REVIEW: Pluripotent Stem Cells for Modelling and Cell Therapy of Parkinson’s Disease
O. S. Lebedeva1 and M. A. Lagarkova1,a*
1Federal Research and Clinical Center of Physical-Chemical Medicine, Federal Medical Biological Agency, 119435 Moscow, Russia* To whom correspondence should be addressed.
Received April 29, 2018; Revision received May 31, 2018
Studying pathogenesis of neurodegenerative diseases, including Parkinson’s disease (PD), requires adequate disease models. The available patient’s material is limited to biological fluids and post mortem brain samples. Disease modeling and drug screening can be done in animal models, although this approach has its own limitations, since laboratory animals do not suffer from many neurodegenerative diseases, including PD. The use of neurons obtained by targeted differentiation from induced pluripotent stem cells (iPSCs) with known genetic mutations, as well as from carriers of sporadic forms of the disease, will allow to elucidate new components of the molecular mechanisms of neurodegeneration. Such neuronal cultures can also serve as unique models for testing neuroprotective compounds and monitoring neurodegenerative changes against a background of various therapeutic interventions. In the future, dopaminergic neurons differentiated from iPSCs can be used for cell therapy of PD.
KEY WORDS: Parkinson’s disease, induced pluripotent stem cells, cell model, cell therapy, dopaminergic neurons, isogenic systemDOI: 10.1134/S0006297918090067
Abbreviations: DNs, dopaminergic neurons; GHSR, growth hormone secretagogue receptor; (h)ESCs, (human) embryonic stem cells; hfVM, human fetal ventral mesencephalic (tissue); iPSCs, induced pluripotent stem cells; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; PD, Parkinson’s disease; SNCA, α-synuclein; SNP, single nucleotide polymorphism.
Parkinson’s disease (PD) is a progressive neurodegenerative
disorder. Despite the fact that it was for the first time described two
hundred years ago, our understanding of its causes, as well as
underlying cellular and molecular pathogenetic mechanisms, remains
incomplete and is continuously updated and modified. PD motor symptoms
result from the death of dopaminergic neurons (DNs) in substantia
nigra. Lewy bodies containing aggregates of α-synuclein and other
proteins are found in most PD patients. At the cellular level, PD
development is associated with impaired mitochondrial and lysosomal
functions, accumulation of aberrantly folded proteins, altered protein
degradation, endoplasmic reticulum stress, oxidative stress,
neuroinflammation, and activation of glial cells presumably involving
some apoptotic components (see reviews [1, 2]).
PD may be caused by environmental (trauma, pesticides, metal ions) [3-6] and genetic factors.
PD pathogenesis is triggered when environmental and/or genetic factors initiate pathological signaling cascades resulting in progressive DN degeneration. The first PD symptoms become evident only after the loss of 70% DNs corresponding to 80% decrease in the dopamine levels in basal ganglia [3].
Around 15% of PD cases are familial. So far, mutations in 19 genes have been linked to PD, but their role in molecular pathogenesis remains only partially characterized [7]. The figure shows major signaling pathways altered as a result of the most studied mutations in PD-related genes. PD usually develops without diagnosable symptoms that manifest themselves at only the terminal disease stage. Moreover, brain tissues of the affected individuals remain unavailable for examination for the entire period of PD development and could be studied only post mortem. Detailed examination of neurodegeneration processes requires the development of proper cellular models carrying relevant genetic mutations. The use of neuronal cell cultures obtained by the induced pluripotent stem cell (iPSC) technology from fibroblasts of patients with sporadic PD or carriers of well characterized mutations will allow to assess the relationship between neuronal degenerative changes and clinical course of the disease.
Pathological changes in cells caused by mutant PD-related genes and triggered by environmental factors and toxins [8]
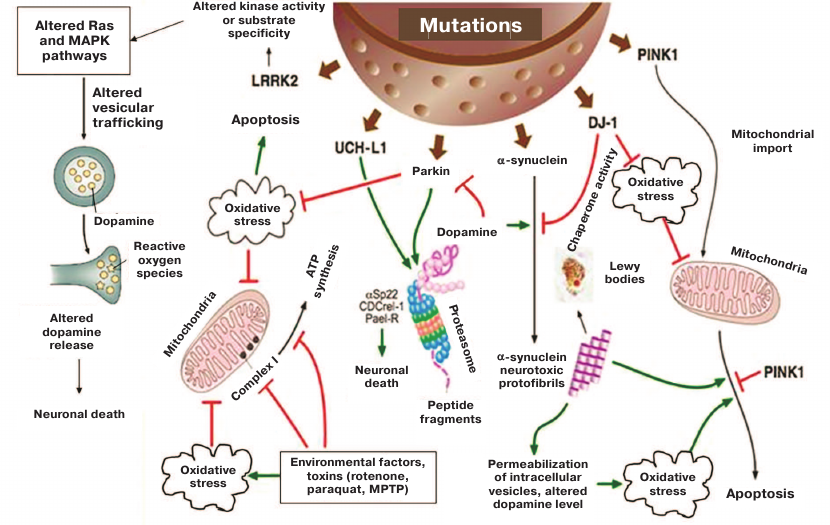
Since 2006, when genetic reprogramming technology was discovered [9], various methods have been developed for delivering reprogramming factors to somatic cells (reviewed in [10]). iPSCs represent virtually limitless source of cell material and may be differentiated into any adult host cell type including neuronal precursors and DNs. Development of the iPSC-based PD model will allow to examine PD mechanisms and functions of mutant gene products in the affected cells. Differentiated iPSC derivatives may be also used to screen for new pharmaceutical agents. An opportunity to reprogram patient’s cells and differentiate pluripotent cells to DNs without modifying cell genome has opened the opportunity for application of these differentiated derivatives in PD cell therapy.
Here, we review iPSC-based PD models with special emphasis on the isogenic systems that are considered more advantageous over the cells obtained from the donor’s biological material. In addition, the historic timeline of development of fetal cell-based of PD treatment is discussed. Finally, approaches to PD cell therapy with iPSCs and human embryonic stem cells (hESCs) are outlined.
iPSCs-BASED MODELS FOR STUDYING PD
A procedure for generation and differentiation of iPSCs for modeling human diseases was for the first time described by Park et al. [11] who successfully generated iPSCs from patients with various hereditary disorders including PD, Duchenne muscular dystrophy, type 1 diabetes mellitus, Down syndrome, and Huntington disease. It was proven that iPSCs can be used as a unique model for investigating molecular and genetic mechanisms of various disorders and a useful research tool for screening potential pharmaceutical agents.
