MINI-REVIEW: Dynamic Microtubules in Alzheimer’s Disease: Association with Dendritic Spine Pathology
E. I. Pchitskaya1,a, V. A. Zhemkov1, and I. B. Bezprozvanny1,2,b*
1Laboratory of Molecular Neurodegeneration, Department of Medical Physics, Peter the Great St. Petersburg Polytechnic University, 195251 St. Petersburg, Russia2Department of Physiology, UT Southwestern Medical Center at Dallas, 75390 Dallas, TX, USA
* To whom correspondence should be addressed.
Received May 7, 2018; Revision received June 7, 2018
Alzheimer’s disease (AD) is the most common incurable neurodegenerative disorder that affects the processes of memory formation and storage. The loss of dendritic spines and alteration in their morphology in AD correlate with the extent of patient’s cognitive decline. Tubulin had been believed to be restricted to dendritic shafts, until recent studies demonstrated that dynamically growing tubulin microtubules enter dendritic spines and promote their maturation. Abnormalities of tubulin cytoskeleton may contribute to the process of dendritic spine shape alteration and their subsequent loss in AD. In this review, association between tubulin cytoskeleton dynamics and dendritic spine morphology is discussed in the context of dendritic spine alterations in AD. Potential implications of these findings for the development of AD therapy are proposed.
KEY WORDS: Alzheimer’s disease, dendritic spines, tubulin, microtubule dynamics, EB3DOI: 10.1134/S0006297918090080
Abbreviations: AD, Alzheimer’s disease; APP, amyloid-precursor protein; EB3, end-binding protein 3; MT, microtubule; PS1 (2), Presenilin 1(2); +TIPs, microtubule plus-end-tracking proteins.
PATHOLOGICAL FEATURES OF ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is the most common incurable neurodegenerative disorder that affects the processes of memory formation and storage and leads to progressive cognitive decline. In most cases, AD appears sporadically with advanced age being the major risk factor. Genetically inherited familial forms of AD (FAD, approximately 1-2% cases) are characterized by the early onset and rapid progression [1]. FAD are caused by mutations in genes encoding amyloid-precursor protein (APP), presenilin 1 (PS1), or presenilin 2 (PS2) proteins [1-3]. Presenilins form catalytic subunits of the γ-secretase complex, which together with β-secretase is responsible for the APP protein cleavage and generation of toxic Aβ peptides [4]. Extracellular plaques composed of Aβ peptide aggregates are one of the key histopathological features of AD. Another feature is neurofibrillary tangles (NFTs) formed by hyperphosphorylated form of the microtubule-associated protein tau. On the cellular level, AD is characterized by the loss of dendritic spines and alteration in their morphology that correlates with the extent of cognitive decline in AD patients [5, 6]. Changes in dendritic spines occur at the early AD stages, prior to neuronal death and amyloid plaque formation [5-8].
Dendritic spines represent the most ubiquitous postsynaptic sites of excitatory inputs in neurons. They control signal transduction in individual synapses and participate in synaptic input compartmentalization [9]. Dendritic spines can be classified into three groups reflecting the key features of their morphology: mushroom, thin, and stubby [10, 11]. Relatively stable mushroom spines have a big head and small neck. They form strong synaptic connections and supposedly act as sites of memory storage [12, 13]. More dynamic thin spines, having a smaller head, are proposed to be “learning spines” responsible for the formation of new memories [12]. Stubby spines are likely to form as a result of mushroom spine elimination [14]. Reduction in the number of mushroom spines is proposed to underlie memory loss observed in AD patients [15-17]. Indeed, the shift from mushroom to stubby spines was observed in cortical biopsies from AD patients [18]. Reduction in the mushroom spine density has been shown in various AD mice models, including PS1-M146V-KI [19], APP-KI [20, 21], and APPPS1 mice [18], in ex vivo hippocampal slice cultures from APPSDL transgenic mice [22, 23], and under conditions of amyloid toxicity in vivo and in vitro [24, 25]. The density of mushroom and thin spines in the pyramidal neuron is significantly decreased in the prefrontal cortex of AD patients with dementia in comparison to cognitively normal individuals with AD pathology [26]. The authors speculate that such organization of dendritic spines makes these individuals more resilient to dementia despite pathological brain lesions.
