REVIEW: Who Needs This Junk, or Genomic Dark Matter
O. I. Podgornaya1,2,3*, D. I. Ostromyshenskii1,3, and N. I. Enukashvily1
1Institute of Cytology, Russian Academy of Sciences, 194064 St. Petersburg, Russia; E-mail: opodg@yahoo.com2St. Petersburg State University, 199034 St. Petersburg, Russia
3Far Eastern Federal University, 690922 Vladivostok, Russia
* To whom correspondence should be addressed.
Received November 13, 2017; Revision received December 26, 2017
Centromeres (CEN), pericentromeric regions (periCEN), and subtelomeric regions (subTel) comprise the areas of constitutive heterochromatin (HChr). Tandem repeats (TRs or satellite DNA) are the main components of HChr forming no less than 10% of the mouse and human genome. HChr is assembled within distinct structures in the interphase nuclei of many species – chromocenters. In this review, the main classes of HChr repeat sequences are considered in the order of their number increase in the sequencing reads of the mouse chromocenters (ChrmC). TRs comprise ~70% of ChrmC occupying the first place. Non-LTR (-long terminal repeat) retroposons (mainly LINE, long interspersed nuclear element) are the next (~11%), and endogenous retroviruses (ERV; LTR-containing) are in the third position (~9%). HChr is not enriched with ERV in comparison with the whole genome, but there are differences in distribution of certain elements: while MaLR-like elements (ERV3) are dominant in the whole genome, intracisternal A-particles and corresponding LTR (ERV2) are prevalent in HChr. Most of LINE in ChrmC is represented by the 2-kb fragment at the end of the 2nd open reading frame and its flanking regions. Almost all tandem repeats classified as CEN or periCEN are contained in ChrmC. Our previous classification revealed 60 new mouse TR families with 29 of them being absent in ChrmC, which indicates their location on chromosome arms. TR transcription is necessary for maintenance of heterochromatic status of the HChr genome part. A burst of TR transcription is especially important in embryogenesis and other cases of radical changes in the cell program, including carcinogenesis. The recently discovered mechanism of epigenetic regulation with noncoding sequences transcripts, long noncoding RNA, and its role in embryogenesis and pluripotency maintenance is discussed.
KEY WORDS: heterochromatin, sequencing, tandem repeats, dispersed repeats, transposable elements, retrotransposons, long noncoding RNA, in situ hybridizationDOI: 10.1134/S0006297918040156
Abbreviations: CEN, centromeres; CENP-B box, CEN-binding site of CENP-B protein; ChrmC, dataset of sequence reads from mouse chromocenters; eDNA, extracellular DNA; ERV, endogenous retrovirus; FISH, fluorescent in situ hybridization; GPG, golden path gap (unfilled gap in assembled genomes with size of 3 Mb reserved for CEN); HAC, human artificial chromosome; HChr, heterochromatin; HOR, high order repeat; HP1, heterochromatic protein 1; HS1-3, human satellites 1-3; HSF-1, heat shock factor-1 (transcription factor); IAPs, intracisternal A-particles; LINE, long interspersed nuclear element; lncRNA, long non-coding RNA; LTR, long terminal repeats in ERV; MaSat, mouse major satellite (periCEN); MiSat, mouse minor satellite (CEN); periCEN, pericentromeric regions; satDNA, satellite DNA; SINE, short interspersed nuclear element; subTel, subtelomeric region; TE, transposable elements; TR, tandem repeats; WGS, Whole Genome Shotgun (dataset of reads assembled to genome contigs).
HETEROCHROMATIN
In 1928 Heitz coined two terms, “euchromatin” and “heterochromatin”, to describe parts of chromosomes with the former subjected to compaction–decompaction processes and the latter being constantly in the compacted state. Heitz considered the heterochromatin regions of chromosome genetically inert [1]. It was established from the very beginning that centromeres (CEN), pericentromeric regions (periCEN), and subtelomeric regions (subTel) – the most important regions in chromosomes – are associated with the regions of constitutive heterochromatin (HChr). HChr enriched with tandem repeats (TRs) remains the most mysterious part of the genome. HChr forms distinctive structures – chromocenters – in the interphase nucleus of many species. It is assumed that formation of one chromocenter could involve HChr from different chromosomes [2].
Proteins characteristic for HChr such as, for example, HP1 protein, have been identified in chromocenters using immunohistochemistry [3, 4]. The genes located in the HChr regions are predominantly in the transcriptionally inactive state, with very few exceptions. Various types of DNA repeats constitute the main part of HChr. Tandem repeats and transposable elements (TEs) of various classes (ERV, LINE, SINE, DNA-transposons) are recognized among the DNA repeats in higher eukaryotes. The combined TEs comprise up to 2/3 of the genome in the genome databases [5].
Tandem repeats (or satellite DNA, satDNA) are among the main components of HChr, making up for example no less than 10% of the human and mouse genome. In recent decades, TR transcripts were found that were specific for early embryonic development and for transformed cells [6-9]. TRs are the most complicated part of the genome for assembly and annotation. Only the most abundant TRs (major) are known for most higher eukaryotes. Even the explosive development of genome sequencing and annotation technologies has provided only limited information on the composition of the chromosome regions formed by TRs due to the lack of suitable assembly algorithms. In the assembled chromosome of most genomes of higher eukaryotes, there is a gap in the place of CEN/periCEN – GPG (golden path gap) – with the size of 3 Mb. Non-mapped on the chromosomes and non-annotated TR fields are present in the contigs of the WGS (Whole Genome Shotgun database). Bioinformatics approaches developed in our laboratory facilitate identification and annotation of large TRs in assembled genomes of different quality.
Because of difficulties in assembling of fragments containing TRs, mapping and annotation of the HChr regions limit the possibilities for investigation of the functional role of HChr and its composition; until the present time, HChr remains the “dark matter” of the genome. The possibility for isolation of chromocenters from mouse nuclei revealed qualitative and quantitative composition of HChr providing reference points for future assembly.
SEQUENCING AND DNA ANALYSIS OF CHROMOCENTERS
Chromocenters in mouse nuclei are associated with HChr protein markers. The protein composition of chromocenters has been investigated quite well [10, 11], but the question on DNA sequences comprising chromocenters and HChr itself remain poorly understood. Chromocenters are brightly stained with DAPI, which is considered as an indicator of enrichment with AT-rich fragments [12, 13]. In this review, we considered the classes of DNA revealed during sequencing of DNA from mouse chromocenters.
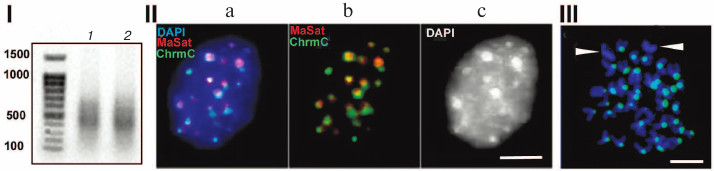
Figure 1 shows characteristics of chromocenters isolated according to the published technique [14, 15]. The method is based on the following procedure: first, nuclei isolated from mouse liver are subjected to mild ultrasound following ultracentrifugation; next, the fraction shown to contain undamaged chromocenters identified using microscopy is used [14, 15]. DNA from chromocenters was labeled using a degenerate primer; the results of FISH (fluorescent in situ hybridization) demonstrated its colocalization with chromocenters (Fig. 1, II and III). Cloning of this preparation showed that it was depleted of TRs, hence the non-amplified preparation was sequenced, thus ruling out all errors associated with amplification. The dataset of all read sequences was termed ChrmC. It was found that TR comprised ~71% of ChrmC, with the known mouse TRs (major mouse satellite MaSat (~66%) and minor mouse satellite MiSat (~4%)) being most represented. The second most frequent type was non-LTR (-long terminal repeats) retroposons (LINE (long interspersed nuclear element) mainly) representing ~11%. Endogenous retroviruses occupied the third place (~9%). Around 6% of ChrmC fragments remained unidentified, which was not surprising considering the number of non-annotated sequences even in the best assembled genomes [16]. In this review we discuss the main classes of HChr sequences in order of increasing of their amount in HChr (sequence reads, ChrmC).
Fig. 1. (I) Agarose gel electrophoresis of products of amplification of DNA from chromocenters (1) and DNA from centromeres (2); (II) FISH of the probe produced with DNA from chromocenters (green, ChrmC) and major satellite (red, MaSat) on the nuclei of the mouse cell culture L929; overlapping image (a) and individual probe images (b, c). (III) FISH of chromocenter DNA (green) on metaphase plates of the CH3 mouse (normal karyotype). Chromosomes without probe (likely sex chromosomes) are marked. Scale bar for II and III – 5 μm (modified from Kuznetsova et al. [46] with permission).
Endogenous retroviruses in heterochromatin. Representation of ERV (endogenous retrovirus) is almost the same in the mouse genome and in ChrmC, but the distribution of the ERV classes is asymmetrical in the assembled genome in comparison with ChrmC.
All contigs containing MiSat, MaSat, and TRPC-21A (TR identified as periCEN) together with the ERV fragment were selected from the WGS database of the mouse genome. This way of selection suggests that all contigs associated with CEN/periCEN including GPG will be present in the selection. The produced selection included ~2000 contigs and more than a half of ERV in the TR fields are represented by the inner part of intracisternal A-particles (IAP) or their individual LTR [17].
Endogenous retroviruses (ERVs) are well represented both in the human and mouse genome. The is no unified classification of ERVs, very often one element either belongs to different groups according to various classifications or has different names. Six families of human ERVs are recognized based on the analysis of similarities of nucleotide sequences of the human pol gene: HERV-K, -H, -W, -L, -F, -I, which are assigned to different types of retroviruses. For example, the HERV-K family is assigned to β-retroviruses, and several families including HERV-H – to γ-retroviruses [18]. ERVs are divided into three main classes depending on structural features, but LTRs are typical for all of them [19]. The ERV3 class includes MaLR (mammalian apparent LTR retrotransposons) elements with 388 thousand distinct copies in the mouse genome. MaLRs are active in the genome and are represented by MERVL, MTA, and ORR1 [20]. We detected the class of TR based on the fragments of the MTA element. Part of the inner MTA fragment and one of the LTRs produce the monomer from which TR fields are assembled [21]. The ERV3 class occupies the second position in the list of ERVs in ChrmC, but the ERV2 (IAP) class represents the major portion of ERVs.
There are 10-time more copies of the ERV2-class elements in the mouse genome in comparison with the human genome. Two groups of elements of this class are the most active in mice: IAP and ETn. It is IAP that is the most abundant ERV in chromocenters (ChrmC). The name of this element – IAP (endoplasmic reticulum intracisternal A particle) indicates that RNAs transcribed from IAP together with proteins can form virus-like particles in the cytoplasm of a cell, which is especially important in the process of embryonic development (see next section).
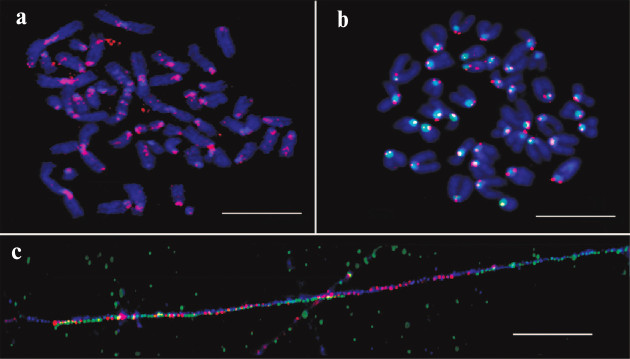
The unexpected enrichment of HChr with IAP was proved experimentally with a synthesized labeled probe designed according to in silico data and FISH test of this probe (Fig. 2). The results confirmed periCEN location of the probe. Hence, the enrichment of HChr with the IAP element was demonstrated both in silico and in situ.