Another study published nearly at the same time demonstrated that transplantation of iPSCs capable of in vitro differentiation into neurons and glial cells restored behavioral activity in rats with induced PD symptoms [12].
iPSCs obtained from patients with sporadic PD were able to differentiate into DNs. Transplantation of these cells in rats with induced parkinsonism attenuated amphetamine-induced rotational behavior of the animals. Immunohistochemical analysis of brain sections from the experimental rats revealed that transplanted neurons sent axonal projections to both adjacent and distant areas; no Lewy bodies were found in the graft [13]. In this study, iPSCs derived from patients with sporadic PD did not display the pathological phenotype in vitro. However, closer examination of DNs from patients with non-hereditary PD revealed some disease symptoms in vitro. DNs from healthy donors and patients with familial and non-hereditary PD forms differed in their genome-wide DNA methylation profiles. Thus, DNA from PD patients was characterized by hypermethylation of regulatory gene regions, e.g., enhancers, resulting in downregulated expression of mRNAs for some PD-related proteins (FOXA1, NR3C1, HNF4A, and FOSL2). Interestingly, DNA methylation profiles in patients with familial and non-hereditary PD forms were similar [14].
In most cases, iPSCs generated from patients with familial PD display typical pathological phenotype and, hence, could be used as a model for studying PD pathogenesis and screening for new pharmaceuticals agents. Mutations in the PARK8 gene encoding the LRRK2 kinase are the most common genetic causes of in PD [15]. Among them, the most frequent is G2019S that underlies 2-7% familial and 1% sporadic PD cases [16]. Unlike neurons generated from healthy cells, neural precursors carrying the G2019S mutation in LRRK2 gene lose their ability to differentiate into MAP2+ neurons after several (15-17) passages [17]. Also, the mutant neurons exhibited an aberrant nuclear architecture. The authors observed defects in cell nuclei in post mortem brain section from patients with clinically diagnosed PD [17], thereby corroborating the relevance of PD model designed in their study.
PD development can be also linked to mutations in the gene coding for the mitochondrial kinase PINK1 localized on the outer mitochondrial membrane and involved in cell protection from oxidative stress by mediating translocation of the PARK2-encoded E3-ubiquitin ligase parkin to dysfunctional mitochondria. This process is essential for the mitophagy of damaged mitochondria in human DNs. Mutations in the PINK1 gene did not affect cell reprogramming and iPSCs differentiation into DNs; however, they impaired stress-induced translocation of parkin in mature neurons, which resulted in the elevated generation and accumulation of dysfunctional mitochondria. At the same time, DNs derived from the wild-type iPSCs did not exhibit these impairments. Overexpression of wild-type PINK1 in the mutant neurons reversed all pathological features of these cells [18].
Sanchez-Danez et al. [19] conducted an extensive study of PD pathogenesis using iPSCs derived from seven patients with sporadic PD, four patients carrying G2019S mutation in the LRRK2 gene, and four volunteers with no neurodegenerative diseases in their history. The efficiency of the iPSC clone development varied between the studied subjects but was unrelated to the presence of PD or age. Using a 30-day protocol, the authors were able to generate adult DNs predominantly of the A9 subtype. These neurons form substantia nigra pars compacta that undergoes profound degeneration in PD. The obtained model allowed to investigate interactions between LRRK2 and α-synuclein (SNCA), another protein associated with PD development. Although the precise role of α-synuclein in vivo is yet unknown, it was suggested that it acts as a molecular chaperone regulating protein–protein and protein–lipid interactions, plays an important role in synaptic vesicle exchange, and modulates storage and compartmentalization of neurotransmitters, primarily dopamine [20]. It is well known that α-synuclein protofibrils are the major component of Lewy bodies in PD, thereby pointing at the important role of α-synuclein aggregation in PD pathogenesis. DNs derived from iPSCs of patients carrying the G2019S mutation abnormally accumulate SNCA as compared to DNs derived from healthy donors and patients with sporadic PD [19]. These results not only corroborate the hypothesis on the mutual effects of mutations in LRRK2 and SNCA, but also prove that that iPSCs from PD patients can be used as a model of the monogenic PD form.
PD development occurs over several years, sometime decades. DNs derived from iPSCs obtained from sporadic PD patients, carriers of mutant LRRK2, and healthy donors were cultured on a mouse postnatal astrocyte monolayer for a long period of time (65-75 days) [19]. DNs from healthy subjects were morphologically homogeneous and displayed mature DN phenotype with well-branched cell processes. At the same time, DNs derived from PD patients exhibited various morphological changes (shortened cell processes, decreased number or completely loss of cell processes, fragmented nuclei, vacuolation). It should be noted that no similar changes were observed in DN cultures after a standard 30-day culturing. A higher percentage of apoptotic cells was found in DNs derived from patients with mutant LRRK2 gene and sporadic PD than in DNs from healthy donors after 75 days of culturing.
iPSCs derived from PD patients have been used to confirm the therapeutic efficacy of suggested pharmaceutical agents. In particular, NAB2 (Nedd4 ubiquitin ligase activator) identified as a potential anti-PD agent in yeast screen was proven to reverse the pathological PD phenotype [21].
Undoubtedly, iPSCs-based PD models have limitations. The onset of PD usually happens at a mature age, while reprogramming takes the cell to the embryonic-like state. It is possible that multiple passages of neuronal precursors, oxidative stress, and other senescence-inducing culturing methods will promote more explicit manifestations of PD phenotype in iPSCs-derived cell models.
Creation of 3D-cultured human brain organoids, particularly, human midbrain organoid differentiation [22] is a promising approach for examining molecular mechanisms underlying parkinsonism in iPSC-based models.
In some studies, iPSC-derived DNs were transplanted into animal PD models. For example, differentiated DNs transplanted into PD rats markedly improved motor functions of the animals [13, 23-26].
ISOGENIC CELL SYSTEMS FOR PD MODELING
The problem to what extent iPSCs are similar to hESCs and whether the reprogramming influences the features of resulting pluripotent cells has been widely debated. De Boni et al. demonstrated that the cell genetic background (the authors compared neurons differentiated from PD patient-derived iPSCs and hESC lines H9 and I3) and variations in the differentiation protocols rather than the reprogramming process itself strongly affect the DNA methylation profiles in both neurons and neuronal precursors [27]. These data prove that isogenic cell lines should be used to reduce variations in the background DNA methylation and expression profiles and to reveal more subtle differences in the normal vs. pathological state. Moreover, isogenic cell lines can be used to confirm the impact of changes previously revealed by genome-wide studies on the PD development.
For instance, CRISPR/Cas9 genome editing in neurons derived from iPSCs confirmed that one type of the SNCA gene enhancer is linked to the PD development [28]. In particular, by overlapping the data of genome-wide associations studies (GWAS) on the single nucleotide polymorphism (SNP) variants in the SNCA gene with epigenetic signatures typical for the enhancer elements of this gene, the authors identified a potential PD-associated risk variant in the non-coding distal enhancer element located in the intron 4. CRISPR/Cas9 genome editing in control hESCs allowed to obtain a series of isogenic cell lines each carrying a specific allele variant of the SNP of interest. Upregulated SNCA expression in neurons was linked to the G allele in the SNP rs356168. Chromatin immunoprecipitation identified two transcription factors (EMX2 and NKX6-1) differing in their binding to the risk alleles. Both of them bound mainly to the A allele, assuming that the G allele in the SNCA enhancer results in the upregulated SNCA expression due to its reduced binding by EMX2/NKX6-1 and therefore, decreased transcriptional repression of this allele [28].