While exact reasons of AD are still debated, it has become clear that therapeutic intervention at earlier disease stages is preferable and more effective. Restoration or stabilization of normal spine morphology may help to preserve human memory and present potential therapeutic approaches for AD treatment [15-17]. Recent studies suggest that abnormalities of tubulin cytoskeleton may contribute to the process of dendritic spine shape alterations and subsequent dendritic spine loss in AD. This review discusses potential mechanisms that link tubulin cytoskeleton dynamics and dendritic spine morphology, as well as ideas for potential AD therapeutic development.
DYNAMICS OF NEURONAL TUBULIN CYTOSKELETON AND DENDRITIC
SPINES
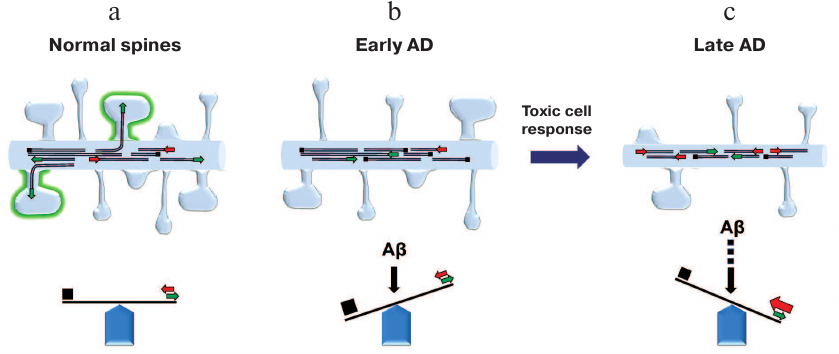
Microtubules (MTs) are polar tubulin polymers that represent exceptionally important cytoskeletal components. In neurons, MTs form the carcass of thin and extremely extended neuronal processes – dendrites and axons – and serve as tracks for cargo trafficking. MT plus-ends undergo repeated process of polymerization/depolymerization (the so-called dynamic instability) [27] that is precisely controlled by various MT-associated proteins. In contrast, MT minus-ends are much more stable [28]. In mature neurons, MTs exist as autonomous structures of various length that are not attached to the centrosome (unlike MTs in non-neuronal cells) [28]. MTs in neurons are more stable than MTs in dividing cells, which helps them to maintain specific shape of neurons. It should be mentioned that neurons also have a fraction of dynamic MTs in dendrites and axons [28]. Another cytoskeletal protein, actin, maintains the structure of dendritic spines that are highly dynamic in comparison to neurites. It had been believed before that tubulin and actin cytoskeleton structures do not overlap in neurons: tubulin forms cytoskeleton in neurites, while actin does the same in dendritic spines. However, recent advances in live cell imaging uncovered transient entry of dynamic MTs into dendritic spines [29-32] (figure, panel (a)). The reasons why this phenomenon had not been observed in previous studies were recently discussed in [29]. Further experiments revealed that transient entry of MTs into the spines lasts on average for a few minutes, and that only small percentage of spines are targeted simultaneously. However, over time, most spines undergo MT invasion that appears to be facilitated by neuronal activity and depends on calcium influx into the spines and actin polymerization [31, 33]. MT entry triggers spine head enlargement [31, 32] and elevates the content of PSD-95 in the spines [34]. Induction of long-term potentiation increases the frequency of MT entry into the spines and the number of targeted spines [31]. Taking together, these results suggest that MT entry into dendritic spines is involved in synaptic plasticity. To corroborate this hypothesis, it was found that the early phase of memory formation (0.5-1 h after learning) is associated with a decrease in MT stability, but the later phase (8 h after learning) is characterized by hyperstable MTs [35]. Therefore, it was proposed that entry of dynamic MTs into dendritic spines is necessary for cargo/signal delivery to the spines and involved in the induction of structural changes in the spines through interaction with actin cytoskeleton as a response to increased activity at the site of synaptic contact [29] (figure, panel (a)).
