MINI-REVIEW: Genome Editing and the Problem of Tetraploidy in Cell Modeling of the Genetic Form of Parkinsonism
V. V. Simonova1, A. S. Vetchinova1, E. V. Novosadova2,a, L. G. Khaspekov1,b, and S. N. Illarioshkin1,c*
1Research Center of Neurology, 125367 Moscow, Russia2Institute of Molecular Genetics, Russian Academy of Sciences, 123182 Moscow, Russia
* To whom correspondence should be addressed.
Received April 19, 2018; Revision received May 8, 2018
The prevalent form of familial parkinsonism is caused by mutations in the LRRK2 gene encoding for the mitochondrial protein kinase. In the review, we discuss possible causes of appearance of tetraploid cells in neuronal precursors obtained from induced pluripotent stem cells from patients with the LRRK2-associated form of parkinsonism after genome editing procedure. As LRRK2 protein participates in cell proliferation and maintenance of the nuclear envelope, spindle fibers, and cytoskeleton, mutations in the LRRK2 gene can affect protein functions and lead, via various mechanisms, to the mitotic machinery disintegration and chromosomal aberration. These abnormalities can appear at different stages of fibroblast reprogramming; therefore, editing of the LRRK2 nucleotide sequence should be done during or before the reprogramming stage.
KEY WORDS: parkinsonism, LRRK2, induced pluripotent stem cells, neuronal precursors, genome editing, tetraploidyDOI: 10.1134/S0006297918090055
Abbreviations: ATM, ataxia telangiectasia mutated; iPSCs, induced pluripotent stem cells; PD, Parkinson’s disease.
Parkinson’s disease (PD) is one of the most prevalent
age-associated neurodegenerative diseases (one case per 120,000
persons; 1% among persons over 60 years old) [1].
The absolute majority of PD cases is sporadic and has a multifactor
nature; the monogenic forms of PD comprise ~10% of all cases [2]. Among the monogenic forms, the most frequent are
those associated with mutations in the LRRK2, PARK2,
GBA, PINK1, DJ-1, and VPS35 genes [3, 4]. Genetic variants of primary
parkinsonism have made it possible to discover key components of PD
pathogenesis, such as mitochondrial dysfunction and impairments in
processing of neuronal proteins [5].
During the last few years, special attention has been given to PD modeling with induced pluripotent stem cells (iPSCs) derived from somatic cells of patients with genetically determined parkinsonism [6, 7]. Using this technique, effects of mutations can be analyzed in vitro in any type of cells purposefully differentiated from iPSCs. This approach allows to study molecular mechanisms of neurodegenerative process in “proper” patient’s neurons with unique genetic profile, which heralds a transition to the cellular level of personalized neurology. Cell templates obtained from iPSCs can be used as a tool for personalized screening of pharmaceutical preparations with a neuroprotective potential [8]. Moreover, iPSCs can be also used in substitutive PD therapy using dopaminergic neurons obtained from patient’s fibroblasts to eliminate complex ethical and immunologic problems of neurotransplantation [9]. Genome editing presents additional possibilities for cell modeling of monogenic PD forms because it allows targeted correction of mutant sequences and assessment of phenotypic expression of different nucleotide variants in isogenic cell lines [10].
As any new technique, cell reprogramming is associated with a number of problems that have to be solved before introducing this technique into clinical practice. Despite all obvious advantages of cell reprogramming, it is important to remember the risks associated with interference into the genome at the stages of reprogramming and genome editing. Moreover, when working with specific molecular targets, one has to consider gene networks, i.e., coordinatively expressed genes and their products whose interaction can influence the final result.