iPSC- and hESC-based isogenic cell lines have been extensively used in the studies of hereditary PD forms caused by identified gene mutations. Autosomal dominant mutation G2019S enhances the activity of LRRK2 kinase, thereby affecting its function [29]. In many cases, survived DNs in substantia nigra of PD patients contain Lewy bodies mainly consisting of α-synuclein [30, 31]. Because 90% α-synuclein in Lewy bodies is phosphorylated at serine 129 (S129P) [32], it is possible that S129P-α-synuclein triggers α-synuclein aggregation and DN degeneration in substantia nigra [33]. Moreover, LRRK2 is also present in Lewy bodies [34]. The role of LRRK2 has not been elucidated yet; it remains unknown whether it phosphorylates α-synuclein [35]. Qing et al. designed a protocol for the generation of footprint-free iPSC isogenic lines by using CRISPR/Cas9 in combination with the piggyBac transposon system that gave rise to the iPSC isogenic line carrying the G2019S mutation in LRRK2 [36]. These iPSCs were differentiated to DNs and examined by cytological and histochemical methods. It was shown that the number of neurites in tyrosine hydroxylase-positive neurons was not affected by the mutation, but the neurite length in the mutant cells was markedly reduced. Similar pattern was also found for neurite branching. Immunocytochemical staining demonstrated that all neurons expressed α-synuclein. Staining with anti-S129P-α-synuclein antibodies showed that phosphorylated α-synuclein was present only in neurons with long neurites and did not co-localize with apoptotic markers (activated caspase 3), thereby implying that S129P-α-synuclein is nontoxic to DNs. The number of tyrosine hydroxylase/S129P-α-synuclein positive neurons was significantly lower in differentiated cells with mutant LRRK2 than in the isogenic control line. Cell manifestations of PD were reversed by adding the LRRK2 kinase inhibitor IN-1. The results of this study confirmed that the generated cell lines could be used for studying PD-related phenotype and testing drug candidates capable of PD phenotype reversion [36].
Mutations in the PARK2 gene encoding E3-ubiquitin ligase parkin are the most common cause of autosomal recessive early-onset (juvenile) PD [37]. Ghrelin as 28-a.a. peptide that acts as an endogenous ligand of the growth hormone secretagogue receptor (GHSR) of the G-protein coupled receptor family. It regulates growth hormone secretion and food intake and is involved in the reward system and memory retrieval [38-41]. Although ghrelin is mainly secreted in the stomach [39], small amounts of this peptide could be found in the brain [42]. GHSR is expressed in various brain regions including substantia nigra [38, 39], where ghrelin activates electrical activity in DNs and increases dopamine concentration in the striatum due to selective blockade of the KCNQ potassium channel [43]. Comparison of iPSC-derived PARK2-mutant cells vs. normal iPSCs revealed decreased amounts of GHSR and its mRNA in the mutant cells. Similar pattern was observed by comparing DNs differentiated from iPSC isogenic cell lines, one of which carried a mutation in the PARK2 gene introduced by CRISPR/Cas9 genome editing; however, in this case, the effect of mutation was much less pronounced than in the cells obtained from a PD patient [44]. All the aforementioned model systems are presented in the table in a chronological order (table).
iPSC-based PD models: brief timeline
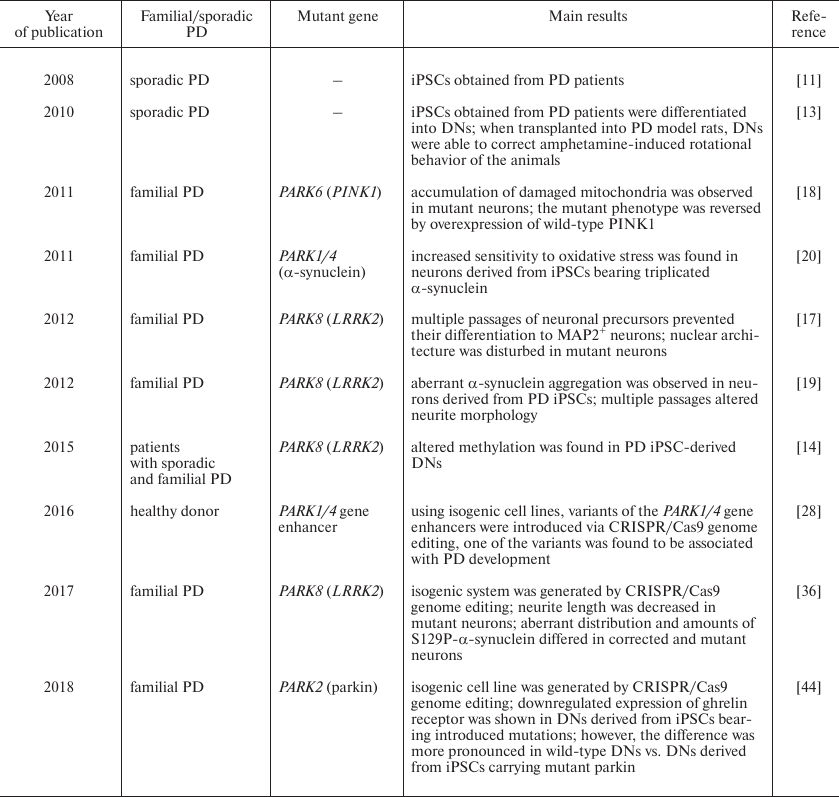
Laboratory experiments with iPSCs are performed in parallel with the clinical studies on the possible application of iPSC derivatives in medical practice. At present, iPSC-derived cell products are awaiting clinical trials for the evaluation of their efficacy in cell therapy.
PROSPECTS OF PD CELL THERAPY WITH iPSC-DERIVED NEURONS
In 1987, the first clinical trials using cell therapy for PD treatment were performed at the Lund University, Sweden [45]. In particular, human fetal ventral mesencephalic (hfVM) neurons were transplanted into PD patients, which resulted in serous improvement in some of the patients. The hfVM allografts were able to release dopamine and survive in the recipient’s brain for a long time. The recipients experienced reduction of some non-motor PD symptoms and improved quality of life (for detailed review, see [46]). However, such positive effects were observed only in some cases; transplantation was sometimes associated with complications (e.g., graft-induced dyskinesia) with typical PD-related pathological symptoms in the grafted cells [46]. Attempts to resolve the problems of previous trials gave rise to TRANSEURO, a new European clinical trial with 11 PD patients, who have been transplanted with hfVM allografts over the last 2.5 years. However, sensitive ethical issues related to the use of human embryonic tissues for cell therapy hinder the development of this area of studies together with technical obstacles related to the problems in obtaining human fetal material and inability to standardize it for clinical application. Thus, only 20 out of 90 scheduled hfVM transplantations have been performed in the TRANSEURO trial due to the deficit of tissues for grafting [47]. To overcome such problems, methods for differentiation of hESCs and iPSCs into midbrain DNs for further use in clinical practice have being extensively developed.