Dendritic spine morphology is regulated by the balance between MT dynamics and stability. a) In normal neurons, dynamic MTs transiently enter dendritic spines in the activity-dependent manner. b) Stabilization of MTs (early response to Aβ) causes simplification of dendritic spine shape. c) Subsequent toxic cellular response to MT stabilization at later AD stages results in the reduction of MT density and length and causes the loss of dendritic spines
Distal ends of growing MTs are decorated with a specific group of MT plus-end-tracking proteins (+TIPs) that regulate MT dynamics and interaction with other cellular components [36]. The group of +TIPs proteins includes end-binding protein 3 (EB3), a neuron-specific member of the EB protein family [37]. EB3 protein is ubiquitously expressed in the nervous system, including dendritic spines, which it enters at the tips of growing MTs [31-33, 38]. EB3 fused to GFP (green fluorescent protein) is a powerful tool for visualization of growing MTs in neurons [39]. Changes in the EB3 expression influence dendritic spine morphology. Thus, EB3 knockdown results in the reduction in the number of mushroom spines, while EB3 overexpression causes their robust increase [32, 40]. Moreover, EB3 overexpression reverses the deficiency of mushroom spines in PS1-M146V-KI mice (AD model) [40]. Another study showed that EB3 silencing results in the reduction of dendritic spine density without changes in the spine shape [30]. These data suggest that EB3 is implicated in the regulation of dendritic spine morphology, although the mechanism of this process still remains unknown. One possible link between EB3 and spine shape is EB3 binding to the p140Cap scaffold protein that affects actin organization in dendritic spines [32]. Another link is EB3 interaction with the STIM2 protein that controls store-operated calcium entry essential for the functioning of mushroom spines [40-42]. Disruption of the STIM2–EB3 interactions leads to the decrease in the relative content of mushroom dendritic spines in neurons [40]. In non-excitable cells, interaction between EB1 and STIM1 proteins (EB3 and STIM2 homologs, respectively) regulates store-operated calcium entry [43] and controls endoplasmic reticulum (ER) movement through the MT tip-attachment complex mechanism, when ER tubule elongates together with a growing MT end [44]. It was suggested that ER tubules may be one of the proposed cargo types delivered by the MT entry into dendritic spines and that tubulin cytoskeleton may be involved in the regulation of calcium homeostasis in the spines [40]. Drebrin, another EB3 binding partner, mediates interactions between F-actin and MTs. It was shown that in neurons, drebrin promotes MT entry into dendritic spines [45]. The authors proposed a model, according to which synaptic calcium influx triggers F-actin polymerization at the base of the spine, where F-actin-bound drebrin interacts then with EB3 to guide polymerizing MT plus-ends into the spines [33]. Alzheimer’s disease is characterized by the loss of drebrin from dendritic spines that occurs prior to the loss of synapses and neurodegeneration [46]. Reduced levels of drebrin may contribute to impaired MT entry into the spines and disturbed synaptic stability in AD [47].
Another insight into relationships between MT dynamics and dendritic spines has been offered by the studies using MT-targeted drugs. It was reported that subnanomolar concentrations of epothilone D (EpoD), which slightly stabilizes MTs, increased the fraction of thin spines and decreased the fraction of mushroom spines without affecting overall spine density [22]. In the same study, it was shown that application of the MT-depolymerizing drug nocodazole (200 nM) from 16 to 20 DIV (days of in vitro culturing) caused spine loss in control cultures without changes in spine morphology [22]. In another study, it was shown that 4-h incubation of CA1 hippocampal slices with 200 nM nocodazole did not affect the total number of dendritic protrusions, but markedly reduced the number of mushroom spines and increased the number of filopodia [32]. Similar effects were observed in dissociated hippocampal cultures after their overnight incubation with nocodazole [32]. It was found that nocodazole (200 nM) inhibited MT dynamics without altering MT density, abolished EB3 accumulation at MT ends, and induced changes in spine morphology similar to those observed in EB3 knockdown [32]. In the same study, incubation with 2 µM MT-stabilizing drug taxol for 4 h induced reduction in protrusion density but did not affect the spines shape. Mice treated with nocodazole and subjected to contextual fear conditioning showed a significantly lower number of both cortical and hippocampal dendritic spines in comparison with vehicle-threated mice; this effect was prevented by MT stabilization with taxol [48]. Another study failed to detect changes in the spine morphology or density in hippocampal cultures after 24-h incubation with either 10 nM taxol or 500 nM nocodazole [30]. However, the same study revealed that taxol facilitated while nocodazole blocked an increase in the spine density after application of brain-derived neurotrophic factor (BDNF) [30]. For yet unknown reasons, EB3 knockdown in this study did not cause alterations in the spine shape [30], which contradicts the results of other publications. Another research group detected reduction in the dendritic spine density and number of mushroom spines in primary neuronal cultures following 3-h incubation with 10 nM taxol [25]. This effect persisted up to 21 h after taxol removal, which allowed the authors to suggest that even acute short-term stabilization of dynamic MTs has long-term effects on synaptic density [25].
