathogenesis. Little evidence exists on a direct role of E. tirucalli in modifying the nuclear organization. However, plant extracts have a high genotoxic potential and can increase DNA breakage and oxidative damage in cell cultures of human leukocytes in a dose-dependent manner [98, 99]. Euphorbia tirucalli can also alter the nuclear organization in B-cells by activating the EBV lytic cycle, as it contains EBV-activating substances such as 4-deoxyphorbol ester [95-97, 100]. When added to EBV-positive B-lymphocytes, E. tirucalli extracts induced chromosomal abnormalities including ones on chromosome 8 [96, 101]. Finally, B-cells treated with the E. tirucalli extracts produced lymphomas when injected into nude mice [97].
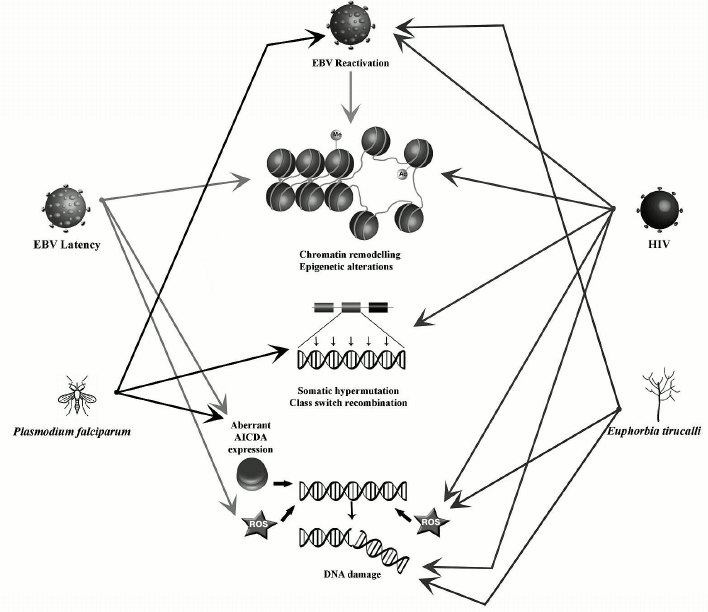
Nuclear organization and chromosome positioning in the nuclear space are characteristic features of different cell types. Their modification by biological processes (e.g. epigenetic modifications, presence of DNA breakage) can result in increased risks of oncogenic translocations. In B-cells, recombination events that naturally occur during the processes of maturation and antibody formation can also alter the nuclear organization. These processes are affected by exogenous factors like EBV, HIV-1, the malaria parasites, or Euphorbia tirucalli. They can induce deregulation of chromatin-related genes, epigenetic changes, and excessive DNA damage provoking large-scale nuclear remodeling, which can potentially lead to the development of BL-specific translocations. The main changes introduced by each of these factors are summarized in the table and figure. However, knowledge of the exact role(s) played by these factors in the generation of BL-specific translocation and BL oncogenesis is still incomplete. Further studies could help in understanding the mechanisms leading to the Burkitt lymphoma etiopathogenesis.
Effects induced on host cells by BL etiological factors
Factors affecting processes implicated in oncogenesis and nuclear architecture remodeling
Acknowledgments
This research was supported by grants from the ANRS, INSERM (ENVIBURKITT), La Ligue Contre le Cancer (M27231) to Y.S.V. and the Russian Science Foundation (14-24-00022) to O.V.I.; A.P.K. was a recipient of a FEBS fellowship.
REFERENCES
1.Cremer, M., Von Hase, J., Volm, T., Brero, A.,
Kreth, G., Walter, J., Ficher, C., Solovei, I., Cremer, C., and Cremer,
T. (2001) Non-random radial higher-order chromatin arrangements in
nuclei of diploid human cells, Chromosome Res., 9,
541-567.
2.Parada, L. A., McQueen, P. G., and Misteli, T.
(2004) Tissue-specific spatial organization of genomes, Genome
Biol., 5, 44.
3.Lin, C., Yang, L., and Rosenfeld, M. G. (2012)
Molecular logic underlying chromosomal translocations, random or
non-random? Adv. Cancer Res., 113, 241-279.
4.Chen, B., Yusuf, M., Hashimoto, T., Estandarte, A.