Advantages and safety of hESC/iPSC-derived DNs should be first justified in animal experiments in vivo. DN-targeting neurotoxins, such as rotenone, 6-hydroxydopamine (6-OHDA), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), are used to develop animal models of parkinsonism. Generally, PD models are developed in rodents and non-anthropoid primates [48]. Due to similarities in the brain structure and PD symptoms between human and monkeys, as well as the possibility to use in both cases high-definition visualization methods (e.g., positron emission tomography and magnetic resonance imaging), studies in non-anthropoid primates substantially promote transition to clinical trials in humans [49].
In 2005, Takagi et al. for demonstrated the first time that primate ESC-derived DNs survive after being transplanted to the putamen of recipient primates with MPTP-induced PD resulting in augmented [18F]DOPA uptake 14 days after the transplantation. Moreover, starting from week 10 after the transplantation, neurological signs improved in DN-grafted vs. sham-surgery monkeys. This study demonstrated for the first time the functional efficacy of primate ESC-derived DNs and opened up an opportunity for their application in PD cell therapy [50].
Human iPSC-derived DNs transplanted to the striatum reversed PD behavioral symptoms in PD model rats [51]. However, there are some problems, e.g., tumorigenicity, that prevent the use of these cells in clinical trials. Tumorigenicity may be caused by the presence in the grafted material of residual pluripotent cells or partially differentiated neuronal precursors capable of teratoma formation and/or unlimited cell proliferation. At the same time, the use of fully mature neuronal cell cultures results in markedly decreased survival rate of the transplanted cells. One of the ways to solve this problem is to determine the optimal period for differentiation of cultured cells. Doi et al. demonstrated that long-term neuronal differentiation of hESC-derived cells (over 28 days) decreases tumorigenesis and/or slows down rapid growth of cells after their grafting in monkeys with parkinsonism. The motor symptoms of PD were relieved in experimental animals after transplantation of hESC-derived DNs differentiated for 42 days [52]. On the other hand, cells with a required phenotype and at a desired maturation stage can be selected by staining for specific surface markers followed by cell sorting. In 2014, Martinez-Morales et al. used anti-Corin antibodies for the enrichment of mesencephalic DN precursors [24]. Transmembrane serine protease Corin is expressed in the floor plate during embryonic brain development. During differentiation of iPSCs to DNs, Corin expression is detected on day 7 and peaks on day 30 of differentiation. Corin-positive cells transplanted to the brain of experimental animals proliferate at a much lower rate than unsorted neurons, resulting in a 2-fold greater amount of survived DNs in the graft.
Kikuchi et al. were the first to showed that DNs derived from iPSCs in the absence of feeder cells and components of animal origin in the culturing medium have survived for 6 months in the brain of monkeys with MPTP-induced PD [53].
The same research group has observed for 2 years primates with MPTP-induced PD after transplantation of iPSC-derived DNs [54]. To examine the efficacy and safety of DNs differentiated in vitro, DN committed precursors were transplanted to the putamen of the animals with MPTP-induced parkinsonism. For this, eight iPSC lines derived from four heathy volunteers and three PD patients were used. iPSC-derived DN precursors were then differentiated using the dual SMAD inhibition procedure and then committed to the floor plate cells as described in [24]. To increase the population homogeneity, the cells were sorted according to the Corin expression levels. Although the percentage of Corin-positive cells on day 12 varied between the cell lines; the content of Corin-positive cells in all sorted cell populations was no less than 90%. After long-term cell differentiation, the cells were able to generate action potentials and release dopamine in response to high potassium-induced membrane depolarization. In addition, no undifferentiated OCT4+ cells and very few proliferating neuronal precursors were found. DN precursors differentiated for 28 days were grafted bilaterally into the putamen of monkeys with MPTP-induced parkinsonism (the animals were treated with immunosuppressing agents). Neurological changes were assessed using the neurological rating scale and video-based analysis of spontaneous movements. It was found that monkeys with grafted DNs displayed gradual improvement in the assayed parameters. No differences were observed between the animals implanted with cells from healthy donors and PD patients. The safety of grafted cells was confirmed by monitoring their survival and proliferation by magnetic resonance imaging (MRI) and positron emission tomography (PET) from 12 to 24 months after transplantation. The animals were then sacrificed, and their brain sections were studied after hematoxylin-eosin staining. The authors found no neural rosette formation, malignant changes, or proliferation of Ki67+ cells in the grafts. Also, differentiation of grafted neuronal precursors was assessed. It was found that 6-54% grafted cells differentiated to tyrosine hydroxylase-positive neurons, which on average corresponds to 6.4·104 cells per hemisphere. These tyrosine hydroxylase-positive neurons expressed the presynaptic dopamine transporter (DAT) (midbrain DN marker) and GIRK2 (A9 type DN marker). DNs derived from PD patients displayed no pathological symptoms such as Lewy bodies. The analysis of functional activity of grafted neurons by PET revealed an increased uptake of [18F]DOPA in the putamen. The obtained results were similar to those found in patients with grafted hfVM [54]. These studies provided a strong evidence that human iPSCs display high therapeutic potential and can be used in future clinical trials.
To use hESC/iPSC-based cell therapy in clinical practice, it is essential to avoid the graft-versus-host reaction. The iPSC-based technology allows to eliminate or significantly reduce problems with immune response, as personalized iPSC lines can be obtained for each patient. Although the brain is believed to be an immune privileged organ, there is a difference between transplantation of autologous cells and allogeneic cells, whose HLA haplotype mismatches that of the recipient [55]. Although autologous cell therapy is ideal in theory, reprogramming of original cells to iPSCs with their subsequent differentiation for each patient is expensive and takes a long time. As an alternative, investigators from the Kyoto University started the iPSCs Stock Project by establishing a bank of iPSC clones from donors with homozygous HLA haplotypes. It was calculated that 50 iPSC lines with homozygous HLA haplotypes would cover 73% of Japanese population at the three HLA loci (HLA-A, B, and DR) [56]. On the other hand, minor HLA loci or innate immune cells, such as macrophages and NK cells, might also contribute to the developing immune response, albeit not as strong as in case of allogeneic transplantation. Clinical trials using iPSCs-derived retinal pigment epithelium (RPE) cells [57] showed that obtaining autologous iPSC derivatives for each patient is laborious and expensive and requires each generated iPSC line to be tested for its quality. Because of this, researchers prefer to use recipient HLA-matching allogeneic hESC and iPSC derivatives in cell therapy.
Although such cell-based products might not fully prevent the development of immune response, they could substantially reduce it, which would allow to apply a milder immunosuppressive therapy. Still, whether allogeneic nervous tissue transplanted to the recipient’s brain undergoes continuous immune-mediated rejection is debated. For instance, live allogeneic fetal cells were found in the brain of recipient PD patients many years after termination of the immunosuppressive therapy [58].
Kyoto University plans to start clinical trials on the transplantation of iPSC-derived DNs to sporadic PD patients 50 to 70 years old. The major objective of this trial is assessment of transplant tumorigenicity and growth by magnetic resonance imaging and positron emission tomography; another goal of this study is evaluation of the neurological state and [18F]DOPA uptake by the grafted cells [59].