Fig. 2. FISH and fiber-FISH with IAP probe on chromatin from M. musculus. a) IAP probe (red) on metaphase plate of the L929 cell culture; b) IAP probe (red) and MiSat probe (green) on metaphase plate from bone marrow (normal karyotype); c) fiber-FISH on chromatin from L929 with IAP probe (red) and MiSat probe (green). Contrasted with DAPI (blue). Scale bar – 10 μm [17].
We did not observe enrichment of HChr with ERVs in comparison with the rest of genome, but asymmetry was observed: while the MaLR-like elements (ERV3) were most frequent in the genome (as a whole), IAP and corresponding LTR (class ERV2) prevailed in HChr. LTRs of ERVs are strong promoters and in some cases can initiate transcription in both directions [22, 23]. The ERV identified in HChr (especially their LTR) can be transcription promoters for the adjacent TR. The ERV transcripts (for mouse – IAP, class ERV2) can support the mechanism of functionally significant breaks in the periCEN region.
ROLE OF ENDOGENOUS RETROVIRUSES IN DEVELOPMENT AND EVOLUTIONARY
INNOVATIONS
It is known that certain classes of TEs (or retroelements), LINEs, and ERVs in particular, mark the sites (breakpoints) of evolutionary innovations [24]. The “evolutionary breakpoints” in the mammalian genome represent certain locations that are used multiple times in the process of karyotype evolution. It can be seen if one follows phylogenetic trajectories of orthologous (syntenic) chromosome segments that many evolutionarily significant breaks coincide with either the extinct centromeric activity or formation of a new CEN [25]. It was shown that ERV is an essential component of CEN [26], which is actively transcribed [23]. The transcripts of TRs and retroviruses comprise a new class of RNAs belonging to long noncoding RNAs (lncRNA). The ERVs from which lncRNA are transcribed are components of the CEN domain in several classes of vertebrates.
The presence of certain classes of ERVs in HChr is gaining attention due to the recently discovered presence of ERV transcripts in the composition of lncRNAs and a key role of lncRNAs in regulation of gene assemblies. For example, it was shown that ERV lncRNAs played an important role in determining which genes and when must be activated in neural stem (progenitor) cells [27, 148].
Intrauterine development is one of the features of placental mammals. More than a thousand genes participate in the process of prenatal development, which prior to that have played absolutely different functions in different parts of the organism. With time these genes became sensitive to female progesterone and this sensitivity, in turn, emerged due to ERV transposons [28-31]. This is how mammals managed to get evolutionary advantage through ERV activity [32]. Now ERVs are included in the system of genome regulation often as domesticated genes, but mainly as transcripts and lncRNA components.
It was shown that HERV-H (one of the classes of human ERVs) is inactive in the differentiated cells in an adult organism, while it plays an important role in the development of embryonic cells: HERV-H represents the key component required for the development of pluripotency. Human stem cells were subjected to the action of RNAs that suppressed HERV-H transcription activity. Following this, stem cells lost the indicators of pluripotency and assumed fibroblast-like phenotype [33, 148].
The ERVs from different classes are systematically transcribed in early embryogenesis and in the different stages of development – different LTRs from various ERVs serve as initiators of stage-specific transcription, thus generating hundreds of lncRNA coexpressed with ERVs. For example, the blastocyst-specific type of ERV activates transformation of human embryonic stem cells into epiblast cells. Change in transcription of certain types of ERVs coordinates the expression of gene assemblies like a conductor conducting an orchestra [34]. The ERV lncRNA: 1) recruit transcription coactivators to regulatory DNA-binding complexes and enhancer parts of the genome; 2) interact with Oct-4 and other protein factors and change chromatin conformation [34-37]. It is demonstrated in Fig. 3 that the increase in transcription of HERV-K and HERV-H is characteristic for certain developmental stages and during pluripotency induction; at these moments, transcription activation could correlate with activation of transcription of gene assemblies (Fig. 3, I; [38]).
Fig. 3. (I) Dynamics of expression of HERV-H (blue line) and HERV-K (red line) during early development (a) and during induction of pluripotency (b); H, high level of expression; L, low level of expression. b) iPSC, induced pluripotent stem cells [34]. (II) a) Particles of HERV-K in endoplasmic reticulum of human embryonic carcinoma cells (hECC): 1) electron microscopy, staining/contrasting with heavy metal salts, virus-like particles (VLP) are inframed; 2) immunoelectron microscopy with antibodies against gag capsid protein; b) scheme summarizing HERV-K transcription dynamics in human embryos and cultivated pluripotent cells (hESC). Dashed lines represent expression levels of OCT4 and HERV-K and DNA methylation in naïve and primed hESC without the data on embryogenesis after implantation. The arrow indicates EGA (early genome activation) in the moment of the activation of the embryo genome [40] (reproduced with permission from the Publisher).
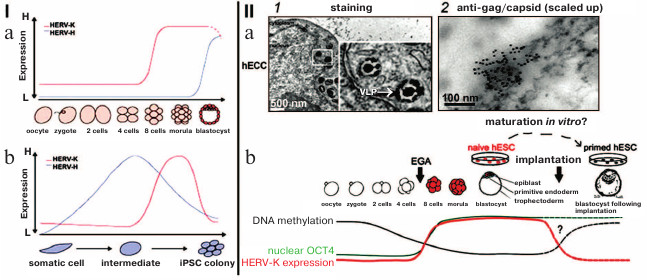
Mouse IAPs belong to the same type of ERVs as human HERV-K [38]. IAP transcripts were found in endoplasmic reticulum particles in numbers correlating with the change in the number of IAP transcripts [39]. Morphology of IAP particles is similar to that of HERV-K (Fig. 3, IIb; [40]).
Data on localization of individual classes of human ERVs in euchromatin or HChr are lacking, but we have observed enrichment of the mouse HChr specifically with IAP (ERV2).
It is likely that ERVs located in HChr maintain the state of pluripotency the same way as is done by HERV-K, an ERV of the same class 2 (Fig. 3, I).
LINEs
It was demonstrated using cytology that the major families of non-LTR TE (LINEs and SINEs) occupy different areas in the mouse and human genomes, which are assigned to G- and R-bands, respectively, in metaphase chromosomes [41]. Sequence analysis of the genome assemblies shows that LINEs are characteristic for AT-rich (G-positive, facultative HChr), while SINEs – for GC-rich (R-positive, euchromatin) chromosome regions [42]. The probes to L1 (most abundant in LINEs) and B1 (most abundant in rodent SINEs) mark facultative HChr (L1) and euchromatin (B1) at the chromosome arms during FISH assay. The probes for full-length L1 or SINEs do not produce any signal in the CEN/periCEN regions [43]. The paradox of the presence of LINEs in HChr but the absence of the LINE probe on hybridization (FISH) can be explained if one suggests that not the full-length LINEs are present in the constitutive HChr, but their separate fragments.
Our results produced using various techniques (analysis of reads in high throughput sequencing of DNA from chromocenters, analysis of clones produced from this DNA following a degenerate oligonucleotide-primed (DOP) amplification, and clone hybridization) demonstrate that chromocenters are enriched with ~2-kb fragment of the LINE 3′-end (Fig. 4b). One of the families of TRs related to TEs from the initial TR classification consists of the LINE fragments (TR-L1) of the same type, hence the monomers consist of the 2-kb fragment that includes 3′-end of the ORF2 and 3′-noncoding region [21]. Mapping of TR-L1 in silico onto the assembled genome showed enrichment in the region of facultative HChr [44]. A similar fragment was found during analysis of the human centromere assembly (Fig. 5). We used the human centromere assembly predicted with bioinformatics methods ([45]; LinearCen 1.1, GCA_000442335.2). Mapping of the LINE fragments identified in centromeres of two human chromosomes for this assembly onto the L1 consensus from RepBase demonstrated enrichment with similar ~2-kb fragments of the LINE 3′-end. Precisely this fragment is characteristic for the mouse and human HChr [46, 47].
Fig. 4. a) Covering of consensus sequences of IAP (ERV2) from RepBase with ChrmC reads. Abscissa axis, length of ERV (bp); ordinate axis, covering with ChrmC reads. b) Covering of consensus sequence of L1 from RepBase with the ChrmC reads. Blue line, sequence reads of chromocenters; red line, reads of genome-wide sequencing with Illumina HiSeq (ERR034297) normalized to the size of the dataset. X-axis, LINE sequence (bp); Y-axis, covering of each nucleotide with reads [17].
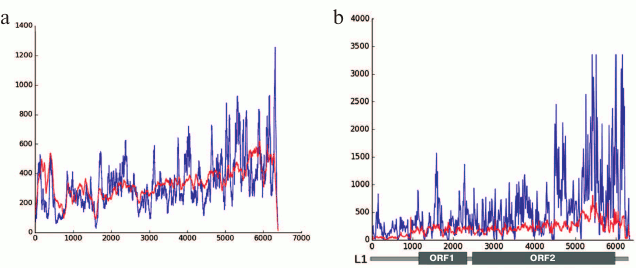
Fig. 5. a) Schematic representation of locations of clones containing LINE fragments on the sequences L1_MM and Lxs; b) LINE fragments found in centromeres of two human chromosomes and mapped on L1 (adapted from Kuznetsova et al. [46]; reproduced with permission).
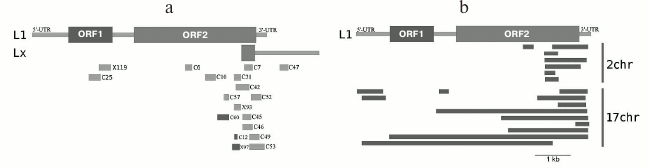
The human artificial chromosome (HAC) is stably maintained in mouse cells only if it is incorporated into the composition of chromocenters [48]. At the same time, the main CEN and periCEN TRs from mouse and human do not exhibit any common elements during alignment except the short 17-bp CENP-B box (binding site of the CENP-B CEN protein) [49]. The common LINE fragment present in the CEN/periCEN regions of the two species can facilitate association of HAC with mouse chromocenters [46].
High content of the particular LINE fragment in chromocenters removes the contradiction between the in silico and in situ (FISH) data. During FISH, HChr is not stained with full-length LINE (amount is too small), but the LINE 3′-fragment is specific for HChr (~11% in ChrmC).
Dynamic equilibrium exists between the lncRNAs originating from LINEs and ERVs during embryonic development; furthermore, higher content of the LINE transcripts corresponds to the later stages [50]. LINEs are the main component of the Cot1 RNA (RNA that hybridizes with high-copy DNA) in differentiated cells; moreover, the transcripts of the same 3′-end of the LINE are most abundant in Cot1 RNA [51]. It is likely that Cot1 RNA is transcribed from the fragments located in HChr. This is how the developing area of science on lncRNAs assigns regulatory functions to components of HChr – dark matter of the genome.