K., Thompson, G., and Robinson, I. (2017) Three-dimensional positioning
and structure of chromosomes in a human prophase nucleus, Sci.
Adv., 3, e1602231.
5.Croft, J. A., Bridger, J. M., Boyle, S., Perry, P.,
Teague, P., and Bickmore, W. A. (1999) Differences in the localization
and morphology of chromosomes in the human nucleus, J. Cell
Biol., 145, 1119-1131.
6.Cavalli, G., and Misteli, T. (2013) Functional
implications of genome topology, Nat. Struct. Mol. Biol.,
20, 290-299.
7.Meaburn, K. J., Misteli, T., and Soutoglou, E.
(2007) Spatial genome organization in the formation of chromosomal
translocations, Semin. Cancer Biol., 17, 80-90.
8.Fanucchi, S., Shibayama, Y., Burd, S., Weinberg, M.
S., and Mhlanga, M. M. (2013) Chromosomal contact permits transcription
between coregulated genes, Cell, 155, 606-620.
9.Therizols, P., Illingworth, R. S., Courilleau, C.,
Boyle, S., Wood, A. J., and Bickmore, W. A. (2014) Chromatin
decondensation is sufficient to alter nuclear organization in embryonic
stem cells, Science, 346, 1238-1242.
10.Gonzalez-Sandoval, A., Towbin, B. D., Kalck, V.,
Cabianca, D. S., Gaidatzis, D., Hauer, M. H., Geng, L., Wang, L., Yang,
T., Wang, X., Zhao, K., and Gasser, S. M. (2015) Perinuclear anchoring
of H3K9-methylated chromatin stabilizes induced cell fate in C.
elegans embryos, Cell, 163, 1333-1347.
11.Bonev, B., and Cavalli, G. (2016) Organization
and function of the 3D genome, Nat. Rev. Genet., 17,
661-678.
12.Jakob, B., Splinter, J., Durante, M., and
Taucher-Scholz, G. (2009) Live cell microscopy analysis of
radiation-induced DNA double-strand break motion, Proc. Natl. Acad.
Sci. USA, 106, 3172-3177.
13.Krawczyk, P. M., Borovski, T., Stap, J., Cijsouw,
T., ten Cate, R., Medema, J. P., Kanaar, R., Franken, N. A., and Aten,
J. A. (2012) Chromatin mobility is increased at sites of DNA
double-strand breaks, J. Cell Sci., 125,
2127-2133.
14.Dion, V., Kalck, V., Horigome, C., Towbin, B. D.,
and Gasser, S. M. (2012) Increased mobility of double-strand breaks
requires Mec1, Rad9 and the homologous recombination machinery, Nat.
Cell Biol., 14, 502-509.
15.Roukos, V., Voss, T. C., Schmidt, C. K., Lee, S.,
Wangsa, D., and Misteli, T. (2013) Spatial dynamics of chromosome
translocations in living cells, Science, 341,
660-664.
16.Kruhlak, M. J., Celeste, A., Dellaire, G.,
Fernandez-Capetillo, O., Müller, W. G., McNally, J. G.,
Bazett-Jones, D. P., and Nussenzweig, A. (2006) Changes in chromatin
structure and mobility in living cells at sites of DNA double-strand
breaks, J. Cell Biol., 172, 823-834.
17.Aten, J. A., Stap, J., Krawczyk, P. M., van Oven,
C. H., Hoebe, R. A., Essers, J., and Kanaar, R. (2004) Dynamics of DNA
double-strand breaks revealed by clustering of damaged chromosome
domains, Science, 303, 92-95.
18.Germini, D., Tsfasman, T., Klibi, M., El-Amine,
R., Pichugin, A., Iarovaia, O. V., Bilhou-Nabera, C., Subra, F., Bou
Saada, Y., Sukhanova, A., Boutboul, D., Raphael, M., Wiels, J., Razin,
S. V., Bury-Mone, S., Oksenhendler, E., Lipinski, M., and Vassetzky, Y.
S. (2017) HIV Tat induces a prolonged MYC relocalization next to IGH in
circulating B-cells, Leukemia, 31, 2515-2522.
19.Roukos, V., and Misteli, T. (2014) The biogenesis
of chromosome translocations, Nat. Cell Biol., 16,
293-300.