Based on these results, we can conclude that spine shape and density depend on a precise balance in the MT dynamics, whereas excessive stabilization or destabilization result in detrimental effects. In the above-mentioned studies, MT-stabilizing agents were used in low concentrations to limit their action only to dynamic MT tips [30, 49]; however, it is still possible that these agents affect basic MT functions, such as axonal transport. Spatially restricted application of MT drugs (e.g., photo-uncaging) in a selected number of spines may help to identify the origin of the observed effects more precisely.
DYNAMICS OF NEURONAL MICROTUBULES IN ALZHEIMER’S
DISEASE
Oligomeric aggregates of Aβ peptides are considered to be major toxic species inducing AD pathogenesis [50]. MT-associated tau protein forms neurofibrillary tangles in AD brain that appear to be linked to neuronal cell death. Does Aβ affect neuronal MT cytoskeleton? Are these effects tau-dependent? The results that were obtained in attempts to answer these questions are contradictory. It was shown that Aβ compromises α-tubulin acetylation, which is an indirect measure of MT stability [51, 52]. This finding was corroborated by the observation that the early effect of the exposure of mature primary hippocampal cultures to Aβ is reduced α-tubulin polymerization, while the delayed effect is reduction in the levels of total and polymerized βIII tubulin that correlates with reduction in neurite length and DNA fragmentation [52]. The decrease in both the number and total length of MTs with no correlation to tau deposition was shown in pyramidal neurons from AD patients [53]. On the other hand, it was reported that oligomeric Aβ42 induces formation of stable detyrosinated microtubules through activation of RhoA-dependent MT stabilization regulated by integrin and formin mDia1 [54]. The same authors showed that stabilization of dynamic MTs by Aβ occurred through decreasing the frequency of MT catastrophe events and promoted tau hyperphosphorylation and tau-dependent loss of dendritic spines [25]. The reduction in the frequency of MT catastrophe events was transient with a peak at approximately 1.5 h after addition of Aβ and then returned to basal level after 8 h. The authors suggested that tau hyperphosphorylation occurs in order to restore normal MT function after it is alteration by Aβ through activation of pathways disrupting MT behavior and/or tubulin posttranslational modification [25]. These effects were inhibited in tau knock-out mice, in which only an insignificant spine loss was observed in response to Aβ [25]. On the contrary, the loss of dendritic spines after pathological NMDA receptor activation by Aβ in organotypic cultures was detected independently on the presence or absence of tau, but in these experiments, longer incubation times with Aβ were used [55].
The results described above suggest that effects of Aβ on MTs are time-dependent. Aβ-driven MT stabilization is the event occurring at the early disease stage that triggers further toxic cell responses. We propose that the early-stage simplification of the spine shape after Aβ treatment occurs due to the inhibition of dynamic MT entry (figure, panel (b)), while subsequent spine loss is induced due to Aβ-dependent massive disruption of other vital processes and structures in neurons, including MT cytoskeleton (figure, panel (c)). This model can explain how microtubule-stabilizing drug EpoD restores spine density in APPSDL transgenic neurons [22]. These results appear to be counterintuitive, as EpoD was shown to induce reduction of mushroom spines in the hippocampal neurons from wild-type slice cultures [22]. It is possible that EpoD prevents MT disassembly at later stages in APPSDL transgenic neurons and therefore supports neuronal morphology; however, it also inhibits MT dynamics required for spine maturation [22]. Interestingly, nocodazole at low doses partially restores the number of mushroom spines and spine density in APPSDL transgenic neurons, possibly by promoting dynamic MT entry into the spines [22]. These observations clearly support an important role of MT entry into the spines in spine maturation, while Aβ disturbs this process.