In an attempt to optimize clinical studies on the use of hESC and iPSC derivatives in PD therapy and characterization of transplanted cells and to promote cooperation in the introduction of PD cell therapy into clinical practice, the scientists created the GForce-PD (http://www.gforce-pd.com), a global consortium on the use of pluripotent stem cells in PD cell therapy, that has united prominent investigators in this research field from Europe, USA, and Japan. Each GForce-PD research group participated in the development of GMP protocols for obtaining DNs with certain in vivo parameters and iPSCs-derived functional midbrain DNs, as well as cryopreservation protocols and quality control assays. Such protocols also include cell differentiation to committed DN precursors that have demonstrated the best results in animal PD models. Cell obtained according to the developed protocols exhibited no tumorigenicity or uncontrolled growth in vivo [54, 60-63]. Importantly, the protocols are reproducible and scalable, although certain problems still exist with choosing the most appropriate genetic assay for both starting material and/or final product. Other issues discussed by the Consortium include the efficacy and quality of products of iPSC differentiation [47]. The problems of in-depth testing and safety of hESCs and iPSCs in cell therapy are also discussed outside of the scope of PD cell therapy. Genetic and epigenetic stability of cultured human pluripotent stem cells, the methods to minimize the emergence of genetic and epigenetic cell variants during cell culturing, and potential effects of these variants on the efficacy of cell therapy have been discussed at the conference organized in 2017 by the International Stem Cell Initiative (ISCI) [64]. Such conferences are attended by the recognized experts on human pluripotent stem cells and cell therapy, which allows us to expect recommendations on the protocols and regulations for using hESCs and iPSCs in cell therapy. In the future, the need for such cell products might significantly increase [47].
Despite numerous unresolved issues, GForce-PD investigators plan to conduct the first clinical trials in 2018-2020. The majority of research teams are expected to cryoconserve large amounts of DN precursors after a single differentiation round. The final cell products will be tested for stability, tumorigenicity, toxicity, and biological functions in accordance with the requirements of national regulatory agencies (e.g., FDA, MHRA, and PMDA) [47].
One of the topics of 2017 GForce-PD meeting was the problems of immunosuppression in PD cell therapy. It was agreed that at least one immunosuppressive agent (e.g., FK506) should be used; however, the duration of immunosuppressive therapy was debated. On average, the period of immunosuppression should last for 1-2 years after the transplantation. These conclusions were in part based on the immunosuppression protocols used in patients with hfVM transplants that showed a long-term graft survival without a need for a lifelong immunosuppression [47].
Another discussed issue was selection of patients for the enrollment in clinical trials. Such patients should respond to Levodopa oral therapy. The question is at what stage should the experimental cell therapy be started? On one hand, cell therapy should be used in patients with the expected response, i.e., younger patients at the early PD stage, without significant motor and cognitive disorders and early-onset dementia, and with good response to Levodopa. Such inclusion criteria were applied to patients enrolled in the TRANSEURO clinical trial [65]. On the other hand, it would be unethical to subject patients at such early PD stage to this new, untested, and not yet commonly accepted therapy; it might be better to use patients with severe motor disorders. At such advanced stages of PD, invasive cell therapy with associated deep brain stimulation and apomorphine therapy becomes justified and necessary. For this reason, the majority of research groups in the GForce-PD Consortium enroll patients at more advanced PD stages that those used in the TRANSEURO study; the enrolled patients also lack such prominent PD symptom as Levodopa-induced dyskinesia [47].
The clinical efficacy of cell therapy is expected to be assessed within 3-5 years after cell transplantation, since the effects of hfVM grafting in PD patients are mostly pronounced during this time interval. However, the effects of cell therapy should be assessed much earlier than 3-5 years after the transplantation, especially in the absence of sham surgery as a control. Therefore, the majority of patients will be assessed at least two years after surgery [47].
Despite a cautious position of global scientific community on the use of hESCs and iPSCs for PD cell therapy, a clinical trial (NCT03119636) with differentiated hESC derivatives has been started in China (Henan, Zhengzhou, First Affiliated Hospital of Zhengzhou University), with phases I/II officially starting in May, 2017. The trial is conducted in 50 PD patients that match the following inclusion criteria: (i) PD manifestation for at least 5 years before the trial start; (ii) age, 50-80 years; (iii) Hoehn and Yahr scale stage 3 or 4; (iv) documented clinical efficacy of dopaminergic drug therapy. Uncommitted neuronal precursors derived from hESCs were transplanted to the striatum of PD patients. However, these studies have risen serious concerns because of potential risk that after transplantation to the striatum, these immature precursors can differentiate into other than DNs cell types, which might not result in expected positive effect [66]. The next planned stage is transplantation of HLA-matching/mismatching neuronal precursors. The accompanying drug therapy will involve the treatment with Levodopa only, depending on the patient’s state. The first results of this study are expected to be published in November, 2018, including MRI and blood assay data.
The unlimited lifespan of cultured pluripotent stem cells and the development of protocols for their targeted differentiation allow generation of promising model systems (e.g., cell models) of neurodegenerative diseases. These models, including isogenic cell lines for investigating familial PD, pave a way for understanding PD-triggering mechanisms that are induced long before the first clinical PD manifestations.
Recent advances in experiments on DNs derived from hESCs and iPSCs for the transplantation into PD animal models give a hope that the clinical trials started in some countries will result in new promising findings in PD cell therapy.
Funding
This study was supported by the Russian Science Foundation (project no. 14-15-00930).
REFERENCES
1.Illarioshkin, S. N. (2003) Conformational Brain
Diseases [in Russian], Yanus-K, Moscow.
2.Levy, O. A., Malagelada, C., and Greene, L. A.
(2009) Cell death pathways in Parkinson’s disease: proximal
triggers, distal effectors, and final steps, Apoptosis,
14, 478-500.
3.Illarioshkin, S. N. (2004) Diseases of nervous
system, Nervnye Bolezni, 4, 14-21.
4.Gardner, R. C., Burke, J. F., Nettiksimmons, J.,
Goldman, S., Tanner, C. M., and Yaffe, K. (2015) Traumatic brain injury
in later life increases risk for Parkinson’s disease, Ann.
Neurol., 77, 987-995.
5.Lee, P. C., Bordelon, Y., Bronstein, J., and Ritz,
B. (2012) Traumatic brain injury, paraquat exposure, and their
relationship to Parkinson’s disease, Neurology, 79,
2061-2066.
6.Ratner, M. H., Farb, D. H., Ozer, J., Feldman, R.
G., and Durso, R. (2014) Younger age at onset of sporadic
Parkinson’s disease among subjects occupationally exposed to
metals and pesticides, Interdiscip. Toxicol., 7,
123-133.
7.Deng, H., Wang, P., and Jankovic, J. (2018) The
genetics of Parkinson’s disease, Ageing Res. Rev.,
42, 72-85.
8.Lab of Neuroscience, The HIT Center for Life
Sciences, http://hcls.hit.edu.cn/hclsen/wabwofwweurowwcience/list.psp.
9.Takahashi, K., and Yamanaka, S. (2006) Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors, Cell, 126, 663-676.
10.Rawat, N., and Singh, M. K. (2017) Induced
pluripotent stem cell: a headway in reprogramming with promising
approach in regenerative biology, Vet. World, 10,
640-649.