20.Roukos, V., Burman, B., and Misteli, T. (2013)
The cellular etiology of chromosome translocations, Curr. Opin. Cell
Biol., 25, 357-364.
21.Burman, B., Zhang, Z. Z., Pegoraro, G., Lieb, J.
D., and Misteli, T. (2015) Histone modifications predispose genome
regions to breakage and translocation, Genes Dev., 29,
1393-1402.
22.Daniel, J. A., and Nussenzweig, A. (2012) Roles
for histone H3K4 methyltransferase activities during immunoglobulin
class-switch recombination, Biochim. Biophys. Acta, 1819,
733-738.
23.Shimazaki, N., Tsai, A. G., and Lieber, M. R.
(2009) H3K4me3 stimulates the V(D)J RAG complex for both nicking
and hairpinning in trans in addition to tethering in cis:
implications for translocations, Mol. Cell, 34,
535-544.
24.Chiarle, R., Zhang, Y., Frock, R. L., Lewis, S.
M., Molinie, B., Ho, Y. J., Myers, D. R., Choi, V. W., Compagno, M.,
Malkin, D. J., Neuberg, D., Monti, S., Giallourakis, C. C., Gostissa,
M., and Alt, F. W. (2011) Genome-wide translocation sequencing reveals
mechanisms of chromosome breaks and rearrangements in B-cells,
Cell, 147, 107-119.
25.Mathas, S., Kreher, S., Meaburn, K. J., Johrens,
K., Lamprecht, B., Assaf, C., Sterry, W., Kadin, M. E., Daibata, M.,
Joos, S., Hummel, M., Stein, H., Janz, M., Anagnostopoulos, I.,
Schrock, E., and Misteli, T. (2009) Gene deregulation and spatial
genome reorganization near breakpoints prior to formation of
translocations in anaplastic large cell lymphoma, Proc. Natl. Acad.
Sci. USA, 106, 5831-5836.
26.Klein, I. A., Resch, W., Jankovic, M., Oliveira,
T., Yamane, A., Nakahashi, H., Di Virgilio, M., Bothmer, A.,
Nussenzweig, A., Robbiani, D. F., Casellas, R., and Nussenzweig, M. C.
(2011) Translocation-capture sequencing reveals the extent and nature
of chromosomal rearrangements in B-lymphocytes, Cell,
147, 95-106.
27.Mathas, S., and Misteli, T. (2009) The dangers of
transcription, Cell, 139, 1047-1049.
28.Lin, C., Yang, L., Tanasa, B., Hutt, K., Ju, B.,
Ohgi, K., Zhang, J., Rose, D. W., Fu, X. D., Glass, C. K., and
Rosenfeld, M. G. (2009) Nuclear receptor-induced chromosomal proximity
and DNA breaks underlie specific translocations in cancer, Cell,
139, 1069-1083.
29.Deaton, A., and Bird, A. (2011) CpG islands and
the regulation of transcription, Genes Dev., 25,
1010-1022.
30.Tsai, A. G., Lu, H., Raghavan, S. C., Muschen,
M., Hsieh, C. L., and Lieber, M. R. (2008) Human chromosomal
translocations at CpG sites and a theoretical basis for their lineage
and stage specificity, Cell, 135, 1130-1142.
31.Nambiar, M., and Raghavan, S. C. (2011) How does
DNA break during chromosomal translocations? Nucleic Acids Res.,
39, 5813-5825.
32.Clapier, C. R., Iwasa, J., Cairns, B. R., and
Peterson, C. L. (2017) Mechanisms of action and regulation of
ATP-dependent chromatin-remodelling complexes, Nat. Rev. Mol.
Cell Biol., 18, 407-422.
33.Neumann, F. R., Dion, V., Gehlen, L. R.,
Tsai-Pflugfelder, M., Schmid, R., Taddei, A., and Gasser, S. M. (2012)
Targeted INO80 enhances subnuclear chromatin movement and ectopic
homologous recombination, Genes Dev., 26, 369-383.
34.Bornkamm, G. W. (2009) Epstein–Barr virus
and the pathogenesis of Burkitt’s lymphoma: more questions than
answers, Int. J. Cancer, 124, 1745-1755.