TANDEM REPEATS OR SATELLITE DNA
The DNA fraction found as an additional peak during centrifugation in density gradients was termed “satellite” DNA [52]. Genome sequencing did not reveal anything that could make this part of the genome a satellite – the massifs of satDNA (TR) in the assembled genome were continuations of the euchromatin regions. We found it inconvenient to use the term “satDNA” when working with genome databases, and it was decided to use the easily formalized term – tandem repeats (TR). The term “large tandem repeats” considers the size of field without inserts between monomers with size higher than a few kilobase pairs (for mouse higher than 3 kb), length of monomer, GC-composition, and degree of variability of monomers in the field. All these characteristics have quantitative expression, which allows identifying the TR closest to the classic satDNA. Classification of TRs is conducted automatically during comparison of the sequences [21], and this is the way in which the degree of similarity of different monomers is considered. The term “satDNA” has been reserved for some TRs cloned in the pre-genome era and historically was used in their names – major and minor mouse satellites (MaSat and MiSat), human satellite 3 (HS3).
TRs represent a DNA class that is found only in eukaryotes – it is absent in prokaryotes. The TR fields consist of short sequences (monomers) that are repeated multiple times. The TR field gains the capacity for nontrivial chromatin packing [53]. The content of TRs in genomes of high eukaryotes can be tens of percent and only rarely can be below 10% of the genome.
Views on this part of the genome have been changing in recent years following discovery of TR transcription, although most TRs belong to the non-annotated “dark” part of the genome. According to the primary sequence, the TRs are different for different species up to being species-specific. As a rule, different TRs are located in CENs and periCEN regions of the chromosome. TRs evolve rapidly, but they maintain their functions in the kinetochore [44]. Despite the differences in the TR sequences from different species, TRs display common features – organization into long homogenous fields and length of a monomer corresponding to the size of nucleosomal DNA, and propensity of TR regions for formation of noncanonical secondary DNA structures [54].
Individual monomers in the composition of TRs could differ in nucleotide sequence displaying substitutions, deletions, and inserts with lengths of several nucleotides. Different variants of high order repeats (HOR) are characteristic for individual chromosomes and can form long blocks in the composition of a single chromosome [55-57].
TRs are the most variable components of the genome, but in the pre-genome era, when only a limited number of cloned satDNAs was available, it was not possible to elucidate the set of TRs even in organisms from the same genus. Now comparison of the TR sets becomes feasible [58, 59].
Expression of the major mouse satellite (MaSat) was found to be required in the two-cell stage of development for formation of chromocenters [6]. Such fundamental discoveries related to the role of TRs are based on the known cloned mouse MaSat sequence. It is impossible for most other TRs to determine their transcriptional status because these TRs are not yet described and classified. Lack of information on TRs hinders their investigation. Tens of mammalian genomes have been sequenced in the last decade, but there have been only a few studies devoted to the analysis of TRs on the genome level [60-62]. In the case of TEs, the analysis on the genome level was shown to be successful in the studies devoted to the attempts to suggest common classification of TEs [63-67]. The attempt has been made to create a TR database using the data from the sequences of the reference genomes – TRDB (Tandem Repeats Data Base) [68]. Unfortunately, TRs were not annotated and only a limited part of the currently available genomes was used. The RepBase database comprising manually annotated DNA repeats is a valuable source of information [69]. Authors actively use this information on identified repeats in the assembled genomes.
The results of a large study were published in 2013, the goal of which was the search for the major TRs for each species with the sequenced genome [70]. Unfortunately, the authors identified the major TRs based on contradictory assumptions. They suggested that the major TR would be the one that would be represented in high copy number in the genome and localized in the region of centromeres. It has been known from pre-genome era knowledge on the cloned and mapped TRs that the major TRs were located in periCEN region (~10% of genome), while the CENP-box containing CEN TRs comprised no more than 1% [21, 71]. The contradictory assumptions of these authors led to an anticipated outcome – none of the three experimentally identified CEN TRs was included in their analysis. Nevertheless, this work represents the most exhaustive analysis of the high-copy TRs in 282 species conducted using methods of bioinformatics. It was shown that the high-copy TRs show similarity only in phylogenetically very close species, while the question on variability of TRs between genera and more so between species was beyond the scope of this study. The main conclusion of the study [70] was that the high-copy TRs in the genomes of different species differed in almost all parameters. At the same time, the proteins common for both CEN and periCEN regions are highly conserved. It is clear that there is no such motif as, for example, CENP-box, for binding these proteins. The question arises, what factor determines binding of these proteins? The main candidate for this role is TR curvature. Structure-specific interaction of helicase p68 (DDX5) and SAF-A proteins with DNA was demonstrated, which depended on the DNA curvature [72-74]. A model was suggested according to which the TRs in centromere region formed a series of sequences alternating in the degree of curvature [75]. The specific folding structures of the chromatin areas containing TRs are based on locus-specific TRs and likely are stabilized by binding of nuclear proteins. It is impossible so far to prove this hypothesis due to insufficient number of assembled CEN/periCEN regions.
Almost all MaSat (periCEN) and MiSat (CEN) of the mouse genome are in the composition of chromocenters (ChrmC). All the rest TR families together comprise only ~1% of the ChrmC composition, and 31 families from 62 TR families have not been found in ChrmC, which suggests their location in the chromosome arms. The availability of TRs in the chromosome arms was reposted for the house mouse [44], human [60, 61], and Tribolium castaneum beetle [54]. In silico analysis of the assembled T. castaneum genome demonstrated the presence of TRs with characteristic monomer length of 170 bp and number of repeats in the field of five and more in the euchromatin part of chromosome arms [54].
The presence of TR fields in euchromatic chromosome arms can impart a structural barcode to the chromosome, i.e. chromosome-specific marking [44]. The marking can be set in motion during morphogenesis due to association of similar sequences. The number of monomers in the field is also important. It was shown in dogs that exactly the number of monomers in the TR fields adjacent to the developmental genes results in the rapid but topologically conservative inheritable change of the skull shape [76]. The role of TR fields in euchromatin will be further clarified during their classification and investigation in higher eukaryotes.
The mechanisms of chromosome motions remain poorly understood [77]. The locations of chromosomes relative to each other can change in the cell cycle only during mitosis at the moment of metaphase plate formation [78]. In the same short time interval, a fiber between chromosomes is synthesized that consists of TRs and proteins bound to them [79, 80]. The chromosome motion can occur due to the synthesis of the fiber containing TRs during mitosis and, as a result, the positions of chromosome territories change.
It can be concluded at this time that the basic MaSat and MiSat (or human alpha and HS1-4, according to our preliminary data their analogs are present in all genomes) form the basis of CEN/periCEN regions in chromosomes and in the interphase nucleus they represent the basis of chromosome territories (immobile but not inert part inside the nucleus), where a “boiling” surface is located on which the processes occur associated with maintaining active cell metabolism [81]. In this manner HChr – “dark matter of the genome” – ensures fundamental 3D structure of interphase chromatin.
TRANSCRIPTION OF TANDEM REPEATS IN NORMAL CELLS
There is no doubt at present that transcriptional activity in HChr regions – in fact noncoding DNA – exists. The first data on transcriptional activity of TRs were reported in 1960-1970 [82-85]. These data seemed so contradictory to the “central dogma of cell biology” that the authors explained their results with inadequate methods resulting in artifacts. Nevertheless, several years later the transcription of satDNA had been fully proven for the lampbrush chromosomes of amphibians [86, 87], and next for birds [88]. TR transcripts were found in many organisms belonging to different phyla and classes – insects, fishes, human, mouse, fission yeasts, cereal grasses, and others [89-94]. The amount of data is sufficient to conclude that we are dealing not with a unique phenomenon, but with a regular event in biology.
The amount of TR transcripts in terminally differentiated cells in the absence of stress conditions varies in the range of 1-5% of the transcriptome [95, 96]. The number of transcripts can increase manifold under certain conditions: during embryogenesis, at the beginning of proliferation, cell aging, cell differentiation, cancerogenesis, and cell stress [95, 97, 98].
The TR transcripts consist of RNAs with length from 20 to 5000 bp. Despite the availability of polyA sequence, the main part of lncRNA TRs is localized in nucleus close to the DNA encoding them [99]. Variability of the found transcripts can be explained by either variability of the TRs in the genome, or lncRNA processing [100, 101]. The periCEN TRs in mouse (MaSat and γ-satellites) and human (HS1-3) are transcribed more often. Transcription of CEN TRs is observed less frequently. Asymmetry is the feature of the periCEN TR transcription in mammals – only one strand is transcribed. Furthermore, switches between the strands are most likely not random. The switch between the strands occurs in strictly defined moments during human and mouse embryogenesis (Fig. 6, I; [6, 7, 102, 103]). Transcriptional activity is often accompanied by significant decondensation of periCEN HChr (Fig. 6, II) and its demethylation [104, 105].
Fig. 6. Transcription and decondensation of periCEN TRs. (I) a) Chromatin organization in the one-cell and four-cell stage of mouse development. Color-coding is presented in the left upper corner. Location of heterochromatin 1 is determined with FISH; heterochromatin 2 displays protein labels of inactivated chromatin; b) strand-specific transcription of MaSat (mouse periCEN TR) in preimplantation mouse embryogenesis; forward – transcription from “sense” (T-rich – GGAAT); reverse – transcription from “antisense” (A-rich – CCTTA) satDNA strands; polyA RNA – increase in total expression (of coding regions). The time scale of development is presented at the bottom from the moment of introduction of human chorionic gonadotropin hormone (hCG, 0; induction of ovulation) and fertilization (F, 12). Figures designate hours from the hCG administration. Cell cycle phases are indicated. Increase in polyA RNA expression coincides with the time point of induction of the parent genome (our scheme). (II) Decondensation of pericentromeric TR HS3-1. Localization of HS3 in chromosome 1 (red, panel (a)) in A431 cells and lung fibroblasts at 11th (M11) and 32nd passage (M32). Nuclei are stained with DAPI (blue). Scale bar 5 μm; b) statistical calculation of the degree of decondensation of HS3-1 in nuclei (black) and in chromosomes (dashed); * significant (p < 0.05) difference from lung fibroblasts from the 11th passage (control) (adapted from Enukashvily et al. [115], reproduced with permission).
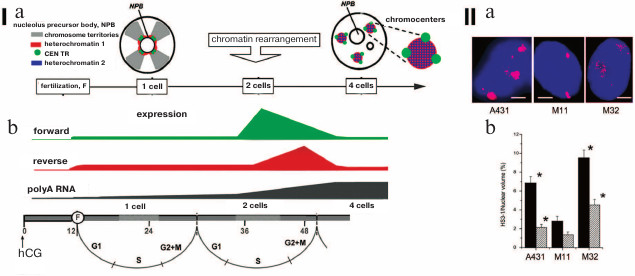
Transcription of many noncoding sequences is characteristic for mammalian embryogenesis. In humans, 90% of transcripts are represented by lncRNA in the blastocyst developmental stage. In mice at the stage of preovulation oocyte, 45% of the transcriptome is not identified as genes with known functions [106]. Tissue-specific expression of the particular repeating elements is higher than for the genes [107].