11.Park, I.-H., Arora, N., Huo, H., Maherali, N.,
Ahfeldt, T., Shimamura, A., Lensch, M. W., Cowan, C., Hochedlinger, K.,
and Daley, G. Q. (2008) Disease-specific induced pluripotent stem (iPS)
cells, Cell, 134, 877-886.
12.Wernig, M., Zhao, J.-P., Pruszak, J., Hedlund,
E., Fu, D., Soldner, F., Broccoli, V., Constantine-Paton, M., Isacson,
O., and Jaenisch, R. (2008) Neurons derived from reprogrammed
fibroblasts functionally integrate into the fetal brain and improve
symptoms of rats with Parkinson’s disease, Proc. Natl. Acad.
Sci. USA, 105, 5856-5861.
13.Hargus, G., Cooper, O., Deleidi, M., Levy, A.,
Lee, K., Marlow, E., Yow, A., Soldner, F., Hockemeyer, D., Hallett, P.
J., Osborn, T., Jaenisch, R., and Isacson, O. (2010) Differentiated
Parkinson patient-derived induced pluripotent stem cells grow in the
adult rodent brain and reduce motor asymmetry in Parkinsonian rats,
Proc. Natl. Acad. Sci. USA, 107, 15921-15926.
14.Fernandez-Santiago, R., Carballo-Carbajal, I.,
Castellano, G., Torrent, R., Richaud, Y., Sanchez-Danes, A.,
Vilarrasa-Blasi, R., Sanchez-Pla, A., Mosquera, J. L., Soriano, J.,
Lopez-Barneo, J., Canals, J. M., Alberch, J., Raya, A., Vila, M.,
Consiglio, A., Martin-Subero, J. I., Ezquerra, M., and Tolosa, E.
(2015) Aberrant epigenome in iPSC-derived dopaminergic neurons from
Parkinson’s disease patients, EMBO Mol. Med., 7,
1529-1546.
15.Martin, I., Kim, J. W., Dawson, V. L., and
Dawson, T. M. (2014) LRRK2 pathobiology in Parkinson’s
disease, J. Neurochem., 131, 554-565.
16.Healy, D. G., Falchi, M., O’Sullivan, S.
S., Bonifati, V., Durr, A., Bressman, S., Brice, A., Aasly, J.,
Zabetian, C. P., Goldwurm, S., Ferreira, J. J., Tolosa, E., Kay, D. M.,
Klein, C., Williams, D. R., Marras, C., Lang, A. E., Wszolek, Z. K.,
Berciano, J., Schapira, A. H., Lynch, T., Bhatia, K. P., Gasser, T.,
Lees, A. J., and Wood, N. W. (2008) Phenotype, genotype, and worldwide
genetic penetrance of LRRK2-associated Parkinson’s
disease: a case-control study, Lancet Neurol., 7,
583-590.
17.Liu, G.-H., Qu, J., Suzuki, K., Nivet, E., Li,
M., Montserrat, N., Yi, F., Xu, X., Ruiz, S., Zhang, W., Wagner, U.,
Kim, A., Ren, B., Li, Y., Goebl, A., Kim, J., Soligalla, R. D., Dubova,
I., Thompson, J., Yates, J., 3rd, Esteban, C. R., Sancho-Martinez, I.,
and Belmonte, J. C. I. (2012) Progressive degeneration of human neural
stem cells caused by pathogenic LRRK2, Nature,
491, 603-607.
18.Seibler, P., Graziotto, J., Jeong, H., Simunovic,
F., Klein, C., and Krainc, D. (2011) Mitochondrial Parkin recruitment
in neurons derived from mutant PINK1 iPS cells, J. Neurosci.,
31, 5970-5976.
19.Sanchez-Danes, A., Richaud-Patin, Y.,
Carballo-Carbajal, I., Jimenez-Delgado, S., Caig, C., Mora, S., Di
Guglielmo, C., Ezquerra, M., Patel, B., Giralt, A., Canals, J. M.,
Memo, M., Alberch, J., Lopez-Barneo, J., Vila, M., Cuervo, A. M.,
Tolosa, E., Consiglio, A., and Raya, A. (2012) Disease-specific
phenotypes in dopamine neurons from human iPS-based models of genetic
and sporadic Parkinson’s disease, EMBO Mol. Med.,
4, 380-395.
20.Byers, B., Cord, B., Nguyen, H. N., Schule, B.,
Fenno, L., Lee, P. C., Deisseroth, K., Langston, J. W., Pera, R. R.,
and Palmer, T. D. (2011) SNCA triplication Parkinson’s
patient’s iPSC-derived DA neurons accumulate α-synuclein
and are susceptible to oxidative stress, PLoS One, 6,
e26159.
21.Chung, C. Y., Khurana, V., Auluck, P. K.,
Tardiff, D. F., Mazzulli, J. R., Soldner, F., Baru, V., Lou, Y.,
Freyzon, Y., Cho, S., Mungenast, A. E., Muffat, J., Mitalipova, M.,
Pluth, M. D., Jui, N. T., Schule, B., Lippard, S. J., Tsai, L. H.,
Krainc, D., Buchwald, S. L., Jaenisch, R., and Lindquist, S. (2013)
Identification and rescue of α-synuclein toxicity in
Parkinson’s patient-derived neurons, Science, 342,
983-987.
22.Monzel, A. S., Smits, L. M., Hemmer, K., Hachi,
S., Moreno, E. L., van Wuellen, T., Jarazo, J., Walter, J., Bruggemann,
I., Boussaad, I., Berger, E., Fleming, R. M. T., Bolognin, S., and
Schwamborn, J. C. (2017) Derivation of human midbrain-specific
organoids from neuroepithelial stem cells, Stem Cell Rep.,
8, 1144-1154.
23.Cai, J., Yang, M., Poremsky, E., Kidd, S.,
Schneider, J. S., and Iacovitti, L. (2010) Dopaminergic neurons derived
from human induced pluripotent stem cells survive and integrate into
6-OHDA-lesioned rats, Stem Cells Dev., 19, 1017-1023.
24.Martinez-Morales, P. L., and Liste, I. (2012)
Stem cells as in vitro model of Parkinson’s disease,
Stem Cells Int., 2012, 980941.
25.Doi, D., Samata, B., Katsukawa, M., Kikuchi, T.,
Morizane, A., Ono, Y., Sekiguchi, K., Nakagawa, M., Parmar, M., and
Takahashi, J. (2014) Isolation of human induced pluripotent stem
cell-derived dopaminergic progenitors by cell sorting for successful
transplantation, Stem Cell Rep., 2, 337-350.
26.Nishimura, K., Murayama, S., and Takahashi, J.
(2015) Identification of neurexophilin 3 as a novel supportive factor
for survival of induced pluripotent stem cell-derived dopaminergic
progenitors, Stem Cells Transl. Med., 4, 932-944.
27.De Boni, L., Gasparoni, G., Haubenreich, C.,
Tierling, S., Schmitt, I., Peitz, M., Koch, P., Walter, J., Wullner,
U., and Brustle, O. (2018) DNA methylation alterations in iPSC- and
hESC-derived neurons: potential implications for neurological disease
modeling, Clin. Epigenetics, 10, 13.