35.Mawson, A. R., and Majumdar, S. (2017) Malaria,
Epstein–Barr virus infection, and the pathogenesis of
Burkitt’s lymphoma, Int. J. Cancer, 141,
1849-1855.
36.Moormann, A. M., and Bailey, J. A. (2016) Malaria
– how this parasitic infection aids and abets EBV-associated
Burkitt lymphomagenesis, Curr. Opin. Virol., 20,
78-84.
37.Brady, G., MacArthur, G. J., and Farrell, P. J.
(2007) Epstein–Barr virus and Burkitt lymphoma, J. Clin.
Pathol., 60, 1397-1402.
38.Allday, M. J. (2009) How does Epstein–Barr
virus (EBV) complement the activation of myc in the pathogenesis
of Burkitt’s lymphoma? Semin. Cancer Biol., 19,
366-376.
39.Torgbor, C., Awuah, P., Deitsch, K., Kalantari,
P., Duca, K. A., and Thorley-Lawson, D. A. (2014) A multifactorial role
for P. falciparum malaria in endemic Burkitt’s lymphoma
pathogenesis, PLoS Pathog., 10, e1004170.
40.Bernard, O., Cory, S., Gerondakis, S., Webb, E.,
and Adams, J. M. (1983) Sequence of the murine and human cellular myc
oncogenes and two modes of myc transcription resulting from
chromosome translocation in B lymphoid tumours, EMBO J.,
2, 2375-2383.
41.Sklyar, I., Iarovaia, O. V., Gavrilov, A. A.,
Pichugin, A., Germini, D., Tsfasman, T., Caron, G., Fest, T., Lipinski,
M., Razin, S. V., and Vassetzky, Y. S. (2016) Distinct patterns of
colocalization of the CCND1 and CMYC genes with their
potential translocation partner IGH at successive stages of
B-cell differentiation, J. Cell. Biochem., 117,
1506-1510.
42.Allinne, J., Pichugin, A., Iarovaia, O., Klibi,
M., Barat, A., Zlotek-Zlotkiewicz, E., Saada, Y., Dib, C., Dmitriev,
P., Hamade, A., and Carnac, G. (2014) Perinucleolar relocalization and
nucleolin as crucial events in the transcriptional activation of key
genes in mantle cell lymphoma, Blood, 123, 2044-2053.
43.Roix, J. J., McQueen, P. G., Munson, P. J.,
Parada, L. A., and Misteli, T. (2003) Spatial proximity of
translocation-prone gene loci in human lymphomas, Nat. Genet.,
34, 287-291.
44.Nikiforova, M. N., Stringer, J. R., Blough, R.,
Medvedovic, M., Fagin, J. A., and Nikiforov, Y. E. (2000) Proximity of
chromosomal loci that participate in radiation-induced rearrangements
in human cell, Science, 290, 138-141.
45.Osborne, C. S., Chakalova, L., Mitchell, J. A.,
Horton, A., Wood, A. L., Bolland, D. J., Corcoran, A. E., and Fraser,
P. (2007) Myc dynamically and preferentially relocates to a
transcription factory occupied by IGH, PLoS Biol.,
5, e192.
46.Pichugin, A., Iarovaia, O. V., Gavrilov, A.,
Sklyar, I., Barinova, N., Barinov, A., Ivashkin, E., Caron, G.,
Aoufouchi, S., Razin, S. V., Fest, T., Lipinski, M., and Vassetzky, Y.
S. (2017) The IGH locus relocalizes to a “recombination
compartment” in the perinucleolar region of differentiating
B-lymphocytes, Oncotarget, 8, 16941.
47.Ramiro, A. R., Jankovic, M., Eisenreich, T.,
Difilippantonio, S., Chen-Kiang, S., Muramatsu, M., Honjo, T.,
Nussenzweig, A., and Nussenzweig, M. C. (2004) AID is required for
c-myc/IGH chromosome translocations in vivo,
Cell, 118, 431-438.
48.Robbiani, D. F., Bothmer, A., Callen, E.,
Reina-San-Martin, B., Dorsett, Y., Difilippantonio, S., Bolland, D. J.,
Chen, H. T., Corcoran, A. E., Nussenzweig, A., and Nussenzweig, M. C.
(2008) AID is required for the chromosomal breaks in c-myc that
lead to c-myc/IGH translocations, Cell,
135, 1028-1038.