The transcription of MaSat in mouse embryogenesis demonstrates pulsed character, i.e. burst-like growth with rapid decline is observed. In the early stage of two blastomeres (32 h), the transcription of the “sense” (in mouse T-rich – GGAAT) strand of periCEN MaSat increases 80-fold, but in the later stage (48 h) the transcription of this strand decreases by 40%, while the transcription of the antisense strand (in mouse A-rich – CCTTA) increases 140-fold. At the stage of 4 cells, transcripts of both strands have not been observed (Fig. 6, I). Inactivation of the early transcription of the periCEN TR results in disruption of formation of the HChr association regions – chromocenters. As a result, the required expression pattern of the coding part of the genome is not formed, which leads to embryo death [6]. Transcription of TRs provides condition for heterochromatization of the respective areas and formation of chromocenters in the stage of 4 cells [108].
In the stage of blastocyst, during the process of losing pluripotency, the transcription of the sense strand of MaSat is renewed. When differentiation of embryonic stem cells (ESCs) is induced by retinoic acid, the number of strand-specific transcripts of periCEN TR increases sharply, and these transcripts are located exclusively in the nucleus. Prior to the induction with retinoic acid, chromocenters in this ESC line are not formed, and their formation begins only after induction with retinoic acid [109].
Complex tissue- and time-specific transcription of periCEN TRs was observed during postimplantation in human and mouse embryogenesis [92, 102]. The switch of HSAT 3-1 transcription from the sense strand to the antisense one occurs in the human embryo at approximately the 9-10th week of pregnancy [102].
A paradox situation is observed in the adult organism – the most significant amounts of TR transcripts has been revealed in intensively proliferating cells [23, 100, 110, 111] and, surprisingly, in aging cells [110, 112, 113]. However, both CEN TR and periCEN TR processed to short fragments are active in the intensively proliferating cells [23, 100, 114]. In aging cells the periCENs are more active [115, 116].
The transcripts of periCEN TRs can be found in proliferating cells during two stages of the cell cycle. Transcripts of the mouse γ-satellite with sizes below 200 bp have been detected only in mitotic cells, and they disappear 1 h after completion of mitosis. The lncRNAs of the same gamma satellite but with more than 1 kb size are present in a maximum amount in the nucleus in G1/early S phase [100]. The length of TR transcripts identified in the CEN region is usually on a small scale – up to 200 pairs, but it is still not clear if such transcripts are the result of lncRNA processing, or they were initially expresses as short transcripts. The transcripts of CEN TRs in the form of a single-strand RNAs bind to CENP-C (one of the key proteins in assembly of the kinetochore), attract CENP-C to the CEN region, and next are identified in the composition of the assembled kinetochore. It is precisely these CEN TRs that are essential for the kinetochore assembly and proliferative activity [110, 117, 118]. Excessive accumulation of the CEN TR transcripts leads to genome instability in a form of chromosome anomalies and accumulation of micronuclei containing periCEN. Transcription of the periCEN TRs increases in aging cells [116]. The transcription is accompanied by the significant decondensation of periCEN HChr in the interphase nucleus, but not on mitotic chromosomes (Fig. 6, II; [115]). The mechanism involved is likely not activation of transcription, but disruption of transcript processing, accumulation of lncRNA, and, consequently, disruption of heterochromatinization of the periCEN [113]. The mechanism of accumulation of the periCEN TR transcripts during aging could be associated with the impaired functioning of deacetylase SIRT6 leading to periCEN TR chromatin acetylation and depression of their transcription [113, 114].
While accumulation of the periCEN TRs during cell aging is the result of the disruption of lncRNA TR processing, sharp transcription activation of some TRs required for heterochromatinization of the respective region is observed during embryogenesis.
TRANSCRIPTION OF TANDEM REPEATS DURING STRESS AND
CARCINOGENESIS
Transcription of the periCEN TRs is observed during pathological cell states. Heat shock and cadmium are strong inducers of cell stress [99, 119]. The transcriptional factor HSF-1 associated with the periCEN DNA of chromosome 9 stimulates transcription during heat shock [120]. This factor recruits acetyltransferases to the periCEN region that hyperacetylate TRs in the composition of the chromatin TRs, which in turn attracts proteins with amino acid chain containing a bromodomain. These proteins are required for the transcription of satellites by RNA-polymerase II [121]. Two groups of researchers independently produced results confirming transcription of the periCEN TRs during heat shock [90, 120]. The main role of the periCEN TR transcripts is participation in the assembly of nuclear stress-bodies [119]. Inactivation of TR transcription or the protein regulators leads to blocking of stress-body assembly and decrease in the activity effector caspases 3/7 from the apoptotic cascade [122]. TR transcription is observed only for the first two hours from the beginning of heat action and depends on the functioning of the chaperon Daxx and RNA-helicase p68 [123]. The transcription of perCEN TR TCAST, which comprises 35% of the red flour beetle T. castaneum genome, was investigated in insects. The authors demonstrated that TCAST transcription also increased in all cells of the organism following heat shock [124].
Many researchers have reported activation of periCEN TR transcription in tumors. The TR transcripts are prognostic indicators for many types of malignant tumors. Initially, the periCEN transcription but not the CEN TRs was established for Wilms tumors, and after that it was found in cells of epithelial carcinoma A431, but not in HeLa [8, 9, 115]. Unlike the TR transcription in normal tissues, the transcription in tumors is not strand-specific. The transcription of periCEN TRs can be very active in some tumors, increasing 130-fold in comparison with the adjacent normal tissues [95]. The decrease in the amount of ubiquitinated histone H2A as a result of mutation in the BRCA1 gene in breast tumor results in the derepression of transcription of mouse periCEN TRs. The increased genome instability, which leads to malignization more often in mouse than in humans, is the result of activation of transcription of periCEN TRs [8, 113, 125].
However, TR transcription is not universal for all tumors. The transcription of perCEN TRs is observed only in the tumor surroundings (tumor stroma) in mouse non-small cell lung cancer, but not in the tumor cells. The phenotype of aging cells is characteristic for tumor-associated fibroblasts that form stroma. Hence, in this case TR transcription is not related to the cell malignization but is due to processes of cell aging and acquiring of tumor-associated phenotype [126].
Investigation of TR transcription (lncRNA TR) in cancer cells facilitated establishing a fundamentally new mechanism of TR proliferation. In colorectal adenocarcinomas, periCEN TR HS2 (human satellite 2) transcripts are required for formation of double-strand DNA–RNA hybrids. These hybrids are formed catalyzed by the virus reverse transcriptases activated in the tumor cells and represent one of the mechanisms of the increase in number of HS2 copies in cells and at the same time increasing genome instability [101]. The mechanism suggests reverse transcription on TR RNAs, existence of the TR RNA–DNA hybrids, and the possibility of insertion of a new copy into the genome. The presence of ERV and its fragments in HChr provides a new hypothesis for explanation of the known variability of TRs (Fig. 7).
Fig. 7. Scheme summarizing the transcription/reverse transcription cycle of periCEN TRs (a, b) and membrane TR (c), and TR cycle in eDNA (d). a) LncRNA transcripts are produced in cancer cells as a result of transcription of periCEN TRs, which are subjected to reverse transcription mediated by the ERV-K reverse transcriptase (?, IAP) followed by reintegration into the genome, resulting in expansion of TRs via DNA–RNA duplexes and double stranded TR dsDNA, or (b) lncRNA is recognized by specific receptors and induces primary inflammatory response (according to [140]); c) DNA TR on a membrane with polymerase II specific to TR (according to [139]); d) fragments of DNA TR are introduced into an extracellular medium and become components of eDNA in complex with proteins; eDNA is recognized by receptors (?), penetrates into cells, and is inserted into the subtelomeric region (according to [135]) (the scheme as a whole is original).
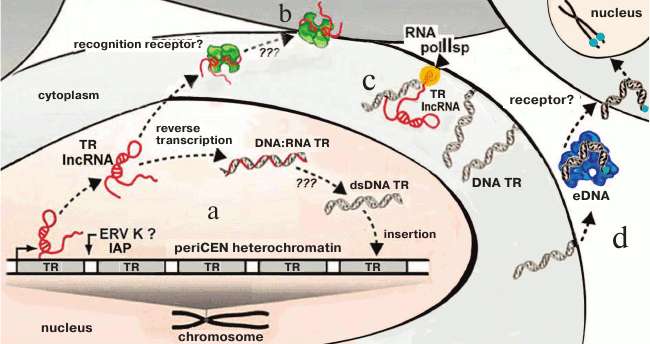
Participation of TR transcripts in cell metabolism is still very poorly understood. But some conclusions can be made based on the available data. The periCEN TR transcripts are required in normal tissues for formation of the periCEN HChr and, hence, for formation of the expression pattern due to trans-effects. The CEN TRs play a role in assembly of the kinetochore. The excessive expression of periCEN TRs could lead to genome instability and associated malignization of cells. The dark genome matter thus gradually reveals its secrets.
VARIABILITY AND MOBILITY OF TANDEM REPEATS
In the few cases when karyotype rearrangements were investigated at the level of genome databases, correlation of the break points with availability of TRs was reported [25, 127], not to mention rapid evolution of CEN/periCEN TRs [70]. Investigation of the role of extracellular DNA in the life of an organism could shed light on the mechanism of rapid TR evolution and their presence in chromosome break points [128].
eDNA (extracellular DNA) circulates in the body liquids of higher vertebrates, but so far only medical professionals rather than biologists consider this as an interesting problem [129-132]. Whole genome sequencing and new sequencing techniques allow presenting the issue of eDNA from another standpoint [133].
The main sources of the eDNA in an organism are: 1) exosomes from resting cells; 2) microvesicles from activated cells; 3) apoptotic cells [134]. The capture of eDNA by cell cultures and its incorporation into the chromatin of the host cell was demonstrated [135]. Both the DNA from cancer patients and from healthy donors was used in the study either in the form of purified DNA (DNAfs) or in the form of chromatin fragments (Cfs); the preparations were labeled and added to the culture medium with the murine cell line NIH3T3 (immortal fibroblasts). It was found that the fragments of both DNAfs and Cfs migrated rapidly to the nucleus and were retained there for so long that it was possible to identify transformed clones. Inserts of human DNA into the mouse genome were monitored using hybridization with total human DNA (contains predominately Alu repeats; class SINE) and with a pancentromeric probe (contains predominately TRs). The genome of the transformed clones was sequenced, and multiple copies of the integrated fragments were detected. Hybridization with the pan-CEN probe demonstrates that TRs represent a significant portion of the plasma DNA. Cfs were integrated better than DNAfs. Both preparations from the cancer patients were integrated more actively than the same ones from healthy donors. The in vivo transformation via injection of human DNA into mice was also reported. The authors conclude that the circulating eDNA is a constant physiological damaging agent inducing apoptosis under normal conditions, but also participating in multiple pathologies including aging and malignization [135]. It is important that a significant portion of the eDNA consists of TRs, which are integrated into genome during natural transformation.
Apoptosis was experimentally induced in human umbilical vein endothelial cells (HUVEC), and the DNA pool present in the culture medium (eDNA) was sequenced and used as a probe for in situ hybridization [136]. Both methods demonstrated enrichment of the eDNA with repeating elements: 1) eDNA was enriched with periCEN TRs, particularly with HS3, but by contrast the CEN TR α-satellite was underrepresented; 2) eDNA was enriched with Alu element (SINE), but the LINEs were underrepresented. Similar features were reported for the blood serum from healthy donors [137, 138].