In this mini-review, we discuss the problem of tetraploidy in genome editing. We have encountered this problem when editing the genome of iPSCs obtained from fibroblasts of a PD patient who was a carrier of the G2019S mutation in the LRRK2 gene. This gene is associated with the development of the most frequent hereditary form of PD recorded in 7% cases of primary parkinsonism in different world populations and in up to 40% cases in the populations from the Middle-East Mediterranean region [11]. Carriers of mutations in LRRK2 inherit the disease by the autosomal-recessive type. The protein product of the LRRK2 gene is a cytoplasmic GTP-dependent kinase presumably involved in autophagy and functioning of mitochondria [12, 13]. The pathological increase in the kinase activity of LRRK2 is believed to be especially important in the neurotoxic effects of LRRK2-associated mutations, and therefore, identification of molecular substrates of LRRK2 is extremely important.
As we have described earlier [14], iPSC culture and neuronal precursors were obtained from fibroblasts of a patient with the LRRK2-associated form of PD; the cells were reprogrammed by the non-integrational approach using transduction with the Sendai virus. To correct the G2019S mutation (G6055A) and to compare structure-functional features of the mutant and “normalized” isogenic cultures of iPSCs and neurons differentiated from them, genome editing of iPSCs was performed with the CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 protein) system [15, 16]. After editing, iPSCs were differentiated by the neuronal type; the resulting isogenic lines of the neuronal precursors carrying the G2019S mutation (the non-edited type) and lacking the mutation (the edited type) were subjected to routine cytogenetic treatment for preparing metaphase plates for structural and quantitative analysis of chromosomes. Karyotyping of edited cells revealed that 24 to 43% cells from different clones contained the 92,XXYY/46,XY mosaic variant of tetraploidy (figure) [14].
Karyotypes of two neuronal precursor clones after genome editing: a) 46,XY; b) 92,XXYY (tetraploidy)
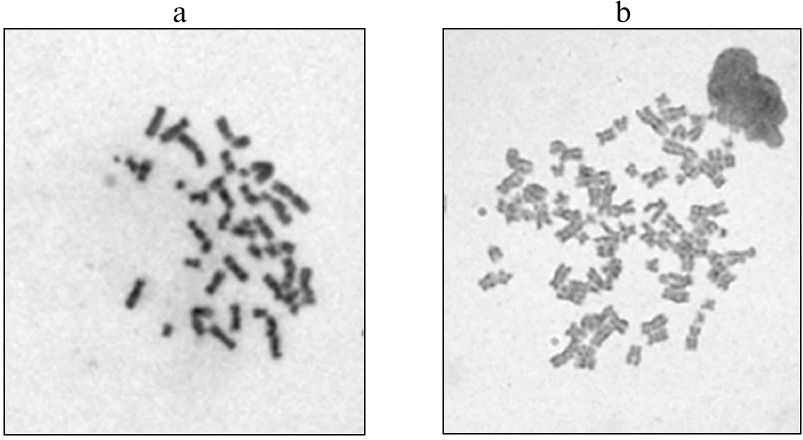
This variant of polyploidy can be a part of normal differentiation of some types of mammalian cells, for instance, keratinocytes [17] or brain neurons [18]. Healthy human brain has ~10% neurons with more than a diploid chromosomal set, and <1% of them are neurons with the tetraploid DNA [19]. Tetraploid cells are also observed in the normal retina of vertebrates [20]. However, despite all the aforementioned examples, association between tetraploidy and various pathologies, in particular, oncological diseases, has been established long ago [21].
The appearance of tetraploidy can be related to impairments in the cell division mechanism. All stages of the mitotic cycle are regulated by specific genes, including genes of the spindle assembly checkpoint (SAC) system that regulates correct segregation of chromosomes and attachment of spindle microtubules [22]. Impairments in the SAC system can disturb cytokinesis (separation of daughter cells due to the formation of cleavage furrow) and result in cell tetraploidy [18, 23]. Tetraploidy is highly probable in case of such mitotic phenomena as mitotic slippage (exit from mitosis without termination of anaphase or cytokinesis) and endoreduplication (two rounds of DNA replication without mitosis) [20, 21].