49.Robbiani, D. F., Deroubaix, S., Feldhahn, N.,
Oliveira, T. Y., Callen, E., Wang, Q., Jankovic, M., Silva, I. T.,
Rommel, P. C., Bosque, D., and Eisenreich, T. (2015) Plasmodium
infectio promotes genomic instability and AID-dependent B-cell
lymphoma, Cell, 162, 727-737.
50.Rocha, P. P., Micsinai, M., Kim, J. R., Hewitt,
S. L., Souza, P. P., Trimarchi, T., Strino, F., Parisi, F., Kluger, Y.,
and Skok, J. A. (2012) Close proximity to IGH is a contributing
factor to AID-mediated translocations, Mol. Cell, 47,
873-885.
51.Epstein, M. A. (1965) Morphological and
biological studies on a virus in cultured lymphoblasts from
Burkitt’s lymphoma, J. Exp. Med., 121, 761-770.
52.Amon, W., and Farrell, P. J. (2005) Reactivation
of Epstein–Barr virus from latency, Rev. Med. Virol.,
15, 149-156.
53.Li, C., Shi, Z., Zhang, L., Huang, Y., Liu, A.,
Jin, Y., Lukasova, E., Kozubek, S., Kozubek, M., Kjeronska, J., Ryznar,
L., Horakova, J., and Krahulcova, E. (2010) Dynamic changes of
territories 17 and 18 during EBV-infection of human lymphocytes,
Mol. Biol. Rep., 37, 2347-2354.
54.Kamranvar, S. A., Gruhne, B., Szeles, A., and
Masucci, M. G. (2007) Epstein–Barr virus promotes genomic
instability in Burkitt’s lymphoma, Oncogene, 26,
5115-5123.
55.Gruhne, B., Sompallae, R., Marescotti, D.,
Kamranvar, S., Gastaldello, S., and Masucci, M. (2009) The
Epstein–Barr virus nuclear antigen-1 promotes genomic instability
via induction of reactive oxygen species, Proc. Natl. Acad. Sci.
USA, 106, 2313-2318.
56.Young, L. S., and Rickinson, A. B. (2004)
Epstein–Barr virus: 40 years on, Nat. Rev. Cancer,
4, 757-768.
57.Sivachandran, N., Wang, X., and Frappier, L.
(2012) Functions of the Epstein–Barr virus EBNA1 protein in viral
reactivation and lytic infection, J. Virol., 86,
6146-6158.
58.Sompallae, R., Callegari, S., Kamranvar, S. A.,
and Masucci, M. G. (2010) Transcription profiling of Epstein–Barr
virus nuclear antigen (EBNA)-1 expressing cells suggests targeting of
chromatin remodeling complexes, PLoS One, 5.
59.Coppotelli, G., Mughal, N., Callegari, S.,
Sompallae, R., Caja, L., Luijsterburg, M. S., Dantuma, N. P.,
Moustakas, A., and Masucci, M. G. (2013) The Epstein–Barr virus
nuclear antigen-1 reprograms transcription by mimicry of high mobility
group A proteins, Nucleic Acids Res., 41, 2950-2962.
60.Wu, D. Y., Kalpana, G. V., Goff, S. P., and
Schubach, W. H. (1996) Epstein–Barr virus nuclear protein 2
(EBNA2) binds to a component of the human SNF–SWI complex, J.
Virol., 70, 6020-6028.
61.Wood, C. D., Veenstra, H., Khasnis, S., Gunnell,
A., Webb, H. M., Shannon-Lowe, C., Andrews, S., Osborne, C. S., and
West, M. J. (2016) Myc activation and BCL2L11 silencing
by a tumour virus through the large-scale reconfiguration of
enhancer–promoter hubs, Elife, 5, e18270.
62.Chen, A., Zhao, B., Kieff, E., Aster, J. C., and
Wang, F. (2006) EBNA-3B- and EBNA-3C-regulated cellular genes in
Epstein–Barr virus-immortalized lymphoblastoid cell lines, J.
Virol., 80, 10139-10150.
63.Kalchschmidt, J. S., Bashford-Rogers, R.,
Paschos, K., Gillman, A. C. T., Styles, C. T., Kellam, P., and Alldae,
M. J. (2016) Epstein–Barr virus nuclear protein EBNA3C directly
induces expression of AID and somatic mutations in B-cells, J. Exp.