The fact that eDNA in apoptotic cells is enriched with TRs has been proven [136], but it is likely that another mechanism involving membrane DNA is in place during formation of exosomes and microvesicles. The cell line WIL2-CG was constructed based on the human diploid B-lymphocyte cell line WIL2, which expressed a chitin-binding domain on the surface. The domain allowed isolating cell membrane without nucleus disruption, and, thus, the problem of contamination of the membrane fraction with the genomic DNA was resolved. When the sequenced membrane DNA was compared with the initial WGS\WGA databases, it became clear that it was precisely the TRs that were attached to the inner side of cytoplasmic membrane as ~6-kb fragments (Fig. 7; [139]). The special type of RNA polymerase II was identified that transcribed membrane DNA, in particular the alphoid satellite DNA [139]. There is no doubt of the existence of the membrane fraction of DNA. The fact that the eDNA was found to be enriched with TRs was unexpected.
Heterochromatin that is enriched with TRs remains the most mysterious portion of the genome; the presence of TRs in the membrane DNA fraction makes the functions of TRs even more significant, and availability of the special polymerase and, likely, DNA–RNA TR duplexes is in agreement with the mechanism of transfer and multiplication of TRs mediated by reverse transcription that was found in cancer cells (Fig. 7; [140]).
The presence of TRs in eDNA can be assumed in relation to the history of discovery of the complex of centromeric proteins CENP-A, CENP-B, and CENP-C [141]. These proteins were identified as main antigens in certain autoimmune disorders [142]. They were detected in CREST serum (Calcinosis, Raynaud’s phenomenon, Esophageal dysmotility, Sclerodactyly, Telangiectasia) in scleroderma. CENP-A and CENP-B are very well understood, their genes have been cloned, and antibodies towards pure proteins are commercially available. CENP-A was found to be the CEN-specific variant of the nucleosome core histone H3 [143]. DNA in the form of nucleosomes of the “beads-on-a-string” type, which is characteristic for TRs, has been found rather frequently in eDNA [138, 144-146]. Hence, the fact of enrichment of the membrane and eDNA with TRs demonstrated with sequencing is in good agreement with previous data.
TRs are present in eDNA, and precisely the TRs are the most variable portion of the genome. The fact that TRs are different even in closely related species [70, 147] indicates that the change of the TR repertoire implies fixation of a new species. The availability of TRs in the eDNA, mechanism of transcription, and reverse transcription of TRs demonstrated in the membrane fraction and in cancer cells suggests that one-step replacement of TR fields is possible during species fixation.
The questions related to the mysteries of constitutive heterochromatin are far from being resolved. It can be concluded based on the facts available at the present time that ERVs represent a vital component of HChr that likely includes certain ERV classes (IAP (ERV2) in mouse) and ~2-kb LINE fragment, transcripts of which in the interphase nucleus are the main components of Cot RNA.
The availability of TRs in the genome is characteristic for eukaryotes. So far we understand that: 1) the main portion of TRs is associated with interphase nucleus, in mice – with chromocenters; 2) TRs located in the euchromatin portion of the genome are likely an underlying morphogenetic program; 3) transcription of TRs is necessary for maintaining heterochromatin status of the HChr portion of the genome, but most important are the bursts of TR transcription accompanying normal embryogenesis and other stages of cardinal changes in the cell cycle including cancerogenesis; 4) the main hurdle in investigation of the role of TRs is lack of their satisfactory classification and annotation; up to the present time, TRs represent the “dark matter of the genome”.
Acknowledgments
This work was financially supported by the Presidium of the Russian Academy of Sciences, grant “Molecular and Cell Biology”, and by the Russian Science Foundation, grant No. 15-15-20026.
REFERENCES
1.Koryakov, D. E., and Zhimulev, I. F. (2009)
Chromosomes. Structure and Functions [in Russian], SO RAN
Publishers, Novosibirsk.
2.Wijchers, P. J., Geeven, G., Eyres, M., Bergsma, A.
J., Janssen, M., Verstegen, M., Zhu, Y., Schell, Y., Vermeulen, C., De
Wit, E., and De Laat, W. (2015) Characterization and dynamics of
pericentromere-associated domains in mice, Genome Res.,
25, 958-969.
3.Guenatri, M., Bailly, D., Maison, C., and Almouzni,
G. (2004) Mouse centric and pericentric satellite repeats form distinct
functional heterochromatin, J. Cell Biol., 166,
493-505.
4.Snapp, R. R., Goveia, E., Peet, L., Bouffard, N.
A., Badger, G. J., and Langevin, H. M. (2013) Spatial organization of
fibroblast nuclear chromocenters: component tree analysis, J.
Anat., 223, 255-261.
5.De Koning, A. J., Gu, W., Castoe, T. A., Batzer, M.
A., and Pollock, D. D. (2011) Repetitive elements may comprise over
two-thirds of the human genome, PLoS Genet., 7,
e1002384.
6.Probst, A. V., Okamoto, I., Casanova, M., Marjou,
F., Le Baccon, P., and Almouzni, G. (2010) A strand-specific burst in
transcription of pericentric satellites is required for chromocenter
formation and early mouse development, Dev. Cell, 19,
625-638.
7.Casanova, M., Pasternak, M., El Marjou, F., Le
Baccon, P., Probst, A. V., and Almouzni, G. (2013) Heterochromatin
reorganization during early mouse development requires a
single-stranded noncoding transcript, Cell Rep., 4,
1156-1167.
8.Zhu, Q., Pao, G. M., Huynh, A. M., Suh, H., Tonnu,
N., Nederlof, P. M., and Verma, I. M. (2011) BRCA1 tumour suppression
occurs via heterochromatin-mediated silencing, Nature,
477, 179-184.
9.Alexiadis, V., Ballestas, M. E., Sanchez, C.,
Winokur, S., Vedanarayanan, V., Warren, M., and Ehrlich, M. (2007)
RNAPol-ChIP analysis of transcription from FSHD-linked tandem repeats
and satellite DNA, Biochim. Biophys. Acta, 1769,
29-40.
10.Elgin, S. C., and Reuter, G. (2013)
Position-effect variegation, heterochromatin formation, and gene
silencing in Drosophila, Cold Spring Harb. Perspect.
Biol., 5, a01778.
11.Shatskikh, A. S., and Gvozdev, V. A. (2013)
Heterochromatin formation and transcription in relation to
trans-inactivation of genes and their spatial organization in the
nucleus, Biochemistry (Moscow), 78, 603-612.
12.Mayer, R., Brero, A., Von Hase, J., Schroeder,
T., Cremer, T., and Dietzel, S. (2005) Common themes and cell type
specific variations of higher order chromatin arrangements in the
mouse, BMC Cell. Biol., 6, 44.
13.Probst, A. V., and Almouzni, G. (2008)
Pericentric heterochromatin: dynamic organization during early
development in mammals, Differentiation, 76, 15-23.
14.Prusov, A. N., and Zatsepina, O. V. (2002)
Isolation of the chromocenter fraction from mouse liver nuclei,
Biochemistry (Moscow), 67, 423-431.
15.Zatsepina, O. V., Zharskaya, O. O., and Prusov,
A. N. (2008) Isolation of the constitutive heterochromatin from mouse
liver nuclei, in The Nucleus. Vol. 1: Nuclei and Subnuclear
Components, Springer, pp. 169-180.
16.Hutchins, A. P., and Pei, D. (2015) Transposable
elements at the center of the crossroads between embryogenesis,
embryonic stem cells, reprogramming, and long non-coding RNAs, Sci.
Bull., 60, 1722-1733.
17.Ostromyshenskii, D. I., Chernyaeva, E. N.,
Kuznetsova, I. S., and Podgornaya, O. I. (2018) Mouse chromocenters DNA
content: sequencing and in silico analysis, BMC Genomics,
19, 151.
18.Van der Kuyl, A. C. (2012) HIV infection and HERV
expression: a review, Retrovirology, 9, 6.
19.Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J.
L., Capy, P., Chalhoub, B., and Paux, E. (2007) A unified
classification system for eukaryotic transposable elements, Nat.
Rev. Genet., 8, 973-982.
20.Mouse Genome Sequencing Consortium; Waterston, R.
H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal,
P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P.,
Antonarakis, S. E., Attwood, J., Baertsch, R., Bailey, J., Barlow, K.,
Beck, S., Berry, E., Birren, B., Bloom, T., Bork, P., Botcherby, M.,
Bray, N., Brent, M. R., Brown, D. G., Brown, S. D., Bult, C., Burton,
J., Butler, J., Campbell, R. D., Carninci, P., Cawley, S., Chiaromonte,
F., Chinwalla, A. T., Church, D. M., Clamp, M., Clee, C., Collins, F.
S., Cook, L. L., Copley, R. R., Coulson, A., Couronne, O., Cuff, J.,
Curwen, V., Cutts, T., Daly, M., David, R., Davies, J., Delehaunty, K.
D., Deri, J., Dermitzakis, E. T., Dewey, C., Dickens, N. J., Diekhans,
M., Dodge, S., Dubchak, I., Dunn, D. M., Eddy, S. R., Elnitski, L.,
Emes, R. D., Eswara, P., Eyras, E., Felsenfeld, A., Fewell, G. A.,
Flicek, P., Foley, K., Frankel, W. N., Fulton, L. A., Fulton, R. S.,
Furey, T. S., Gage, D., Gibbs, R. A., Glusman, G., Gnerre, S., Goldman,
N., Goodstadt, L., Grafham, D., Graves, T. A., Green, E. D., Gregory,
S., Guigo, R., Guyer, M., Hardison, R. C., Haussler, D., Hayashizaki,
Y., Hillier, L. W., Hinrichs, A., Hlavina, W., Holzer, T., Hsu, F.,
Hua, A., Hubbard, T., Hunt, A., Jackson, I., Jaffe, D. B., Johnson, L.
S., Jones, M., Jones, T. A., Joy, A., Kamal, M., Karlsson, E. K.,
Karolchik, D., Kasprzyk, A., Kawai, J., Keibler, E., Kells, C., Kent,
W. J., Kirby, A., Kolbe, D. L., Korf, I., Kucherlapati, R. S.,
Kulbokas, E. J., Kulp, D., Landers, T., Leger, J. P., Leonard, S.,
Letunic, I., Levine, R., Li, J., Li, M., Lloyd, C., Lucas, S., Ma, B.,
Maglott, D. R., Mardis, E. R., Matthews, L., Mauceli, E., Mayer, J. H.,
McCarthy, M., McCombie, W. R., McLaren, S., McLay, K., McPherson, J.
D., Meldrim, J., Meredith, B., Mesirov, J. P., Miller, W., Miner, T.
L., Mongin, E., Montgomery, K. T., Morgan, M., Mott, R., Mullikin, J.
C., Muzny, D. M., Nash, W. E., Nelson, J. O., Nhan, M. N., Nicol, R.,
Ning, Z., Nusbaum, C., O’Connor, M. J., Okazaki, Y., Oliver, K.,
Overton-Larty, E., Pachter, L., Parra, G., Pepin, K. H., Peterson, J.,
Pevzner, P., Plumb, R., Pohl, C. S., Poliakov, A., Ponce, T. C.,
Ponting, C. P., Potter, S., Quail, M., Reymond, A., Roe, B. A., Roskin,
K. M., Rubin, E. M., Rust, A. G., Santos, R., Sapojnikov, V., Schultz,
B., Schultz, J., Schwartz, M. S., Schwartz, S., Scott, C., Seaman, S.,
Searle, S., Sharpe, T., Sheridan, A., Shownkeen, R., Sims, S., Singer,
J. B., Slater, G., Smit, A., Smith, D. R., Spencer, B., Stabenau, A.,
Stange-Thomann, N., Sugnet, C., Suyama, M., Tesler, G., Thompson, J.,
Torrents, D., Trevaskis, E., Tromp, J., Ucla, C., Ureta-Vidal, A.,
Vinson, J. P., Von Niederhausern, A. C., Wade, C. M., Wall, M., Weber,
R. J., Weiss, R. B., Wendl, M. C., West, A. P., Wetterstrand, K.,
Wheeler, R., Whelan, S., Wierzbowski, J., Willey, D., Williams, S.,
Wilson, R. K., Winter, E., Worley, K. C., Wyman, D., Yang, S., Yang, S.