MICROTUBULES AS POTENTIAL TARGET FOR AD DRUG DEVELOPMENT
For the reasons discussed above it is clear that normalization of altered MT dynamics in AD neurons helps to rescue dendritic spines and prevents the loss of synaptic connectivity. Indeed, several brain-penetrating MT-stabilizing compounds, including EpoD [56], CNDR-51657 [56], and dictyostatin [57] were reported to show neuroprotective effects in mouse models of AD and tauopathies. Phase 1 clinical trial for EpoD was conducted by Bristol-Myers Squibb, but because of insufficient effect on cerebrospinal fluid biomarkers (tau N-terminal fragments), the trial was stopped after 9 weeks. The results obtained with AD mouse models suggest that 9-week treatment is perhaps too short to observe significant positive effects [29]. The use of MT-stabilizing drugs in cancer therapy showed that they have a number of adverse side effects [58] that may restrict their application in AD treatment. As discussed above, both stabilization and destabilization of MTs have detrimental effects on dendritic spines. Therefore, successful MT-targeting anti-AD drug should be neuron-specific (to reduce side effects) and have a mild effect on MT dynamics without major disturbance of tubulin cytoskeleton. Targeting MT entry into the spines is a promising approach to satisfy these requirements.
Funding
This work was supported by the Russian Science Foundation Grant (I.B.) (project 14-25-00024-П; Pathological features of Alzheimer’s disease), federal grant 17.991.2017/4.6 (I.B.) (Microtubule dynamics as a target for anti-AD drug discovery), Russian Foundation for Basic Research (E.P.) (project 18-34-00183; Tubulin cytoskeleton dynamicity and dendritic spines, Microtubule dynamics in Alzheimer’s disease). E. I. Pchitskaya is a holder of the Russian Presidential Stipend SP–1929.2018.4.
I. B. Bezprozvanny is the Carl J. and Hortense M. Thomsen Professor in Alzheimer’s Disease Research.
REFERENCES
1.Hardy, J., and Selkoe, D. J. (2002) The amyloid
hypothesis of Alzheimer’s disease: progress and problems on the
road to therapeutics, Science, 297, 353-356.
2.Hardy, J. (2009) The amyloid hypothesis for
Alzheimer’s disease: a critical reappraisal, J.
Neurochem., 110, 1129-1134.
3.Bergmans, B. A., and De Strooper, B. (2010)
Gamma-secretases: from cell biology to therapeutic strategies,
Lancet Neurol., 9, 215-226.
4.Duggan, S. P., and McCarthy, J. V. (2016) Beyond
gamma-secretase activity: the multifunctional nature of presenilins in
cell signalling pathways, Cell. Signal., 28, 1-11.
5.DeKosky, S. T., and Scheff, S. W. (1990) Synapse
loss in frontal cortex biopsies in Alzheimer’s disease:
correlation with cognitive severity, Ann. Neurol., 27,
457-464.
6.Terry, R. D., Masliah, E., Salmon, D. P., Butters,
N., DeTeresa, R., Hill, R., Hansen, L. A., and Katzman, R. (1991)
Physical basis of cognitive alterations in Alzheimer’s disease:
synapse loss is the major correlate of cognitive impairment, Ann.
Neurol., 30, 572-580.
7.Koffie, R. M., Hyman, B. T., and Spires-Jones, T.
L. (2011) Alzheimer’s disease: synapses gone cold, Mol.
Neurodegener., 6, 63.
8.Selkoe, D. J. (2002) Alzheimer’s disease is a
synaptic failure, Science, 298, 789-791.
9.Chen, Y., and Sabatini, B. L. (2012) Signaling in
dendritic spines and spine microdomains, Curr. Opin. Neurobiol.,
22, 389-396.
10.Bourne, J. N., and Harris, K. M. (2008) Balancing
structure and function at hippocampal dendritic spines, Annu. Rev.
Neurosci., 31, 47-67.
11.Kasai, H., Matsuzaki, M., Noguchi, J., Yasumatsu,
N., and Nakahara, H. (2003) Structure-stability-function relationships
of dendritic spines, Trends Neurosci., 26, 360-368.
12.Bourne, J., and Harris, K. M. (2007) Do thin
spines learn to be mushroom spines that remember? Curr. Opin.
Neurobiol., 17, 381-386.
13.Hayashi, Y., and Majewska, A. K. (2005) Dendritic
spine geometry: functional implication and regulation, Neuron,
46, 529-532.
14.Hering, H., and Sheng, M. (2001) Dentritic
spines: structure, dynamics and regulation, Nat. Rev. Neurosci.,
2, 880-888.
15.Tackenberg, C., Ghori, A., and Brandt, R. (2009)
Thin, stubby or mushroom: spine pathology in Alzheimer’s disease,
Curr. Alzheimer Res., 6, 261-268.
16.Popugaeva, E., Supnet, C., and Bezprozvanny, I.
(2012) Presenilins, deranged calcium homeostasis, synaptic loss and
dysfunction in Alzheimer’s disease, Messenger, 1,
53-62.