Med., 213, 921-928.
64.Leonard, S., Wei, W., Anderton, J., Vockerodt,
M., Rowe, M., Murray, P. G., and Woodman, C. B. (2011) Epigenetic and
transcriptional changes which follow Epstein–Barr virus infection
of germinal center B-cells and their relevance to the pathogenesis of
Hodgkin’s lymphoma, J. Virol., 85, 9568-9577.
65.Chang, Y. N., Dong, D. L., Hayward, G. S., and
Hayward, S. D. (1990) The Epstein–Barr virus Zta transactivator:
a member of the bZIP family with unique DNA-binding specificity and a
dimerization domain that lacks the characteristic heptad leucine zipper
motif, J. Virol., 64, 3358-3369.
66.Kgatle, M. M., Spearman, C. W., Kalla, A. A., and
Hairwadzi, H. N. (2017) DNA oncogenic virus-induced oxidative stress,
genomic damage, and aberrant epigenetic alterations, Oxid. Med.
Cell. Longev., 3179421.
67.Chen, X., Kamranvar, S. A., and Masucci, M. G.
(2016) Oxidative stress enables Epstein–Barr virus-induced B-cell
transformation by posttranscriptional regulation of viral and cellular
growth-promoting factors, Oncogene, 35, 3807-3816.
68.Chiu, Y. F., Sugden, A. U., and Sugden, B. (2013)
Epstein–Barr viral productive amplification reprograms nuclear
architecture, DNA replication, and histone deposition, Cell Host
Microbe, 14, 607-618.
69.Asai, R., Kato, A., Kato, K., Kanamori-Koyama,
M., Sugimoto, K., Sairenji, T., Nishiyama, Y., and Kawaguchi, Y. (2006)
Epstein–Barr virus protein kinase BGLF4 is a virion tegument
protein that dissociates from virions in a phosphorylation-dependent
process and phosphorylates the viral immediate-early protein BZLF1,
J. Virol., 80, 5125-5134.
70.Lee, C.-P., Chen, J.-Y., Wang, J.-T., Kimura, K.,
Takemoto, A., Lu, C.-C., and Chen, M. R. (2007) Epstein–Barr
virus BGLF4 kinase induces premature chromosome condensation through
activation of condensin and topoisomerase II, J. Virol.,
81, 5166-5180.
71.Achkar, E., Gerbault-Seureau, M., Muleris, M.,
Dutrillaux, B., and Debatisse, M. (2005) Premature condensation induces
breaks at the interface of early and late replicating chromosome bands
bearing common fragile sites, Proc. Natl. Acad. Sci. USA,
102, 18069-18074.
72.Moquin, S. A., Thomas, S., Whalen, S., Warburton,
A., Fernanadez, S. G., McBride, A. A., Katherine, S., Pollard, J. J.,
and Miranda, L. (2017) The Epstein–Barr virus episome maneuvers
between nuclear chromatin compartments during reactivation, J.
Virol., doi: 10.1128/JVI.01413-17.
73.Hurley, E., Agger, S., McNeil, J., Lawrence, J.
B., Calendar, A., Lenoir, G., and Thorley-Lawson, D. A. (1991) When
Epstein–Barr virus persistently infects B-cell lines, it
frequently integrates, J. Virol., 65, 1245-1254.
74.Xiao, K., Yu, Z., Li, X., Li, X., Tang, K., Tu,
C., Qi, P., Liao, Q., Chen, P., Zeng, Z., Li, G., and Xiong, W. (2016)
Genome-wide analysis of Epstein–Barr virus (EBV) integration and
strain in C666-1 and Raji cells, J. Cancer, 7,
214-224.
75.Nunnari, G., Smith, J. A., and Daniel, R. (2008)
HIV-1 Tat and AIDS-associated cancer: targeting the cellular
anti-cancer barrier? J. Exp. Clin. Cancer Res., 27,
3.
76.Musinova, Y. R., Sheval, E. V., Dib, C., Germini,
D., and Vassetzky, Y. S. (2016) Functional roles of HIV-1 Tat protein
in the nucleus, Cell. Mol. Life Sci., 73, 589-601.