P., Zdobnov, E. M., Zody, M. C., and Lander, E. S. (2002) Initial
sequencing and comparative analysis of the mouse genome, Nature,
420, 520-562.
21.Komissarov, A. S., Gavrilova, E. V., Demin, S.
J., Ishov, A. M., and Podgornaya, O. I. (2011) Tandemly repeated DNA
families in the mouse genome, BMC Genomics, 12, 531.
22.Dunn, C. A., Romanish, M. T., Gutierrez, L. E.,
Van de Lagemaat, L. N., and Mager, D. L. (2006) Transcription of two
human genes from a bidirectional endogenous retrovirus promoter,
Gene, 366, 335-342.
23.Carone, D. M., Longo, M. S., Ferreri, G. C.,
Hall, L., Harris, M., Shook, N., and O’Neill, R. J. (2009) A new
class of retroviral and satellite encoded small RNAs emanates from
mammalian centromeres, Chromosoma, 118, 113-125.
24.Longo, M. S., Carone, D. M., Green, E. D.,
O’Neill, M. J., and O’Neill, R. J. (2009) Distinct
retroelement classes define evolutionary breakpoints demarcating sites
of evolutionary novelty, BMC Genomics, 10, 334.
25.Ruiz-Herrera, A., Farre, M., and Robinson, T. J.
(2012) Molecular cytogenetic and genomic insights into chromosomal
evolution, Heredity, 108, 28-36.
26.Ferreri, G. C., Brown, J. D., Obergfell, C., Jue,
N., Finn, C. E., O’Neill, M. J., and O’Neill, R. J. (2011)
Recent amplification of the kangaroo endogenous retrovirus, KERV,
limited to the centromere, J. Virol., 85, 4761-4771.
27.Brattas, P. L., Jonsson, M. E., Fasching, L.,
Wahlestedt, J. N., Shahsavani, M., Falk, R., and Jakobsson, J. (2017)
TRIM28 controls a gene regulatory network based on endogenous
retroviruses in human neural progenitor cells, Cell Rep.,
18, 1-11.
28.Chuong, E. B., Rumi, M. K., Soares, M. J., and
Baker, J. C. (2013) Endogenous retroviruses function as
species-specific enhancer elements in the placenta, Nat. Genet.,
45, 325-329.
29.Lynch, V. J., Nnamani, M. C., Kapusta, A.,
Brayer, K., Plaza, S. L., Mazur, E. C., and Graf, A. (2015) Ancient
transposable elements transformed the uterine regulatory landscape and
transcriptome during the evolution of mammalian pregnancy, Cell
Rep., 10, 551-561.
30.Roberts, R. M., Green, J. A., and Schulz, L. C.
(2016) The evolution of the placenta, Reproduction, 152,
179-189.
31.Imakawa, K., and Nakagawa, S. (2017) The
phylogeny of placental evolution through dynamic integrations of
retrotransposons, Prog. Mol. Biol. Transl. Sci., 145,
89-109.
32.Mager, D. L., and Stoye, J. P. (2015) Mammalian
endogenous retroviruses, Microbiol. Spectrum, 3, No. 1,
MDNA3-0009-2014; doi: 10.1128/microbiolspec.MDNA3-0009-2014.
33.Lu, X., Sachs, F., Ramsay, L., Jacques, P. E.,
Goke, J., Bourque, G., and Ngoh, H. (2014) The retrovirus HERVH is a
long noncoding RNA required for human embryonic stem cell identity,
Nat. Struct. Mol. Biol., 21, 423-425.
34.Goke, J., Lu, X., Chan, Y. S., Ng, H. H., Ly, L.
H., Sachs, F., and Szczerbinska, I. (2015) Dynamic transcription of
distinct classes of endogenous retroviral elements marks specific
populations of early human embryonic cells, Cell Stem Cell,
16, 135-141.
35.Schoorlemmer, J., Perez-Palacios, R., Climent,
M., Guallar, D., and Muniesa, P. (2014) Regulation of mouse
retroelement MuERV-L/MERVL expression by REX1 and epigenetic control of
stem cell potency, Front. Oncol., doi:
10.3389/fonc.2014.00014.
36.Robbez-Masson, L., and Rowe, H. M. (2015)
Retrotransposons shape species-specific embryonic stem cell gene
expression, Retrovirology, 12, 45.
37.Crichton, J. H., Dunican, D. S., MacLennan, M.,
Meehan, R. R., and Adams, I. R. (2014) Defending the genome from the
enemy within: mechanisms of retrotransposon suppression in the mouse
germline, Cell. Mol. Life Sci., 71, 1581-1605.
38.Gerdes, P., Richardson, S. R., Mager, D. L., and
Faulkner, G. J. (2016) Transposable elements in the mammalian embryo:
pioneers surviving through stealth and service, Genome Biol.,
17, 100.
39.Wong, C., Chen, A. A., Behr, B., and Shen, S.
(2013) Time-lapse microscopy and image analysis in basic and clinical
embryo development research, Reprod. Biomed. Online, 26,
120-129.
40.Grow, E. J., Flynn, R. A., Chavez, S. L.,
Bayless, N. L., Wossidlo, M., Wesche, D. J., and Pera, R. A. R. (2015)
Intrinsic retroviral reactivation in human preimplantation embryos and
pluripotent cells, Nature, 522, 221-225.
41.Boyle, A. L., Ballard, S. G., and Ward, D. C.
(1990) Differential distribution of long and short interspersed element
sequences in the mouse genome: chromosome karyotyping by fluorescence
in situ hybridization, Proc. Natl. Acad. Sci. USA,
87, 7757-7761.
42.Waterston, R. H., Lander, E. S., and Sulston, J.
E. (2002) On the sequencing of the human genome, Proc. Natl. Acad.
Sci. USA, 99, 3712-3716.
43.Solovei, I., Kreysing, M., Lanctot, C., Kosem,
S., Peichl, L., Cremer, T., and Joffe, B. (2009) Nuclear architecture
of rod photoreceptor cells adapts to vision in mammalian evolution,
Cell, 137, 356-368.
44.Podgornaya, O., Gavrilova, E., Stephanova, V.,
Demin, S., and Komissarov, A. (2013) Large tandem repeats make up the
chromosome bar code: a hypothesis, Adv. Protein Chem. Struct.
Biol., 90, 1-30.
45.Miga, K. H. (2015) Completing the human genome:
the progress and challenge of satellite DNA assembly, Chromosome
Res., 23, 421-426.
46.Kuznetsova, I. S., Ostromyshenskii, D. I.,
Komissarov, A. S., Prusov, A. N., Waisertreiger, I. S., Gorbunova, A.
V., Trifonov, V. A., Ferguson-Smith, M., and Podgornaya, O. I. (2016)
LINE-related component of mouse heterochromatin and complex
chromocenters’ composition, Chromosome Res., 24,
309-323.
47.Ostromyshenskii, D. I., Komissarov, A. S.,
Kuznetsova, I. S., Chernyaeva, E. N., Vaysertreyger, I. R., and
Podgornaya, O. I. (2016) The structure of DNA chromocentres in mouse
in silico and in situ. LINE and ERV fragments are an
obligatory components of DNA chromocentres besides tandem repeats,
Tsitologiya, 58, 389-392.
48.Van de Werken, H. J. G., De Haan, J. C.,
Feodorova, Y., Bijos, D., Weuts, A., Theunis, K., and Kumar, P. (2017)
Small chromosomal regions position themselves autonomously according to
their chromatin class, Genome Res., 27, 922-933.
49.Choo, K. H. (1997) Centromeres, John Wiley
& Sons, Ltd.
50.Fadloun, A., Le Gras, S., Jost, B.,
Ziegler-Birling, C., Takahashi, H., Gorab, E., and Torres-Padilla, M.
E. (2013) Chromatin signatures and retrotransposon profiling in mouse
embryos reveal regulation of LINE-1 by RNA, Nat. Struct. Mol.
Biol., 20, 332-338.
51.Hall, L. L., Carone, D. M., Gomez, A. V., Kolpa,
H. J., Byron, M., Mehta, N., and Lawrence, J. B. (2014) Stable C0 T-1
repeat RNA is abundant and is associated with euchromatic interphase
chromosomes, Cell, 156, 907-919.
52.Kit, S. (1961) Equilibrium sedimentation in
density gradients of DNA preparations from animal tissues, J. Mol.
Biol., 3, 711-716.
53.Vogt, P. (1990) Potential genetic functions of
tandem repeated DNA sequence blocks in the human genome are based on a
highly conserved “chromatin folding code”, Hum.
Genet., 84, 301-336.
54.Pavlek, M., Gelfand, Y., Plohl, M., and
Mestrovic, N. (2015) Genome-wide analysis of tandem repeats in
Tribolium castaneum genome reveals abundant and highly dynamic
tandem repeat families with satellite DNA features in euchromatic
chromosomal arms, DNA Res., 22, 387-401.
55.Wevrick, R., and Willard, H. F. (1991) Physical
map of the centromeric region of human chromosome 7: relationship
between two distinct alpha satellite arrays, Nucleic Acids Res.,
19, 2295-2301.
56.Ikeno, M., Masumoto, H., and Okazaki, T. (1994)
Distribution of CENP-B boxes reflected in CREST centromere antigenic
sites on long-range α-satellite DNA arrays of human chromosome
21, Hum. Mol. Genetics, 3, 1245-1257.
57.He, D., Zeng, C., Woods, K., Zhong, L., Turner,
D., Busch, R. K., and Busch, H. (1998) CENP-G: a new centromeric
protein that is associated with the α-1 satellite DNA subfamily,
Chromosoma, 107, 189-197.
58.Miheev, D. Yu., Podgornaya, O. I., and
Ostromyshenskii, D. I. (2015) Large tandem repeats of Mesocricetus
auratus in silico and in situ, Tsitologiya,
57, 95-101.
59.Ostromyshenskii, D. I., Kuznetsova, I. S.,
Komissarov, A. S., Kartavtseva, I. V., and Podgornaya, O. I. (2015)
Tandem repeats in rodents genome and their mapping, Tsitologiya,
57, 102-110.
60.Ames, D., Murphy, N., Helentjaris, T., Sun, N.,
and Chandler, V. (2008) Comparative analyses of human single- and
multilocus tandem repeats, Genetics, 179, 1693-1704.
61.Warburton, P. E., Hasson, D., Guillem, F.,
Lescale, C., Jin, X., and Abrusan, G. (2008) Analysis of the largest
tandemly repeated DNA families in the human genome, BMC
Genomics, 9, 533.
62.Alkan, C., Cardone, M. F., Catacchio, C. R.,
Antonacci, F., O’Brien, S. J., Ryder, O., and Ventura, M. (2011)
Genome-wide characterization of centromeric satellites from multiple
mammalian genomes, Genome Res., 21, 137-145.