17.Popugaeva, E., and Bezprozvanny, I. (2013) Role
of endoplasmic reticulum Ca2+ signaling in the pathogenesis
of Alzheimer disease, Front. Mol. Neurosci., 6, 29.
18.Androuin, A., Potier, B., Nagerl, U. V.,
Cattaert, D., Danglot, L., Thierry, M., Youssef, I., Triller, A.,
Duyckaerts, C., El Hachimi, K. H., Dutar, P., Delatour, B., and Marty,
S. (2018) Evidence for altered dendritic spine compartmentalization in
Alzheimer’s disease and functional effects in a mouse model,
Acta Neuropathol., 135, 839-854.
19.Sun, S., Zhang, H., Liu, J., Popugaeva, E., Xu,
N. J., Feske, S., White, C. L., 3rd, and Bezprozvanny, I. (2014)
Reduced synaptic STIM2 expression and impaired store-operated calcium
entry cause destabilization of mature spines in mutant presenilin mice,
Neuron, 82, 79-93.
20.Zhang, H., Wu, L., Pchitskaya, E., Zakharova, O.,
Saito, T., Saido, T., and Bezprozvanny, I. (2015) Neuronal
store-operated calcium entry and mushroom spine loss in amyloid
precursor protein knock-in mouse model of Alzheimer’s disease,
J. Neurosci., 35, 13275-13286.
21.Saito, T., Matsuba, Y., Mihira, N., Takano, J.,
Nilsson, P., Itohara, S., Iwata, N., and Saido, T. C. (2014) Single App
knock-in mouse models of Alzheimer’s disease, Nat.
Neurosci., 17, 661-663.
22.Penazzi, L., Tackenberg, C., Ghori, A.,
Golovyashkina, N., Niewidok, B., Selle, K., Ballatore, C., Smith Iii,
A. B., Bakota, L., and Brandt, R. (2016) Ab-mediated spine changes in
the hippocampus are microtubule-dependent and can be reversed by a
subnanomolar concentration of the microtubule-stabilizing agent
epothilone D, Neuropharmacology, 105, 84-95.
23.Tackenberg, C., and Brandt, R. (2009) Divergent
pathways mediate spine alterations and cell death induced by
amyloid-beta, wild-type tau, and R406W tau, J. Neurosci.,
29, 14439-14450.
24.Popugaeva, E., Pchitskaya, E., Speshilova, A.,
Alexandrov, S., Zhang, H., Vlasova, O., and Bezprozvanny, I. (2015)
STIM2 protects hippocampal mushroom spines from amyloid
synaptotoxicity, Mol. Neurodegener., 10, 37.
25.Qu, X., Yuan, F. N., Corona, C., Pasini, S.,
Pero, M. E., Gundersen, G. G., Shelanski, M. L., and Bartolini, F.
(2017) Stabilization of dynamic microtubules by mDia1 drives
Tau-dependent Abeta1-42 synaptotoxicity, J. Cell. Biol.,
216, 3161-3178.
26.Boros, B. D., Greathouse, K. M., Gentry, E. G.,
Curtis, K. A., Birchall, E. L., Gearing, M., and Herskowitz, J. H.
(2017) Dendritic spines provide cognitive resilience against
Alzheimer’s disease, Ann. Neurol., 82, 602-614.
27.Mitchison, T., and Kirschner, M. (1984) Dynamic
instability of microtubule growth, Nature, 312,
237-242.
28.Baas, P. W., Rao, A. N., Matamoros, A. J., and
Leo, L. (2016) Stability properties of neuronal microtubules,
Cytoskeleton (Hoboken, N.J.), 73, 442-460.
29.Dent, E. W. (2017) Of microtubules and memory:
implications for microtubule dynamics in dendrites and spines, Mol.
Biol. Cell, 28, 1-8.
30.Gu, J., Firestein, B. L., and Zheng, J. Q. (2008)
Microtubules in dendritic spine development, J. Neurosci.,
28, 12120-12124.
31.Hu, X., Viesselmann, C., Nam, S., Merriam, E.,
and Dent, E. W. (2008) Activity-dependent dynamic microtubule invasion
of dendritic spines, J. Neurosci., 28, 13094-13105.
32.Jaworski, L., Kapitein, L. C., Gouveia, S. M.,
Dortland, B. R., Wulf, P. S., Grigoriev, I., Camera, P., Spangler, S.