28.Soldner, F., Stelzer, Y., Shivalila, C. S.,
Abraham, B. J., Latourelle, J. C., Barrasa, M. I., Goldmann, J., Myers,
R. H., Young, R. A., and Jaenisch, R. (2016) Parkinson-associated risk
variant in distal enhancer of α-synuclein modulates target gene
expression, Nature, 533, 95-99.
29.Li, J. Q., Tan, L., and Yu, J. T. (2014) The role
of the LRRK2 gene in parkinsonism, Mol. Neurodegener.,
9, 47.
30.Pollanen, M. S., Dickson, D. W., and Bergeron, C.
(1993) Pathology and biology of the Lewy body, J. Neuropathol. Exp.
Neurol., 52, 183-191.
31.Spillantini, M. G., Schmidt, M. L., Lee, V. M.,
Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997) α-Synuclein
in Lewy bodies, Nature, 388, 839-884.
32.Anderson, J. P., Walker, D. E., Goldstein, J. M.,
de Laat, R., Banducci, K., Caccavello, R. J., Barbour, R., Huang, J.,
Kling, K., Lee, M., Diep, L., Keim, P. S., Shen, X., Chataway, T.,
Schlossmacher, M. G., Seubert, P., Schenk, D., Sinha, S., Gai, W. P.,
and Chilcote, T. J. (2006) Phosphorylation of Ser-129 is the dominant
pathological modification of α-synuclein in familial and
sporadic Lewy body disease, J. Biol. Chem., 281,
29739-29752.
33.Oueslati, A. (2016) Implication of
α-synuclein phosphorylation at S129 in synucleinopathies: what
have we learned in the last decade? J. Parkinson’s Dis.,
6, 39-51.
34.Alegre-Abarrategui, J., Ansorge, O., Esiri, M.,
and Wade-Martins, R. (2008) LRRK2 is a component of granular
α-synuclein pathology in the brainstem of Parkinson’s
disease, Neuropathol. Appl. Neurobiol., 34, 272-283.
35.Martin, I., Dawson, V. L., and Dawson, T. M.
(2011) Recent advances in the genetics of Parkinson’s disease,
Annu. Rev. Genomics Hum. Genet., 12, 301-325.
36.Qing, X., Walter, J., Jarazo, J.,
Arias-Fuenzalida, J., Hillje, A. L., and Schwamborn, J. C. (2017)
CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in
reveals neuronal complexity phenotypes and α-synuclein modulation
in dopaminergic neurons, Stem Cell Res., 24, 44-50.
37.Ferreia, M., and Massano, J. (2017) An updated
review of Parkinson’s disease genetics and clinicopathological
correlations, Acta Neurol. Scand., 135, 273-284.
38.Abizaid, A., Liu, Z. W., Andrews, Z. B.,
Shanabrough, M., Borok, E., Elsworth, J. D., Roth, R. H., Sleeman, M.
W., Picciotto, M. R., Tschop, M. H., Gao, X. B., and Horvath, T. L.
(2006) Ghrelin modulates the activity and synaptic input organization
of midbrain dopamine neurons while promoting appetite, J. Clin.
Invest., 116, 3229-3239.
39.Diano, S., Farr, S. A., Benoit, S. C., McNay, E.
C., da Silva, I., Horvath, B., Gaskin, F. S., Nonaka, N., Jaeger, L.
B., Banks, W. A., Morley, J. E., Pinto, S., Sherwin, R. S., Xu, L.,
Yamada, K. A., Sleeman, M. W., Tschop, M. H., and Horvath, T. L. (2006)
Ghrelin controls hippocampal spine synapse density and memory
performance, Nat. Neurosci., 9, 381-388.
40.Kojima, M., Hosoda, H., Date, Y., Nakazato, M.,
Matsuo, H., and Kangawa, K. (1999) Ghrelin is a
growth-hormone-releasing acylated peptide from stomach, Nature,
402, 656-660.
41.Nakazato, M., Murakami, N., Date, Y., Kojima, M.,
Matsuo, H., Kangawa, K., and Matsukura, S. (2001) A role for ghrelin in
the central regulation of feeding, Nature, 409,
194-198.
42.Cowley, M. A., Smith, R. G., Diano, S., Tschop,
M., Pronchuk, N., Grove, K. L., Strasburger, C. J., Bidlingmaier, M.,
Esterman, M., Heiman, M. L., Garcia-Segura, L. M., Nillni, E. A.,
Mendez, P., Low, M. J., Sotonyi, P., Friedman, J. M., Liu, H., Pinto,
S., Colmers, W. F., Cone, R. D., and Horvath, T. L. (2003) The
distribution and mechanism of action of ghrelin in the CNS demonstrates
a novel hypothalamic circuit regulating energy homeostasis,
Neuron, 37, 649-661.
43.Shi, L., Bian, X., Qu, Z., Ma, Z., Zhou, Y.,
Wang, K., Jiang, H., and Xie, J. (2013) Peptide hormone ghrelin
enhances neuronal excitability by inhibition of Kv7/KCNQ channels,
Nat. Commun., 4, 1435.
44.Suda, Y., Kuzumaki, N., Sone, T., Narita, M.,
Tanaka, K., Hamada, Y., Iwasawa, C., Shibasaki, M., Maekawa, A.,
Matsuo, M., Akamatsu, W., Hattori, N., Okano, H., and Narita, M. (2018)
Down-regulation of ghrelin receptors on dopaminergic neurons in the
substantia nigra contributes to Parkinson’s disease-like motor
dysfunction, Mol. Brain, 11, 6.
45.Brundin, P., Strecker, R. E., Lindvall, O.,
Isacson, O., Nilsson, O. G., Barbin, G., Prochiantz, A., Forni, C.,
Nieoullon, A., Widner, H., Gage, F. H., and Bjorklund, A. (1987)
Intracerebral grafting of dopamine neurons. Experimental basis for
clinical trials in patients with Parkinson’s disease, Ann. N.
Y. Acad. Sci., 495, 473-496.
46.Barker, R. A., Drouin-Ouellet, J., and Parmar, M.
(2015) Cell-based therapies for Parkinson’s disease – past
insights and future potential, Nat. Rev. Neurol., 11,
492-503.
47.Barker, R. A., Parmar, M., Studer, L., and
Takahashi, J. (2017) Human trials of stem cell-derived dopamine neurons
for Parkinson’s disease: dawn of a new era, Cell Stem
Cell, 21, 569-573.
48.Shimohama, S., Sawada, H., Kitamura, Y., and
Taniguchi, T. (2003) Disease model: Parkinson’s disease,
Trends Mol. Med., 9, 360-365.
49.Saiki, H., Hayashi, T., Takahashi, R., and
Takahashi, J. (2010) Objective and quantitative evaluation of motor
function in a monkey model of Parkinson’s disease, J.
Neurosci. Methods, 190, 198-204.