63.Capy, P. (2005) Classification and nomenclature
of retrotransposable elements, Cytogenet. Genome Res.,
110, 457-461.
64.Kronmiller, B. A., and Wise, R. P. (2008) Tenest:
automated chronological annotation and visualization of nested plant
transposable elements, Plant Physiol., 146, 45-59.
65.Feschotte, C., Keswani, U., Ranganathan, N.,
Guibotsy, M. L., and Levine, D. (2009) Exploring repetitive DNA
landscapes using REPCLASS, a tool that automates the classification of
transposable elements in eukaryotic genomes, Genome Biol. Evol.,
1, 205-220.
66.Seberg, O., and Petersen, G. (2009) A unified
classification system for eukaryotic transposable elements should
reflect their phylogeny, Nat. Rev. Genet., 10,
276-276.
67.Vassetzky, N. S., and Kramerov, D. A. (2012)
SINEBase: a database and tool for SINE analysis, Nucleic Acids
Res., 41, 83-89.
68.Gelfand, Y., Rodriguez, A., and Benson, G. (2006)
TRDB – the tandem repeats database, Nucleic Acids Res.,
35, 80-87.
69.Jurka, J., Kapitonov, V. V., Pavlicek, A.,
Klonowski, P., Kohany, O., and Walichiewicz, J. (2005) Repbase Update,
a database of eukaryotic repetitive elements, Cytogenet. Genome
Res., 110, 462-467.
70.Melters, D. P., Bradnam, K. R., Young, H. A.,
Telis, N., May, M. R., Ruby, J. G., and Garcia, J. F. (2013)
Comparative analysis of tandem repeats from hundreds of species reveals
unique insights into centromere evolution, Genome Biol.,
14, R10.
71.Kuznetsova, I., Podgornaya, O., and
Ferguson-Smith, M. A. (2006) High-resolution organization of mouse
centromeric and pericentromeric DNA, Cytogenet. Genome Res.,
112, 248-255.
72.Lobov, I. B., Tsutsui, K., Mitchell, A. R., and
Podgornaya, O. (2000) Specific interaction of mouse major satellite
with MAR-binding protein SAF-A, Europ. J. Cell Biol., 79,
839-849.
73.Lobov, I. B., Tsutsui, K., Mitchell, A. R., and
Podgornaya, O. I. (2001) Specificity of SAF‐A and lamin B
binding in vitro correlates with the satellite DNA bending
state, J. Cell. Biochem., 83, 218-229.
74.Enukashvily, N., Donev, R., Sheer, D., and
Podgornaya, O. (2005) Satellite DNA binding and cellular localisation
of RNA helicase P68, J. Cell Sci., 118, 611-622.
75.Podgornaya, O. I., Voronin, A. P., Enukashvily,
N., Matveev, I. V., and Lobov, I. B. (2003) Structure-specific
DNA-binding proteins as the foundation for three-dimensional chromatin
organization, Int. Rev. Cytol., 224, 227-296.
76.Fondon, J. W., and Garner, H. R. (2004) Molecular
origins of rapid and continuous morphological evolution, Proc. Natl.
Acad. Sci. USA, 101, 18058-18063.
77.Politz, J. C. R., Scalzo, D., and Groudine, M.
(2013) Something silent this way forms: the functional organization of
the repressive nuclear compartment, Ann. Rev. Cell Develop.
Biol., 29, 241-270.
78.Cremer, M., Solovei, I., Schermelleh, L., and
Cremer, T. (2003) Chromosomal arrangement during different phases of
the cell cycle, in Nature Encyclopedia of the Human Genome,
Macmillan Publishers Ltd., Nature Publishing Group, pp. 451-457.
79.Kuznetsova, I. S., Enukashvily, N. I.,
Noniashvili, E. M., Shatrova, A. N., Aksenov, N. D., Zenin, V. V.,
Dyban, A. P., and Podgornaya, O. I. (2007) Evidence for the existence
of satellite DNA‐containing connection between metaphase
chromosomes, J. Cell. Biochem., 101, 1046-1061.
80.Wang, L. H.-C., Schwarzbraun, T., Speicher, M.
R., and Nigg, E. A. (2008) Persistence of DNA threads in human anaphase
cells suggests late completion of sister chromatid decatenation,
Chromosoma, 117, 123-135.
81.Dreissig, S., Schiml, S., Schindele, P., Weiss,
O., Rutten, T., Schubert, V., and Houben, A. (2017) Live cell
CRISPR‐imaging in plants reveals dynamic telomere movements,
Plant J., 91, 565-573.
82.Cohen, A. K., Huh, T. Y., and Helleiner, C. W.
(1973) Transcription of satellite DNA in mouse L-cells, Can. J.
Biochem., 51, 529-532.
83.Cohen, A. K., Rode, H. N., and Helleiner, C. W.
(1972) The time of synthesis of satellite DNA in mouse cells (L cells),
Can. J. Biochem., 50, 229-231.
84.Seidman, M. M., and Cole, R. D. (1977) Chromatin
fractionation related to cell type and chromosome condensation but
perhaps not to transcriptional activity, J. Biol. Chem.,
252, 2630-2639.
85.Haaf, T., and Ward, D. C. (1996) Inhibition of
RNA polymerase II transcription causes chromatin decondensation, loss
of nucleolar structure, and dispersion of chromosomal domains, Exp.
Cell Res., 224, 163-173.
86.Macgregor, H. C. (1979) In situ
hybridization of highly repetitive DNA to chromosomes of Triturus
cristatus, Chromosoma, 71, 57-64.
87.Varley, J. M., Macgregor, H. C., Nardi, I.,
Andrews, C., and Erba, H. P. (1980) Cytological evidence of
transcription of highly repeated DNA sequences during the lampbrush
stage in Triturus cristatus carnifex, Chromosoma,
80, 289-307.
88.Krasikova, A. V., Vasilevskaia, E. V., and
Gaginskaia, E. R. (2010) Chicken lampbrush chromosomes: transcription
of tandemly repetitive DNA sequences, Genetika, 46,
1329-1334.
89.Rouleux-Bonnin, F., Bigot, S., and Bigot, Y.
(2004) Structural and transcriptional features of Bombus
terrestris satellite DNA and their potential involvement in the
differentiation process, Genome, 47, 877-888.
90.Rizzi, N., Denegri, M., Chiodi, I., Corioni, M.,
Valgardsdottir, R., Cobianchi, F., and Biamonti, G. (2004)
Transcriptional activation of a constitutive heterochromatic domain of
the human genome in response to heat shock, Mol. Biol. Cell,
15, 543-551.
91.Lehnertz, B., Ueda, Y., Derijck, A. A.,
Braunschweig, U., Perez-Burgos, L., Kubicek, S., and Peters, A. H.
(2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA
methylation to major satellite repeats at pericentric heterochromatin,
Curr. Biol., 13, 1192-1200.
92.Rudert, F., Bronner, S., Garnier, J. M., and
Dolle, P. (1995) Transcripts from opposite strands of gamma satellite
DNA are differentially expressed during mouse development, Mamm.
Genome, 6, 76-83.
93.Lorite, P., Renault, S., Rouleux-Bonnin, F.,
Bigot, S., Periquet, G., and Palomeque, T. (2002) Genomic organization
and transcription of satellite DNA in the ant Aphaenogaster
subterranea (Hymenoptera, Formicidae), Genome, 45,
609-616.
94.Lee, H. R., Neumann, P., Macas, J., and Jiang, J.
(2006) Chromosomal localization, copy number assessment, and
transcriptional status of BamHI repeat fractions in water buffalo
Bubalus bubalis, Mol. Biol. Evol., 23,
2505-2520.
95.Ting, D. T., Lipson, D., Paul, S., Brannigan, B.
W., Akhavanfard, S., Coffman, E. J., and Rivera, M. N. (2011) Aberrant
overexpression of satellite repeats in pancreatic and other epithelial
cancers, Science, 331, 593-596.
96.Kuznetsova, I. S., Thevasagayam, N. M., Sridatta,
P. S., Komissarov, A. S., Saju, J. M., Ngoh, S. Y., Jiang, J., Shen,
X., and Orban, L. (2014) Primary analysis of repeat elements of the
Asian seabass (Lates calcarifer) transcriptome and
genome, Front. Genet., doi: 10.3389/fgene.2014.00223.
97.Saksouk, N., Simboeck, E., and Dejardin, J.
(2015) Constitutive heterochromatin formation and transcription in
mammals, Epigenet. Chromatin, doi: 10.1186/1756-8935-8-3.
98.Enukashvily, N. I., and Ponomartsev, N. V. (2013)
Mammalian satellite DNA: a speaking dumb, Adv. Protein Chem. Struct.
Biol., 90, 31-65.
99.Valgardsdottir, R., Chiodi, I., Giordano, M.,
Rossi, A., Bazzini, S., Ghigna, C., Riva, S., and Biamonti, G. (2008)
Transcription of satellite III non-coding RNAs is a general stress
response in human cells, Nucleic Acids Res., 36,
423-434.
100.Lu, J., and Gilbert, D. M. (2007)
Proliferation-dependent and cell cycle-regulated transcription of mouse
pericentric heterochromatin, J. Cell Biol., 179,
411-421.
101.Bersani, F., Leeb, E., Kharchenko, P. V., Xu,
A. W., Liu, M., Xega, K., MacKenzie, O. C., Brannigan, B. W., Wittner,
B. S., Jung, H., Ramaswamy, S., Park, P. J., Maheswaran, S., Ting, D.
T., and Haber, D. A. (2015) Pericentromeric satellite repeat expansions
through RNA-derived DNA intermediates in cancer, Proc. Natl. Acad.
Sci. USA, 112, 15148-15153.
102.Kuznetsova, T. V., Enukashvily, N. I.,
Trofimova, I. L., Gorbunova, A., Vashukova, E. S., and Baranov, V. S.
(2012) Localization and transcription of human chromosome 1
pericentromeric heterochromatin in embryonic and extraembryonic tis
sues, Med. Genet. (Moscow), 11, 19-24.
103.Trofimova, I. L., Enukashvily, N. I.,
Kuznetsova, T. V., and Baranov, V. S. (2018) Transcription of satellite
DNA in human embryogenesis: literature review and our own data, Med.
Genet. (Moscow), 17, 3-7.
104.Suzuki, T., Fujii, M., and Ayusawa, D. (2002)
Demethylation of classical satellite 2 and 3 DNA with chromosomal
instability in senescent human fibroblasts, Exp. Gerontol.,
37, 1005-1014.
105.Tessadori, F., Schulkes, R. K., Van Driel, R.,
and Fransz, P. (2007) Light-regulated large-scale reorganization of
chromatin during the floral transition in Arabidopsis, Plant
J., 50, 848-857.
106.Zhang, P., Kerkela, E., Skottman, H., Levkov,
L., Kivinen, K., Lahesmaa, R., and Kere, J. (2007) Distinct sets of
developmentally regulated genes that are expressed by human oocytes and
human embryonic stem cells, Fertil. Steril., 87,
677-690.
107.Gerrard, D. T., Berry, A. A., Jennings, R. E.,
Hanley, K. P., Bobola, N., and Hanley, N. A. (2016) An integrative
transcriptomic atlas of organogenesis in human embryos, Elife,
pii: e15657.