Cell tetraploidization can be caused by telomere exhaustion and represents a response to the loss of chromosomal ends. In the norm, continuous proliferation of somatic cells in the absence of telomerase activity leads to the gradual shortening of telomeres and apoptosis. However, under certain conditions, telomere dysfunction can cause the appearance of tetraploid cells. This process can proceed by two different pathways. One of them is associated with the dicentric chromosome formation at the expense of unprotected telomeric regions. In this case, if two centromeres of such chromosome are pulled to the opposite poles of the cell during mitosis, the delayed chromosome can interrupt cell division and prevent cytokinesis [18, 21, 23]. The other mechanism involves activation of ATM (ataxia telangiectasia mutated) and ATM- and Rad3-related (ATR) serine/threonine protein kinases in response to continuous DNA damage signal caused by telomere dysfunction. This causes cell cycle arrest followed by apoptosis [24]. However, in the absence of p53 protein (transcription factor and tumor growth suppressor) in the cells, the cell cycle proceeds – mitosis is bypassed, and the cell repeatedly enters the S-phase, which results in tetraploidy. This pathway does not require genetic alterations, except the loss of the p53 gene function [24, 25]. The LRRK2 gene associated with PD and studied by us in genome editing experiments interacts with ATM and actively participates in the ATM-Mdm2-p53 pathway regulating cell proliferation in response to DNA damage [26], which can be the cause of changes in the number of chromosomes.
Tetraploidy can result from cell fusion, that is sometimes observed for the brain cells. Cell fusion can be caused by viral infection, inflammation, chemicals, and ionizing radiation [23] and is believed to provide neuroprotection and defense of damaged neurons [27].
Impairments in spindle assembly and disorders in chromosomal segregation can be caused by dysfunctions of mitochondria. Close correlation between neurodegeneration and mutations in mitochondrial DNA has been already established [28, 29] that can be directly related to the aforementioned LRRK2-associated pathology. Using iPSCs, it was shown that various mutations in the LRRK2 gene in PD models are accompanied by a decrease in the functional activity of mitochondria and increase in the neuronal susceptibility to oxidative stress [30]. Developing oxidative stress can promote pathological polyploidization. Reactive oxygen species affect a number of proliferation-regulating signaling pathways via contributing to changes in ploidy and chromosomal instability [31, 32]. Dopaminergic neurons differentiated from iPSCs and carrying the G2019S mutation displayed impaired cell morphology, including shorter neurites [33]. As compared to normal dopaminergic neurons, the neurons differentiated from iPSCs with the G2019S mutation are more susceptible to such stress agents as proteasome inhibitor MG-132 and 6-hydroxydopamine, as well as to caspase-3 activation and apoptosis [34]. Mutations in the LRRK2 gene decrease the ability of cell substrates for oxidative phosphorylation, impair mitochondrial motility, and cause mitochondrial DNA damage in patient-specific iPSCs [35]. These impairments can be reversed or prevented by genome editing [30]. It is interesting that PD patients who are carriers of the G2019S mutation in the LRRK2 gene also exhibit changes in the mitochondrial function [36].
Another important cell component is nuclear envelope, whose major components, lamins A and B, may be called “guardians of the genome”. Lamin A/C (LMNA gene), lamin B1 (LMNB1 gene), and lamin B receptor (LBR) are extremely important for the chromosomal stability and structural integrity of cell nucleus by regulating DNA transcription and replication. That is why changes in lamins impair DNA repair and can induce malignant transformation of somatic cells [37, 38]. These processes are also influenced by the G2019S mutation in the LRRK2 gene: sections from PD patients with this mutation demonstrated changed nuclear morphology in the dentate gyrus cells. Another characteristic feature was hyperphosphorylation and disorders in functioning of lamins B1 and B2 regulating nuclear structure in neuronal derivatives of iPSCs [39]. It was suggested that defects of the nuclear structure are caused by the LRRK2 interaction with the protein complexes of lamins B1 and B2. The G2019S mutation stimulates kinase activity of LRRK2 that directly or indirectly promotes phosphorylation of B-type lamins.