50.Takagi, Y., Takahashi, J., Saiki, H., Morizane,
A., Hayashi, T., Kishi, Y., Fukuda, H., Okamoto, Y., Koyanagi, M.,
Ideguchi, M., Hayashi, H., Imazato, T., Kawasaki, H., Suemori, H.,
Omachi, S., Iida, H., Itoh, N., Nakatsuji, N., Sasai, Y., and
Hashimoto, N. (2005) Dopaminergic neurons generated from monkey
embryonic stem cells function in a Parkinson’s primate model,
J. Clin. Invest., 115, 102-109.
51.Lebedeva, O. S., Lagar’kova, M. A.,
Kiselev, S. L., Mukhina, I. V., Vedunova, M. V., Usova, O. V.,
Stavrovskaya, A. V., Yamshchikova, N. G., Fedotova, E. Yu.,
Grivennikov, I. A., Khaspekov, L. G., and Illarioshkin, S. N. (2013)
Morphofunctional properties of induced pluripotent stem cells derived
from human skin fibroblasts and differentiated to dopaminergic neurons,
Neirokhimiya, 3, 233-241.
52.Doi, D., Morizane, A., Kikuchi, T., Onoe, H.,
Hayashi, T., Kawasaki, T., Motono, M., Sasai, Y., Saiki, H., Gomi, M.,
Yoshikawa, T., Hayashi, H., Shinoyama, M., Refaat, M. M., Suemori, H.,
Miyamoto, S., and Takahashi, J. (2012) Prolonged maturation culture
favors a reduction in the tumorigenicity and the dopaminergic function
of human ESC-derived neural cells in a primate model of
Parkinson’s disease, Stem Cells, 30, 935-945.
53.Kikuchi, T., Morizane, A., Doi, D., Onoe, H.,
Hayashi, T., Kawasaki, T., Saiki, H., Miyamoto, S., and Takahashi, J.
(2011) Survival of human induced pluripotent stem cell-derived midbrain
dopaminergic neurons in the brain of a primate model of
Parkinson’s disease, J. Parkinson’s Dis., 1,
395-412.
54.Kikuchi, T., Morizane, A., Doi, D., Magotani, H.,
Onoe, H., Hayashi, T., Mizuma, H., Takara, S., Takahashi, R., Inoue,
H., Morita, S., Yamamoto, M., Okita, K., Nakagawa, M., Parmar, M., and
Takahashi, J. (2017) Human iPS cell-derived dopaminergic neurons
function in a primate Parkinson’s disease model, Nature,
548, 592-596.
55.Morizane, A., Doi, D., Kikuchi, T., Okita, K.,
Hotta, A., Kawasaki, T., Hayashi, T., Onoe, H., Shiina, T., Yamanaka,
S., and Takahashi, J. (2013) Direct comparison of autologous and
allogeneic transplantation of iPSC-derived neural cells in the brain of
a non-human primate, Stem Cell Rep., 1, 283-292.
56.Okita, K., Matsumura, Y., Sato, Y., Okada, A.,
Morizane, A., Okamoto, S., Hong, H., Nakagawa, M., Tanabe, K., Tezuka,
K., Shibata, T., Kunisada, T., Takahashi, M., Takahashi, J., Saji, H.,
and Yamanaka, S. (2011) A more efficient method to generate
integration-free human iPS cells, Nat. Methods, 8,
409-412.
57.Mandai, M., Watanabe, A., Kurimoto, Y., Hirami,
Y., Morinaga, C., Daimon, T., Fujihara, M., Akimaru, H., Sakai, N.,
Shibata, Y., Terada, M., Nomiya, Y., Tanishima, S., Nakamura, M.,
Kamao, H., Sugita, S., Onishi, A., Ito, T., Fujita, K., Kawamata, S.,
Go, M. J., Shinohara, C., Hata, K. I., Sawada, M., Yamamoto, M., Ohta,
S., Ohara, Y., Yoshida, K., Kuwahara, J., Kitano, Y., Amano, N.,
Umekage, M., Kitaoka, F., Tanaka, A., Okada, C., Takasu, N., Ogawa, S.,
Yamanaka, S., and Takahashi, M. (2017) Autologous induced
stem-cell-derived retinal cells for macular degeneration, N. Engl.
J. Med., 376, 1038-1046.
58.Li, W., Englund, E., Winder, H., Mattsson, B.,
van Westen, D., Latt, D., Rehncrona, S., Brundin, P., Bjorklund, A.,
Lindvall, O., and Li, J. Y. (2016) Extensive graft-derived dopaminergic
innervation is maintained 24 years after transplantation in the
degenerating parkinsonian brain, Proc. Natl. Acad. Sci. USA,
113, 6544-6549.
59.Morizane, A., and Takahashi, J. (2016) Cell
therapy for Parkinson’s disease, Neurol. Med. Chir.
(Tokyo), 56, 102-109.
60.Grealish, S., Diguet, E., Kirkeby, A., Mattsson,
B., Heuer, A., Bramoulle, Y., Van Camp, N., Perrier, A. L., Hantraye,
P., Bjorklund, A., and Parmar, M. (2014) Human ESC-derived dopamine
neurons show similar preclinical efficacy and potency to fetal neurons
when grafted in a rat model of Parkinson’s disease, Cell Stem
Cell, 15, 653-665.
61.Kirkeby, A., Grealish, S., Wolf, D. A., Nelander,
J., Wood, J., Lundblad, M., Lindvall, O., and Parmar, M. (2012)
Generation of regionally specified neural progenitors and functional
neurons from human embryonic stem cells under defined conditions,
Cell Rep., 1, 703-714.
62.Kriks, S., Shim, J.-W., Piao, J., Ganat, Y. M.,
Wakeman, D. R., Xie, Z., Carrillo-Reid, L., Auyeung, G., Antonacci, C.,
Buch, A., Yang, L., Beal, M. F., Surmeier, D. J., Kordower, J. H.,
Tabar, V., and Studer, L. (2011) Dopamine neurons derived from human ES
cells efficiently engraft in animal models of Parkinson’s
disease, Nature, 480, 547-553.
63.Steinbeck, J. A., Choi, S. J., Mrejeru, A.,
Ganat, Y., Deisseroth, K., Sulzer, D., Mosharov, E. V., and Stude, L.
(2015) Optogenetics enables functional analysis of human embryonic stem
cell-derived grafts in a Parkinson’s disease model, Nat.
Biotechnol., 33, 204-209.
64.Andrews, P. W., Ben-David, U., Benvenisty, N.,
Coffey, P., Eggan, K., Knowles, B. B., Nagy, A., Pera, M., Reubinoff,
B., Rugg-Gunn, P. J., and Stacey, G. N. (2017) Assessing the safety of
human pluripotent stem cells and their derivatives for clinical
applications, Stem Cell Rep., 9, 1-4.
65.Barker, R. A., Parmar, M., Kirkeby, A.,
Bjorklund, A., Thompson, L., and Brundin, P. (2016) Are stem cell-based
therapies for Parkinson’s disease ready for the clinic in 2016?
J. Parkinson’s Dis., 6, 57-63.
66.Cyranoski, D. (2017) Trials of embryonic stem
cells to launch in China, Nature, 546, 15-16.