108.Santenard, A., Ziegler-Birling, C., Koch, M.,
Tora, L., Bannister, A. J., and Torres-Padilla, M. E. (2010)
Heterochromatin formation in the mouse embryo requires critical
residues of the histone variant H3.3, Nat. Cell Biol.,
12, 853-862.
109.Enukashvily, N. I., Malashicheva, A. B., and
Waisertreiger, I. S. (2009) Satellite DNA spatial localization and
transcriptional activity in mouse embryonic E-14 and IOUD2 stem cells,
Cytogenet. Genome Res., 124, 277-287.
110.Kuhn, G. C. S. (2015) Satellite DNA transcripts
have diverse biological roles in Drosophila, Heredity,
115, 1-2.
111.Bouzinba-Segard, H., Guais, A., and Francastel,
C. (2006) Accumulation of small murine minor satellite transcripts
leads to impaired centromeric architecture and function, Proc. Natl.
Acad. Sci. USA, 103, 8709-8714.
112.Gaubatz, J. W., and Cutler, R. G. (1990) Mouse
satellite DNA is transcribed in senescent cardiac muscle, J. Biol.
Chem., 265, 17753-17758.
113.Pastor, B. M., and Mostoslavsky, R. (2016)
SIRT6: a new guardian of mitosis, Nat. Struct. Mol. Biol.,
23, 360-362.
114.Chan, D. L., Moralli, D., Khoja, S., and
Monaco, Z. L. (2017) Noncoding centromeric RNA expression impairs
chromosome stability in human and murine stem cells, Dis.
Markers, 7506976.
115.Enukashvily, N. I., Donev, R., Waisertreiger,
I. S.-R., and Podgornaya, O. I. (2007) Human chromosome 1 satellite 3
DNA is decondensed, demethylated and transcribed in senescent cells and
in A431 epithelial carcinoma cells, Cytogenet. Genome Res.,
118, 42-54.
116.De Cecco, M., Criscione, S. W., Peckham, E. J.,
Hillenmeyer, S., Hamm, E. A., Manivannan, J., Peterson, A. L.,
Kreiling, J. A., Neretti, N., and Sedivy, J. M. (2013) Genomes of
replicatively senescent cells undergo global epigenetic changes leading
to gene silencing and activation of transposable elements, Aging
Cell, 12, 247-256.
117.Wong, L. H., Brettingham-Moore, K. H., Chan,
L., Quach, J. M., Anderson, M. A., Northrop, E. L., Hannan, R.,
Saffery, R., Shaw, M. L., Williams, E., and Choo, K. A. (2007)
Centromere RNA is a key component for the assembly of nucleoproteins at
the nucleolus and centromere, Genome Res., 17,
1146-1160.
118.Du, Y., Topp, C. N., and Dawe, R. K. (2010) DNA
binding of centromere protein C (CENPC) is stabilized by
single-stranded RNA, PLoS Genet., 6, e1000835.
119.Eymery, A., Souchier, C., Vourc’h, C.,
and Jolly, C. (2010) Heat shock factor 1 binds to and transcribes
satellite II and III sequences at several pericentromeric regions in
heat-shocked cells, Exp. Cell Res., 316, 1845-1855.
120.Jolly, C., Metz, A., Govin, J., Vigneron, M.,
Turner, B. M., Khochbin, S., and Vourc’h, C. (2004)
Stress-induced transcription of satellite III repeats, J. Cell.
Biol., 164, 25-33.
121.Col, E., Hoghoughi, N., Dufour, S., Penin, J.,
Koskas, S., Faure, V., Ouzounova, M., Hernandez-Vargash, H., Reynoird,
N., Daujat, S., Folco, E., Vigneron, M., Schneider, R., Verdel, A.,
Khochbin, S., Herceg, Z., Caron, C., and Vourc’h, C. (2017)
Bromodomain factors of BET family are new essential actors of
pericentric heterochromatin transcriptional activation in response to
heat shock, Nature Sci. Rep., 7, 5418.
122.Sengupta, S., Parihar, R., and Ganesh, S.
(2009) Satellite III non-coding RNAs show distinct and stress-specific
patterns of induction, Biochem. Biophys. Res. Commun.,
382, 102-107.
123.Morozov, V. M., Gavrilova, E. V., Ogryzko, V.
V., and Ishov, A. M. (2012) Dualistic function of Dax at centromeric
and pericentromeric heterochromatin in normal and stress conditions,
Nucleus, 3, 276-285.
124.Pezer, Z., and Ugarkovic, D. (2012) Satellite
DNA-associated siRNAs as mediators of heat shock response in insects,
RNA Biol., 9, 587-595.
125.Wang, Y., Zhang, Z., Chi, Y., Zhang, Q., Xu,
F., Yang, Z., Meng, L., Yang, S., Yan, S., Mao, A., Zhang, J., Yang,
Y., Wang, S., Cui, J., Liang, L., Ji, Y., Han, Z.-B., Fang, X., and
Han, Z. C. (2013) Long-term cultured mesenchymal stem cells frequently
develop genomic mutations but do not undergo malignant transformation,
Cell Death Dis., 4, 950.
126.Ponomartsev, N., Bulavin, D., Enukashvily, N.,
and Brichkina, A. (2016) Transcription of pericentromeric major
satellite DNA in lung cancer, Cytogenet. Genome Res.,
148, 146.
127.Ruiz-Herrera, A., Castresana, J., and Robinson,
T. J. (2006) Is mammalian chromosomal evolution driven by regions of
genome fragility? Genome Biol., 7, 115.
128.Podgornaya, O. I. (2016) Extracellular DNA for
the unsolved evolutionary problems, Tsitologiya, 58,
385-388.
129.Anker, P., Stroun, M., and Maurice, P. A.
(1975) Spontaneous release of DNA by human blood lymphocytes as shown
in vitro system, Cancer Res., 35, 2375-2382.
130.Vasyukhin, V. I., Lipskaya, L. A., Tsvetkov, A.
G., and Podgornaya, O. I. (1991) DNA excreted by human lymphocytes
contains sequences homologous to Ck gene, Mol. Biol.
(Moscow), 25, 405-412.
131.Vasioukhin, V., Anker, P., Maurice, P.,
Lyautey, J., Lederrey, C., and Stroun, M. (1994) Point mutations of the
N‐ras gene in the blood plasma DNA of patients with
myelodysplastic syndrome or acute myelogenous leukaemia, Brit. J.
Haematol., 86, 774-779.
132.Thierry, A. R., Mouliere, F., El Messaoudi, S.,
Mollevi, C., Lopez-Crapez, E., Rolet, F., Gillet, B., Gongora, C.,
Dechelotte, P., Robert, B., Del Rio, M., Lamy, P.-J., Bibeau, F.,
Nouaille, M., Loriot, V., Jarrousse, A.-S., Molina, F., Mathonnet, M.,
Pezet, D., and Ychou, M. (2014) Clinical validation of the detection of
KRAS and BRAF mutations from circulating tumor DNA, Nat. Med.,
20, 430-435.
133.Murtaza, M., Dawson, S. J., Tsui, D. W., Gale,
D., Forshew, T., Piskorz, A. M., Parkinson, C., Chin, S.-F., Kingsbury,
Z., Wong, A. S. C., Marass, F., Humphray, S., Hadfield, J., Bentley,
D., Chin, T. M., Brenton, J. D., Caldas, C., and Rosenfeld, N. (2013)
Non-invasive analysis of acquired resistance to cancer therapy by
sequencing of plasma DNA, Nature, 497, 108-112.
134.Rykova, E. Y., Morozkin, E. S., Ponomaryova, A.
A., Loseva, E. M., Zaporozhchenko, I. A., Cherdyntseva, N. V., Vlasov,
V. V., and Laktionov, P. P. (2012) Cell-free and cell-bound circulating
nucleic acid complexes: mechanisms of generation, concentration and
content, Expert. Opin. Biol. Ther., 12, 141-153.
135.Mittra, I., Khare, N. K., Raghuram, G. V.,
Chaubal, R., Khambatti, F., Gupta, D., Gaikwad, A., Prasannan, P.,
Singh, A., Iyer, A., Singh, A., Upadhyay, P., Nair, N. K., Mishra, P.
K., and Dutt, A. (2015) Circulating nucleic acids damage DNA of healthy
cells by integrating into their genomes, J. Biosci., 40,
91-111.
136.Morozkin, E. S., Loseva, E. M., Morozov, I. V.,
Kurilshikov, A. M., Bondar, A. A., Rykova, E. Y., Rubtsov, N. B.,
Vlasov, V. V., and Laktionov, P. P. (2012) A comparative study of
cell-free apoptotic and genomic DNA using FISH and massive parallel
sequencing, Expert Opin. Biol. Ther., 12, 141-153.
137.Beck, J., Urnovitz, H. B., Riggert, J.,
Clerici, M., and Schutz, E. (2009) Profile of the circulating DNA in
apparently healthy individuals, Clin. Chem., 55,
730-738.
138.Vasil’eva, I. N., Podgornaya, O. I., and
Bespalov, V. G. (2015) Nucleosome fraction of extracellular DNA as the
index of apoptosis, Tsitologiya, 57, 87-94.
139.Cheng, J., Torkamani, A., Peng, Y., Jones, T.
M., and Lerner, R. A. (2012) Plasma membrane associated transcription
of cytoplasmic DNA, Proc. Natl. Acad. Sci. USA, 109,
10827-10831.
140.Youngera, S. T., and Rinna, J. L. (2015) Silent
pericentromeric repeats speak out, Proc. Natl. Acad. Sci. USA,
112, 15008-15009.
141.Podgornaya, O. I., Vasilyeva, I. N., and
Bespalov, V. G. (2016) Heterochromatic tandem repeats in the
extracellular DNA, in Circulating Nucleic Acids in Serum and Plasma
– CNAPS IX, pp. 85-89.
142.Earnshaw, W. C., Halligan, N., Cooke, C., and
Rothfield, N. (1984) The kinetochore is part of the metaphase
chromosome scaffold, J. Cell. Biol., 98, 352-357.
143.Zasadzinska, E., and Foltz, D. R. (2017)
Orchestrating the specific assembly of centromeric nucleosomes, in
Centromeres and Kinetochores, Springer, Cham, pp. 165-192.
144.Van Helden, P. D. (1985) Potential
Z-DNA-forming elements in serum DNA from human systemic lupus
erythematosus, J. Immunol., 134, 177-179.
145.Herrman, M., Leitmann, W., Krapf, E. F., and
Kalden, J. R. (1989) Molecular characterization and in vitro
effects of nucleic acids from plasma of patients with systemic lupus
erythematosus, in Molecular and Cellular Mechanisms of Human
Hypersensitivity and Autoimmunity, Wiley, New York, pp.
147-157.
146.Winter, O., Musiol, S., Schabowsky, M., Cheng,
Q., Khodadadi, L., and Hiepe, F. (2015) Analyzing pathogenic
(double-stranded (ds) DNA-specific) plasma cells via immunofluorescence
microscopy, Arthritis Res. Ther., 17, 293.
147.Ostromyshenskii, D. I., Kuznetsova, I. S.,
Golenishchev, F. N., Malikov, V. G., and Podgornaya, O. I. (2011)
Satellite DNA as a phylogenetic marker: case study of three genera of
the murine subfamily, Cell Tiss. Biol., 5, 543-550.
148.Carey, N. (2015) Junk DNA: A Journey through
the Dark Matter of the Genome, Icon Books.