The LRRK2 protein is involved in the regulation of F-actin via modulating the ERM (ezrin, radixin, moesin) proteins through phosphorylation [40]. F-actin is a polymerized microfilament form of the major cytoskeleton protein actin. There are more than 50 proteins interacting with actin in its F- and G-forms; activity of these proteins is regulated by Ca2+ ions and protein kinases. Impairments in actin polymerization lead to disorders in cytokinesis [41, 42]. LRRK2 is also involved in microtubule stabilization – it interacts with α- and β-tubulins, phosphorylates β-tubulin, and thus controls microtubule stability [41-43]. Obviously, proper functioning of spindle microtubules is important for the normal course of mitosis.
Oncological diseases are often accompanied with mitotic cycle disorders. An increased risk of development of different types of cancer (breast, prostate, kidney, hormone-dependent, and some other) was revealed in carriers of the G2019S mutation in the LRRK2 gene [44, 45]. It was suggested that an increased risk of oncological diseases in such patients is a result of excessive kinase activity of the mutant LRRK2 protein [45] and that this acquired “toxic” function underlie the failure of mitotic mechanisms in carriers of the G2019S mutation. It is interesting that some small kinase inhibitors suggested as anti-tumor agents are also considered promising candidates for modulation of LRRK2 kinase in the LRRK2-associated form of PD [46, 47].
Therefore, LRRK2 actively participates in cell proliferation, maintenance of the nuclear envelope structure, and formation cell membrane, spindle fibers, and cytoskeleton. The G2019S mutation can affect normal course of these processes and provoke systemic disturbances in the mitotic mechanisms thereby causing abnormalities in chromosomal ploidy. As shown above, major and best studied causes of such abnormalities are as follows:
– impairments in the involvement of mutant LRRK2 protein in the ATM-dependent mechanism of cell proliferation;
– dysfunctions of the division spindle and disorders in chromosomal segregation resulting from mitochondrial pathologies and energy deficiency in neurons;
– destabilization of spindle microtubules as a result of phosphorylation of the ERM family proteins and tubulins by abnormal LRRK2 protein;
– disorders in the interaction of mutant LRRK2 with lamin complexes regulating nuclear architecture, DNA transcription, chromatin stability, and DNA replication.
The above-described disorders can originate at different stages of reprogramming of fibroblasts from PD patients with the LRRK2 mutations [48]. Therefore, genome editing for correcting nucleotide sequences in neurobiological experiments and development of neurotransplantation methods should be performed before or during the reprogramming stage [48]. It must be remembered that cell culturing by itself could impair genome apparatus integrity [49-51]. The exact influence of tetraploidy on specific functions of neurons derived from iPSCs still remains unclear and requires further studies [20, 25]. However, in any case, it is necessary to remember that the properties of target gene and its protein product can essentially influence the results of cell reprogramming and genome editing.
Funding
The work was supported by the Russian Science Foundation (project no. 14-15-01047-П).
REFERENCES
1.De Lau, L. M. L., and Breteler, M. M. B. (2006)
Epidemiology of Parkinson’s disease, Lancet
Neurol., 5, 525-535.
2.Hernandez, D. G., Reed, X., and Singleton, A. B.
(2016) Genetics in Parkinson disease: Mendelian versus non-Mendelian
inheritance, J. Neurochem., 139 (Suppl. 1), 59-74.
3.Singleton, A. B., Farrer, M. J., and Bonifati, V.
(2013) The genetics of Parkinson’s disease: progress and
therapeutic implications, Mov. Disord., 28, 14-23.
4.Poewe, W., Seppi, K., and Tanner, C. M. (2017)
Parkinson disease, Nat. Rev. Dis. Primers, 3, 17013.