A., DiStefano, P., Demmers, L., Krugers, H., Defilippi, P., Akhmanova,
A., and Hoogenraad, C. C. (2009) Dynamic microtubules regulate
dendritic spine morphology and synaptic plasticity, Neuron,
61, 85-100.
33.Merriam, E. B., Millette, M., Lumbard, D. C.,
Saengsawang, W., Fothergill, T., Hu, X., Ferhat, L., and Dent, E. W.
(2013) Synaptic regulation of microtubule dynamics in dendritic spines
by calcium, F-actin, and drebrin, J. Neurosci., 33,
16471-16482.
34.Hu, X., Ballo, L., Pietila, L., Viesselmann, C.,
Ballweg, J., Lumbard, D., Stevenson, M., Merriam, E., and Dent, E. W.
(2011) BDNF-induced increase of PSD-95 in dendritic spines requires
dynamic microtubule invasions, J. Neurosci., 31,
15597-15603.
35.Uchida, S., Martel, G., Pavlowsky, A., Takizawa,
S., Hevi, C., Watanabe, Y., Kandel, E. R., Alarcon, J. M., and
Shumyatsky, G. P. (2014) Learning-induced and stathmin-dependent
changes in microtubule stability are critical for memory and disrupted
in ageing, Nat. Commun., 5, 4389-4389.
36.Akhmanova, A., and Steinmetz, M. O. (2010)
Microtubule +TIPs at a glance, J. Cell Sci., 123,
3415-3419.
37.Nakagawa, H., Koyama, K., Murata, Y., Morito, M.,
Akiyama, T., and Nakamura, Y. (2000) EB3, a novel member of the EB1
family preferentially expressed in the central nervous system, binds to
a CNS-specific APC homologue, Oncogene, 19, 210-216.
38.Merriam, E. B., Lumbard, D. C., Viesselmann, C.,
Ballweg, J., Stevenson, M., Pietila, L., Hu, X., and Dent, E. W. (2011)
Dynamic microtubules promote synaptic NMDA receptor-dependent spine
enlargement, PLoS One, 6, e27688.
39.Stepanova, T., Slemmer, J., Hoogenraad, C. C.,
Lansbergen, G., Dortland, B., De Zeeuw, C. I., Grosveld, F., van
Cappellen, G., Akhmanova, A., and Galjart, N. (2003) Visualization of
microtubule growth in cultured neurons via the use of EB3-GFP
(end-binding protein 3-green fluorescent protein), J. Neurosci.,
23, 2655-2664.
40.Pchitskaya, E., Kraskovskaya, N., Chernyuk, D.,
Popugaeva, E., Zhang, H., Vlasova, O., and Bezprozvanny, I. (2017)
Stim2-Eb3 association and morphology of dendritic spines in hippocampal
neurons, Sci. Rep., 7, 17625.
41.Zhang, H., Sun, S., Wu, L., Pchitskaya, E.,
Zakharova, O., Fon Tacer, K., and Bezprozvanny, I. (2016)
Store-operated calcium channel complex in postsynaptic spines: a new
therapeutic target for Alzheimer’s disease treatment, J.
Neurosci., 36, 11837-11850.
42.Pchitskaya, E., Popugaeva, E., and Bezprozvanny,
I. (2018) Calcium signaling and molecular mechanisms underlying
neurodegenerative diseases, Cell Calcium, 70, 87-94.
43.Chang, C. L., Chen, Y. J., Quintanilla, C. G.,
Hsieh, T. S., and Liou, J. (2018) EB1 binding restricts STIM1
translocation to ER-PM junctions and regulates store-operated
Ca2+ entry, J. Cell Biol., 6, 2047-2058.
44.Grigoriev, I., Gouveia, S. M., van der Vaart, B.,
Demmers, J., Smyth, J. T., Honnappa, S., Splinter, D., Steinmetz, M.
O., Putney, J. W., Hoogenraad, C. C., and Akhmanova, A. (2008) STIM1 is
a microtubule plus end tracking protein involved in remodeling of the
endoplasmic reticulum, Curr. Biol., 18, 177-182.
45.Geraldo, S., Khanzada, U. K., Parsons, M.,
Chilton, J. K., and Gordon-Weeks, P. R. (2008) Targeting of the
F-actin-binding protein drebrin by the microtubule plus-tip protein EB3
is required for neuritogenesis, Nat. Cell Biol., 10,
1181-1189.