77.Gibson, T. M., Morton, L. M., Shiels, M. S.,
Clarke, C. A., and Engels, E. A. (2014) Risk of non-Hodgkin lymphoma
subtypes in HIV-infected people during the HAART era: a
population-based study, AIDS, 28, 2313-2318.
78.Mutalima, N., Molyneux, E., Jaffe, H., Kamiza,
S., Borgstein, E., Mkandawire, N., Liomba, G., Batumba, M., Lagos, D.,
Gratrix, F., Boshoff, C., Casabonne, D., Carpenter, L. M., and Newton,
R. (2008) Associations between Burkitt lymphoma among children in
Malawi and infection with HIV, EBV and malaria: results from a
case-control study, PLoS One, 3, e2505.
79.Dolcetti, R., Gloghini, A., Caruso, A., and
Carbone, A. (2016) A lymphomagenic role for HIV beyond immune
suppression? Blood, 127, 1403-1409.
80.Robbiani, D. F., Bunting, S., Feldhahn, N.,
Bothmer, A., Camps, J., Deroubaix, S., Klein, I. A., Stone, G.,
Eisenreich, T. R., Ried, T., Nussenzweig, A., and Nussenzweig, M. C.
(2009) AID produces DNA double-strand breaks in non-Ig genes and mature
B cell lymphomas with reciprocal chromosome translocations, Mol.
Cell, 36, 631-641.
81.Sneller, M., and Lane, H. (2014) HIV/IL-2 and
EBV-associated lymphoproliferative diseases: cause and effect or
coincidence? HIV Med., 15, 1-2.
82.Mbulaiteye, S. M., Biggar, R. J., Goedert, J. J.,
and Engels, E. A. (2003) Immune deficiency and risk for malignancy
among persons with AIDS, J. Acquir. Immune Defic. Syndr.,
32, 527-533.
83.Engels, E. A., Pfeiffer, R. M., Landgren, O., and
Moore, R. D. (2010) Immunologic and virologic predictors of
AIDS-related non-Hodgkin lymphoma in the highly active antiretroviral
therapy era, J. Acquir. Immune Defic. Syndr., 54,
78-84.
84.El-Amine, R., Germini, D., Zakharova, V. V.,
Tsfasman, T., Sheval, E. V., Louzada, R. A. N., Dupuy, C.,
Bilhou-Nabera, C., Hamade, A., Najjar, F., Oksenhendler, E., Lipinski,
M., Chernyak, B. V., and Vassetzky, Y. S. (2017) HIV-1 Tat protein
induces DNA damage in human peripheral blood B-lymphocytes via
mitochondrial ROS production, Redox Biol., in press.
85.Rochford, R., and Moormann, A. M. (2015)
Burkitt’s lymphoma, Curr. Top. Microbiol. Immunol.,
390, 267-285.
86.Singh, B., Sung, L. K., Matusop, A.,
Radhakrishnan, A., Shamsul, S. G., Cox-Singh, J., Thomas, A., and
Conway, D. J. (2004) A large focus of naturally acquired Plasmodium
knowlesi infections in human beings, Lancet, 363,
1017-1024.
87.Petter, M., and Duffy, M. F. (2015)
Pathogen–Host Interactions: Antigenic Variation v. Somatic
Adaptations, (Hsu, E., Du Pasquier, L., eds.) Springer.
88.Biggs, B., Anders, R. F., Dillon, H. E., Davern,
K. M., Martin, M., Petersen, C., Carlson, J., Helmby, H., Hill, A. V.
S., Brewster, D., Greenwood, B. M., and Wahlgren, M. (1992) Adherence
of infected erythrocytes to venular endothelium selects for antigenic
variants of Plasmodium falciparum, J. Immunol.,
149, 2047-2054.
89.Chyne, A., Donati, D., Guerreiro-Cacais, A. O.,
Levitsky, V., Chen, Q., Falk, K., Iorem, J., Kironde, F., Wahlgren, M.,
and Bejarano, M. T. (2007) A molecular link between malaria and
Epstein–Barr virus reactivation, PLoS Pathog., 3,
e80.