5.Jenner, P., Morris, H. R., Robbins, T. W., Goedert,
M., Hardy, J., Ben-Shlomo, Y., Bolam, P., Burn, D., Hindle, J. V., and
Brooks, D. (2013) Parkinson’s disease – the debate on
the clinical phenomenology, etiology, pathology and pathogenesis, J.
Parkinson’s Dis., 3, 1-11.
6.Kang, J. F., Tang, B. S., and Guo, J. F. (2016) The
progress of induced pluripotent stem cells as models of
Parkinson’s disease, Stem Cells Int., 2016,
4126214.
7.Holmqvist, S., Lehtonen, S., Chumarina, M.,
Puttonen, K. A., Azevedo, C., Lebedeva, O., Ruponen, M., Oksanen, M.,
Djelloul, M., Collin, A., Goldwurm, S., Meyer, M., Lagarkova, M.,
Kiselev, S., Koistinaho, J., and Roybon, L. (2016) Creation of a
library of induced pluripotent stem cells from Parkinsonian patients,
NPJ Parkinson’s Dis., 2, 16009.
8.Illarioshkin, S. N., Khaspekov, L. G., and
Grivennikov, I. A. (2017) Modeling of Parkinson’s Disease with
Induced Pluripotent Stem Cells [in Russian], ZAO RKI Sovero Press,
Moscow.
9.Hargus, G., Cooper, O., Deleidi, M., Levy, A., Lee,
K., Marlow, E., Yow, A., Soldner, F., Hockemeyer, D., Hallett, P. J.,
Osborn, T., Jaenisch, R., and Isacson, O. (2010) Differentiated
Parkinson patient-derived induced pluripotent cells grow in the adult
rodent brain and reduce motor asymmetry in Parkinsonian rats, Proc.
Natl. Acad. Sci. USA, 107, 15921-15926.
10.Cornu, T., Mussolino, C., and Cathomen, T. (2017)
Refining strategies to translate genome editing to the clinic,
Nature Med., 23, 415-423.
11.Healy, D. G., Falchi, M., O’Sullivan, S.
S., Bonifati, V., Durr, A., Bressman, S., Brice, A., Aasly, J.,
Zabetian, C. P., Goldwurm, S., Ferreira, J. J., Tolosa, E., Kay, D. M.,
Klein, C., Williams, D. R., Marras, C., Lang, A. E., Wszolek, Z. K.,
Berciano, J., Schapira, A. H., Lynch, T., Bhatia, K. P., Gasser, T.,
Lees, A. J., and Wood, N. W. (2008) International LRRK2 Consortium.
Phenotype, genotype, and worldwide genetic penetrance of
LRRK2-associated Parkinson’s disease: a case-control study,
Lancet Neurol., 7, 583-590.
12.Dachsel, J. C., Behrouz, B., Yue, M., Beevers, J.
E., Melrose, H. L., and Farrer, M. J. (2010) A comparative study of
LRRK2 function in primary neuronal cultures, Parkinsonism Relat.
Disord., 16, 650-655.
13.Tong, Y., Giaime, E., Yamaguchi, H., Ichimura,
T., Liu, Y., Si, H., Cai, H., Bonventre, J. V., and Shen, J. (2012)
Loss of leucine-rich repeat kinase 2 causes age-dependent biphasic
alterations of the autophagy pathway, Mol. Neurodegener.,
7, 2.
14.Vetchinova, A. S., Simonova, V. V., Novosadova,
E. V., Manuilova, E. S., Nenasheva, V. V., Tarantul, V. Z.,
Grivennikov, I. A., Khaspekov, L. G., and Illarioshkin, S. N. (2018)
Cytogenetic analysis of results of the genome editing on the cell model
of the Parkinson’s disease, Byul. Eksp. Biol. Med.,
3, 355-359.