46.Ishizuka, Y., and Hanamura, K. (2017) in
Drebrin. Advances in Experimental Medicine and Biology
(Shirao T., and Sekino, Y., eds.), Vol. 1006, Springer, Tokyo, pp.
203-223.
47.Gordon-Weeks, P. R. (2016) The role of the
drebrin/EB3/Cdk5 pathway in dendritic spine plasticity, implications
for Alzheimer’s disease, Brain Res. Bull., 126,
293-299.
48.Fanara, P., Husted, K. H., Selle, K., Wong, P.
Y., Banerjee, J., Brandt, R., and Hellerstein, M. K. (2010) Changes in
microtubule turnover accompany synaptic plasticity and memory formation
in response to contextual fear conditioning in mice,
Neuroscience, 168, 167-178.
49.Buck, K. B., and Zheng, J. Q. (2002) Growth cone
turning induced by direct local modification of microtubule dynamics,
J. Neurosci., 22, 9358-9367.
50.Selkoe, D. J., and Hardy, J. (2016) The amyloid
hypothesis of Alzheimer’s disease at 25 years, EMBO Mol.
Med., 8, 595-608.
51.Zempel, H., Luedtke, J., Kumar, Y., Biernat, J.,
Dawson, H., Mandelkow, E., and Mandelkow, E.-M. (2013) Amyloid-b
oligomers induce synaptic damage via tau-dependent microtubule severing
by TTLL6 and spastin, EMBO J., 32, 2920-2937.
52.Mota, S. I., Ferreira, I. L., Pereira, C.,
Oliveira, C. R., and Rego, A. C. (2012) Amyloid-beta peptide 1-42
causes microtubule deregulation through N-methyl-D-aspartate receptors
in mature hippocampal cultures, Curr. Alzheimer Res., 9,
844-856.
53.Cash, A. D., Aliev, G., Siedlak, S. L., Nunomura,
A., Fujioka, H., Zhu, X., Raina, A. K., Vinters, H. V., Tabaton, M.,
Johnson, A. B., Paula-Barbosa, M., Avila, J., Jones, P. K., Castellani,
R. J., Smith, M. A., and Perry, G. (2003) Microtubule reduction in
Alzheimer’s disease and aging is independent of tau filament
formation, Am. J. Pathol., 162, 1623-1627.
54.Pianu, B., Lefort, R., Thuiliere, L., Tabourier,
E., and Bartolini, F. (2014) The Abeta(1)(-)(4)(2) peptide regulates
microtubule stability independently of tau, J. Cell Sci.,
127, 1117-1127.
55.Tackenberg, C., Grinschgl, S., Trutzel, A.,
Santuccione, A. C., Frey, M. C., Konietzko, U., Grimm, J., Brandt, R.,
and Nitsch, R. M. (2013) NMDA receptor subunit composition determines
beta-amyloid-induced neurodegeneration and synaptic loss, Cell Death
Dis., 25, 129.
56.Kovalevich, J., Cornec, A.-S., Yao, Y., James,
M., Crowe, A., Lee, V. M. Y., Trojanowski, J. Q., Smith, A. B.,
Ballatore, C., and Brunden, K. R. (2016) Characterization of
brain-penetrant pyrimidine-containing molecules with differential
microtubule-stabilizing activities developed as potential therapeutic
agents for Alzheimer’s disease and related tauopathies, J.
Pharmacol. Exp. Ther., 357, 432-450.
57.Makani, V., Zhang, B., Han, H., Yao, Y.,
Lassalas, P., Lou, K., Paterson, I., Lee, V. M., Trojanowski, J. Q.,
Ballatore, C., Smith, A. B., 3rd, and Brunden, K. R. (2016) Evaluation
of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in
the PS19 tau transgenic mouse model of tauopathy, Acta Neuropathol.
Commun., 4, 106.
58.Lou, K., Yao, Y., Hoye, A. T., James, M. J.,
Cornec, A. S., Hyde, E., Gay, B., Lee, V. M., Trojanowski, J. Q.,
Smith, A. B., 3rd, Brunden, K. R., and Ballatore, C. (2014)
Brain-penetrant, orally bioavailable microtubule-stabilizing small
molecules are potential candidate therapeutics for Alzheimer’s
disease and related tauopathies, J. Med. Chem., 57,
6116-6127.