90.Reynaldi, A., Schlub, T. E., Chelimo, K., Sumba,
P. O., Piriou, E., Ogolla, S., Moormann, A. M., Rochford, R., and
Davenport, M. P. (2016) Impact of plasmodium falciparum coinfection on
longitudinal Epstein–Barr virus kinetics in kenyan children,
J. Infect. Dis., 213, 985-991.
91.Moormann, A. M., Chelimo, K., Sumba, O. P.,
Lutzke, M. L., Ploutz-Snyder, R., Newton, D., Kazura, J., and Rochford,
R. (2005) Exposure to holoendemic malaria results in elevated
Epstein–Barr virus loads in children, J. Infect. Dis.,
191, 1233-1238.
92.Moormann, A. M., Chelimo, K., Sumba, P. O.,
Tisch, D. J., Rochford, R., and Kazura, J. W. (2007) Exposure to
holoendemic malaria results in suppression of Epstein–Barr
virus-specific T cell immunosurveillance in Kenyan children, J.
Infect. Dis., 195, 799-808.
93.Njie, R., Bell, A. I., Jia, H., Croom-Carter, D.,
Chaganti, S., Hislop, A. D., Whittle H., and Rickinson, A. B. (2009)
The effects of acute malaria on Epstein–Barr virus (EBV) load and
EBV-specific T-cell immunity in Gambian children, J. Infect.
Dis., 199, 31-38.
94.Chattopadhyay, P. K., Chelimo, K., Embury, P. B.,
Mulama, D. H., Sumba, P. O., Gostick, E., Ladell, K., Brodie, T. M.,
Vulule, J., Roederer, M., Moormann, A. M., and Prece, D. A. (2013)
Holoendemic malaria exposure is associated with altered
Epstein–Barr virus-specific CD8+ T-cell
differentiation, J. Virol., 87, 1779-1788.
95.Gupta, N., Vishnoi, G., Wal, A., and Wal, P.
(2013) Medicinal value of Euphorbia tirucalli, Syst. Rev.
Pharm., 4, 40.
96.Mannucci, S., Luzzi, A., Carugi, A., Gozzetti,
A., Lazzi, S., Malagnino, V., Monique, S., Cusi, M. G., Leoncini, L.,
Van den Bosch, C. A., and De Falco, G. (2012) EBV reactivation and
chromosomal polysomies: Euphorbia tirucalli as a possible
cofactor in endemic Burkitt lymphoma, Adv. Hematol., 149780.
97.Van den Bosch, C., Griffin, B. E., Kazembe, P.,
Dziweni, C., and Kadzamira, L. (1993) Are plant factors a missing link
in the evolution of endemic Burkitt’s lymphoma? Br. J.
Cancer, 68, 1232-1235.
98.Machado, M. M., De Oliveira, L. F. S., Zuravski,
L., De Souza, R. O., Fischer, P., Duarte, J. A., Jonathaline, A.,
Manoelly, O. R., Camila, M. G., Boligon, A. A., and Margareth, A. L.
(2016) Evaluation of genotoxic and cytotoxic effects of hydroalcoholic
extract of Euphorbia tirucalli (Euphorbiaceae) in cell cultures
of human leukocytes, An. Acad. Bras. Cienc., 88,
17-28.
99.Waczuk, E., Kamdem, J., Ablaji, A., Meinerz, D.,
Bueno, D., Do Nascimento Gonzaga, T., Scotti do Canto Dorow, S.,
Boligon, A. A., Athayde, M. L., and Avila, D. S. (2015) Euphorbia
tirucalli aqueous extract induces cytotoxicity, genotoxicity and
changes in antioxidant gene expression in human leukocytes, Toxicol.
Res. (Camb.), 4, 739-748.
100.MacNeil, A., Sumba, O. P., Lutzke, M. L.,
Moormann, A., and Rochford, R. (2003) Activation of the
Epstein–Barr virus lytic cycle by the latex of the plant
Euphorbia tirucalli, Br. J. Cancer, 88,
1566-1569.
101.Aya, T., Kinoshita, T., Imai, S., Koizumi, S.,
Mizuno, F., Osato, T., Saton, C., Oikawa, T., Kuzumaki, N., and
Ohigashi, H. (1991) Chromosome translocation and c-MYC
activation by Epstein–Barr virus and Euphorbia tirucalli
in B lymphocytes, Lancet, 337, 1190.