15.Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto,
R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., and
Zhang, F. (2013) Multiplex genome engineering using CRISPR/Cas systems,
Science, 339, 819-823.
16.Vasil’eva, E. A., Melino, D., and Barlev,
N. A. (2015) Application of the directed genomic editing system
CRISPR/Cas to pluripotent stem cells, Tsitologiya, 1,
19-30.
17.Zanet, J., Freije, A., Ruiz, M., Coulon, V.,
Sanz, J. R., Chiesa, J., and Gandarillas, A. (2010) A mitosis block
links active cell cycle with human epidermal differentiation and
results in endoreplication, PLoS One, 5, e15701.
18.Davoli, T., and de Lange, T. (2011) The causes
and consequences of polyploidy in normal development and cancer,
Annu. Rev. Cell Dev. Biol., 27, 585-610.
19.Mosch, B., Morawski, M., Mittag, A., Lenz, D.,
Tarnok, A., and Arendt, T. (2007) Aneuploidy and DNA replication in the
normal human brain and Alzheimer’s disease, J. Neurosci.,
27, 6859-6867.
20.Frade, J. M. (2010) Somatic tetraploidy in
vertebrate neurons: implications in physiology and pathology,
Commun. Integr. Biol., 3, 201-203.
21.Ganem, N. J., Storchova, Z., and Pellman, D.
(2007) Tetraploidy: aneuploidy and cancer, Curr. Opin. Genet.
Dev., 17, 157-162.
22.Joglekar, A. P. (2016) A cell biological
perspective on past, present and future investigations of the spindle
assembly checkpoint, Biology (Basel), 5, E44.
23.Holland, A. J., and Cleveland, D. W. (2012)
Losing balance: the origin and impact of aneuploidy in cancer, EMBO
Rep., 13, 501-514.
24.Marechal, A., and Zou, L. (2013) DNA damage
sensing by the ATM and ATR kinases, Cold Spring Harb. Perspect.
Biol., 5, a012716.
25.Davoli, T., Denchi, E. L., and de Lange, T.
(2010) Persistent telomere damage induces bypass of mitosis and
tetraploidy, Cell, 41, 81-93.
26.Chen, Z., Cao, Z., Zhang, W., Gu, M., Dong Zhou,
Z., Li, B., Li, J., King Tan, E., and Zeng, L. (2017) LRRK2 interacts
with ATM and regulates Mdm2–p53 cell proliferation axis in
response to genotoxic stress, Hum. Mol. Genet., 26,
4494-4505.
27.Kemp, K., Wilkins, A., and Scolding, N. (2014)
Cell fusion in the brain: two cells forward, one cell back, Acta
Neuropathol., 128, 629-638.
28.Coskun, P. E., and Busciglio, J. (2012) Oxidative
stress and mitochondrial dysfunction in Down’s syndrome:
relevance to aging and dementia, Curr. Gerontol. Geriatr. Res.,
2012, 383170.
29.Greaves, L. C., Reeve, A. K., Taylor, R. W., and
Turnbul, D. M. (2012) Mitochondrial DNA and disease, J. Pathol.,
226, 274-286.
30.Sanders, L. H., Laganiere, J., Cooper, O., Mak,
S. K., Vu, B. J., Huang, Y. A., Paschon, D. E., Vangipuram, M.,
Sandarajan, R., Urnov, F. D., Langston, J. W., Gregory, P. D., Zhang,
H. S., Greenamyre, J. T., Isacson, O., and Schule, B. (2014) LRRK2
mutations cause mitochondrial DNA damage in iPS-derived neural cells
from Parkinson’s disease patients: reversal by gene correction,
Neurobiol. Dis., 62, 381-386.
31.Gentric, G., Maillet, V., Paradis, V., Couton,
D., L’Hermitte, A., Panasyuk, G., Fromenty, B., Celton-Morizur,
S., and Desdouets, C. (2015) Oxidative stress promotes pathologic
polyploidization in nonalcoholic fatty liver disease, J. Clin.
Invest., 125, 981-992.
32.Dephoure, N., Hwang, S., O’Sullivan, C.,
Dodgson, S. E., Gygi, S. P., Amon, A., and Torres, E. M. (2014)
Quantitative proteomic analysis reveals post-translational responses to
aneuploidy in yeast, Elife, 3, e03023.
33.Orenstein, S. J., Kuo, S.-H., Tasset, I., Arias,
E., Koga, H., Fernandez-Carasa, I., Cortes, E., Honig, L. S., Dauer,
W., Consiglio, A., Raya, A., Sulzer, D., and Cuervo, A. M. (2013)
Interplay of LRRK2 with chaperone-mediated autophagy, Nat.
Neurosci., 16, 394-406.
34.Nguyen, H. N., Byers, B., Cord, B.,
Shcheglovitov, A., Byrne, J., Gujar, P., Kee, K., Schule, B.,
Dolmetsch, R. E., Langston, W., Palmer, T. D., and Pera, R. R. (2011)
LRRK2 mutant iPSC-derived DA neurons demonstrate increased
susceptibility to oxidative stress, Cell Stem Cell, 8,
267-280.
35.Cooper, O., Seo, H., Andrabi, S.,
Guardia-Laguarta, C., Graziotto, J., Sundberg, M., McLean, J. R.,
Carrillo-Reid, L., Xie, Z., Osborn, T., Hargus, G., Deleidi, M.,
Lawson, T., Bogetofte, H., Perez-Torres, E., Clark, L., Moskowitz, C.,
Mazzulli, J., Chen, L., Volpicelli-Daley, L., Romero, N., Jiang, H.,
Uitti, R. J., Huang, Z., Opala, G., Scarffe, L. A., Dawson, V. L.,
Klein, C., Feng, J., Ross, O. A., Trojanowski, J. Q., Lee, V. M.,
Marder, K., Surmeier, D. J., Wszolek, Z. K., Przedborski, S., Krainc,
D., Dawson, T. M., and Isacson, O. (2012) Pharmacological rescue of
mitochondrial deficits in iPSC-derived neural cells from patients with
familial Parkinson’s disease, Sci. Transl. Med., 4,
141ra90.
36.Padlesnig, P., Vilas, D., Taylor, P., Shaw, L.
M., Tolosa, E., and Trullas, R. (2016) Mitochondrial DNA in CSN
distinguishes LRRK2 from idiopathic Parkinson’s disease,
Neurobiol. Dis., 94, 10-17.
37.Ho, C. Y., and Lammerding, J. (2012) Lamins at a
glance, J. Cell Sci., 125, 2087-2093.
38.Gonzalo, S. (2014) DNA damage and lamins, Adv.
Exp. Med. Biol., 773, 377-399.
39.Liu, G.-H., Qu, J., Suzuki, K., Nive, E., Li, M.,
Montserrat, N., Yi, F., Xu, X., Ruiz, S., Zhang, W., Wagner, U., Kim,
A., Ren, B., Li, Y., Goebl, A., Kim, J., Soligalla, R. D., Dubova, I.,
Thompson, J., Yates, J., 3rd, Esteban, C. R., Sancho-Martinez, I., and
Belmonte, J. C. I. (2012) Progressive degeneration of human neural stem
cells caused by pathogenic LRRK2 mutations, Nature, 491,
603-607.
40.McClatchey, A. I. (2014) ERM proteins at a
glance, J. Cell Sci., 127, 3199-3204.
41.Gandhi, P. N., Wang, X., Zhu, X., Chen, S. G.,
and Wilson-Delfosse, A. L. (2008) The Roc domain of leucine-rich repeat
kinase 2 is sufficient for interaction with microtubules, J.
Neurosci. Res., 86, 1711-1720.
42.Gillardon, F. (2009) Leucine-rich repeat kinase 2
phosphorylates brain tubulin-beta isoforms and modulates microtubule
stability — a point of convergence in parkinsonian
neurodegeneration? J. Neurochem., 110, 1514-1522.
43.Parisiadou, L., and Cai, H. (2010) LRRK2 function
on actin and microtubule dynamics in Parkinson disease, Commun.
Integr. Biol., 3, 396-400.
44.Agalliu, I., San Luciano, M., Mirelman, A.,
Giladi, N., Waro, B., Aasly, J., Inzelberg, R., Hassin-Baer, S.,
Friedman, E., Ruiz-Martinez, J., Marti-Masso, J. F., Orr-Urtreger, A.,
Bressman, S., and Saunders-Pullman, R. (2015) Higher frequency of
certain cancers in LRRK2 G2019S mutation carriers with
Parkinson’s disease: a pooled analysis, JAMA Neurol.,
72, 58-65.
45.Saunders-Pullman, R., Barrett, M. J., Stanley,
K., Luciano, M. S., Shanker, V., Severt, L., Hunt, A., Raymond, D.,
Ozelius, L. J., and Bressman, S. B. (2010) LRRK2 G2019S mutations are
associated with an increased cancer risk in Parkinson disease, Mov.
Disord., 25, 2536-2541.
46.Nichols, R. J., Dzamko, N., Hutti, J. E.,
Cantley, L. C., Deak, M., Moran, J., Bamborough, P., Reith, A. D., and
Alessi, D. R. (2009) Substrate specificity and inhibitors of LRRK2, a
protein kinase mutated in Parkinson’s disease, Biochem.
J., 424, 47-60.
47.Liou, G. Y., and Gallo, K. A. (2009) New
biochemical approaches towards understanding the Parkinson’s
disease-associated kinase, LRRK2, Biochem. J., 424,
1-3.
48.Lee, S.-Y., and Chung, S.-K. (2016) Integrating
gene correction in the reprogramming and transdifferentiation
processes: a one-step strategy to overcome stem cell-based gene therapy
limitations, Stem Cells Intern., 2016, 2725670.
49.Dekel-Naftali, M., Aviram-Goldring, A.,
Litmanovich, T., Shamash, J., Reznik-Wolf, H., Laevsky, I., Amit, M.,
Itskovitz-Eldor, J., Yung, Y., Hoyrvitz, A., Schiff, E., and Rienstein,
S. (2012) Screening of human pluripotent stem cells using CGH and FISH
reveals low-grade mosaic aneuploidy and a recurrent amplification of
chromosome 1q, Eur. J. Hum. Genet., 20, 1248-1255.
50.Gore, A., Li, Z., Fung, H.-L., Young, J. E.,
Agarwal, S., Antosiewicz-Bourget, J., Canto, I., Giorgetti, A., Israel,
M. A., Kiskinis, E., Lee, J. H., Loh, Y. H.,, Manos, P. D., Montserrat,
N., Panopoulos, A. D., Ruiz, S., Wilbert, M. L., Yu, J., Kirkness, E.
F., Izpisua Belmonte, J. C., Rossi, D. J., Thomson, J. A., Eggan, K.,
Daley, G. Q., Goldstein, L. S., and Zhang, K. (2011) Somatic coding
mutation in human induced pluripotent stem cells, Nature,
471, 63-67.
51.Hussein, S. M., Batada, N. N., Vuoristo, S.,
Ching, R. W., Autio, R., Narva, E., Ng, S., Sourour, M., Hamalainen,
R., Olsson, C., Lundin, K., Mikkola, M., Trokovic, R., Peitz, M.,
Brustle, O., Bazett-Jones, D. P., Alitalo, K., Lahesmaa, R., Nagy, A.,
and Otonkoski, T. (2012) Copy number variation and selection during
reprogramming to pluripotency, Nature, 471, 58-62.