Mitochondria with Morphology Characteristic for Alzheimer’s Disease Patients Are Found in the Brain of OXYS Rats
M. A. Tyumentsev1, N. A. Stefanova1, E. V. Kiseleva1, and N. G. Kolosova1,2,a*
1Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia2Novosibirsk State University, 630090 Novosibirsk, Russia
* To whom correspondence should be addressed.
Received May 7, 2018; Revision received June 7, 2018
Growing evidence suggests that mitochondrial dysfunction is closely linked to the pathogenesis of sporadic Alzheimer’s disease (AD). One of the key contributors to various aspects of AD pathogenesis, along with metabolic dysfunction, is mitochondrial dynamics, involving balance between fusion and fission, which regulates mitochondrial number and morphology in response to changes in cellular energy demand. Recently, Zhang et al. ((2016) Sci. Rep., 6, 18725) described a previously unknown mitochondrial phenotype manifesting as elongated chain-linked mitochondria termed “mitochondria-on-a-string” (MOAS) in brain tissue from AD patients and mouse models of AD. The authors associated this phenotype with fission arrest, but implications of MOAS formation in AD pathogenesis remain to be understood. Here we analyze the presence and number of MOAS in the brain of OXYS rats simulating key signs of sporadic AD. Using electron microscopy, we found MOAS in OXYS prefrontal cortex neuropil in all stages of AD-like pathology, including manifestation (5-month-old rats) and progression (12-18-month-old rats). The most pronounced elevation of MOAS content (8-fold) in OXYS rats compared to Wistar controls was found at the preclinical stage (20 days) on the background of decreased numbers of non-MOAS elongated mitochondria. From the age of 20 days through 18 months, the percentage of MOAS-containing neuronal processes increased from 1.7 to 8.3% in Wistar and from 13.9 to 16% in OXYS rats. Our results support the importance of the disruption of mitochondrial dynamics in AD pathogenesis and corroborate the existence of a causal link between impaired mitochondrial dynamics and formation of the distinctive phenotype of “mitochondria-on-a-sting”.
KEY WORDS: Alzheimer’s disease, mitochondrial dynamics, OXYS rats, electron microscopyDOI: 10.1134/S0006297918090109
Abbreviations: Aβ, amyloid beta; AD, Alzheimer’s disease; MOAS, mitochondria-on-a-string; ROS, reactive oxygen species.
According to the dominating “amyloid cascade” hypothesis the
central event in the pathogenesis of the Alzheimer’s disease (AD)
– the most common form of senile dementia – is accumulation
of neurotoxic forms of the amyloid-beta peptide (Aβ), which leads
to formation of amyloid plaques, hyperphosphorylation of tau-protein
and formation of neurofibrillary tangles, synaptic loss, neuron death,
inflammation, mitochondrial dysfunction, and oxidative stress [1]. However, this hypothesis is fully justified only
for the early-onset hereditary form of AD (~5% of cases), and in the
case of a late-onset sporadic form of the disease (further – AD)
accounting for ~95% of cases hyperproduction of Aβ is a secondary
event. More evidence is available now favoring the mitochondrial
dysfunction as an initiating factor for AD development. According to
the “mitochondrial cascade” hypothesis [2], oxidative stress and decrease in ATP synthesis
result in overproduction of Aβ that exhibits toxic effect on
mitochondria, thus aggravating neurodegenerative processes. At the same
time, it became clear in recent years that the enhanced generation of
reactive oxygen species (ROS) is neither the initiator no the main
cause of aging [3]. It has been also demonstrated
that mitochondrial dysfunction could facilitate aging and development
of age-related diseases independently on formation of ROS, and that
their development is associated not only with the metabolic dysfunction
but also with the disruption of mitochondrial dynamics [4] – balance between fission and fusion, which
regulates mitochondrial morphology in response to changes in cellular
energy demand [5]. It was demonstrated on the
animal AD models and during investigation of the patients’ brain
samples that the imbalance of mitochondrial dynamics is one of the
early signs of the disease [6, 7]. Zhang et al. [8], who
discovered in the neuropil of the brain samples from AD patients and
from a number of transgenic mice lines – AD models (3xTgAD,
APP/PS1, Tau, and others) the elongated (>10 μm) mitochondria
with previously unknown morphology characterized by an alternation of
areas of normal thickness with narrowed segments (60-100 nm), link
this phenomenon with the change in mitochondrial dynamics. The authors
termed these mitochondria “mitochondria-on-a-string”
(MOAS). Their formation as a result of fission arrest, which was
observed in the brain of wild type mice in response to hypoxia and in
the brain of aging animals, has been considered by the authors as a
compensatory reaction aimed at the neuron survival under conditions of
energy deficit ensuring protection from mitophagy. Mitochondria with
such morphology were detected in another study in the brain cortex of
old rhesus monkeys [9], but the authors associate
their emergence with age-related disorder of mitochondrial dynamics
leading to the disruption of axonal transport [10]. Hence, the question on the role of this
phenomenon remains open and requires further investigation using
various models and organisms.
We have shown recently that the structural-functional changes of mitochondria in hippocampal neurons including the changes in their dynamics precede the development of AD symptoms in OXYS rats – a model of the sporadic form of this disease created in the Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences. Manifestation and progression of all key signs of AD (destructive changes and death of neurons, synaptic loss, hyperphosphorylation of tau-protein, enhanced accumulation of Aβ1-42 and formation of amyloid plaques in the brain, memory loss, and diminished cognitive ability [11-13]) occur in OXYS rats on the background of increasing mitochondrial dysfunction and significant reduction in the specific content of mitochondria in hippocampal neurons. The changes of mitochondrial dynamics in the hippocampal neurons from OXYS rats occur simultaneously with the changes in the content and/or ratio of the levels of proteins directly participating in fusion and fission of mitochondria – MFN1, MFN2, and DRP1 [14]. The increasing signs of structural changes of microtubules and their disorganization as well as accumulation of the large mitochondria in axons have been observed in OXYS rats with aging, which indicates the disruption of axonal transport [12, 13, 15, 16]; these phenomena play a critical role in AD pathogenesis [17].
The objective of this study was to analyze the occurrence and to evaluate the content of MOAS (observed in AD patients) in the axons of the prefrontal cortex from OXYS rats at different stages of the disease development: at the age of 20 days – the period prior to the development of AD signs, at the age of 5 months – the period of manifestation of AD signs, and at the age of 12 and 18 months – the period of their active progression. Wistar rats of the same age served as a control.
MATERIALS AND METHODS
Animals. The work was carried out on 12 male OXYS rats and on 12 male Wistar rats (control) aged 20 days, 5, 12, and 18 months based in the Center of Genetic Resources of Laboratory Animals at the Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences (RFMEFI61914X0005 and RFMEFI61914X0010). Animals were kept in a vivarium under standard conditions with fixed illumination mode (12 h light/12 h dark) and free access to water and food (granulated animal feed Chara; ZAO Assortment-Agro, Russia).
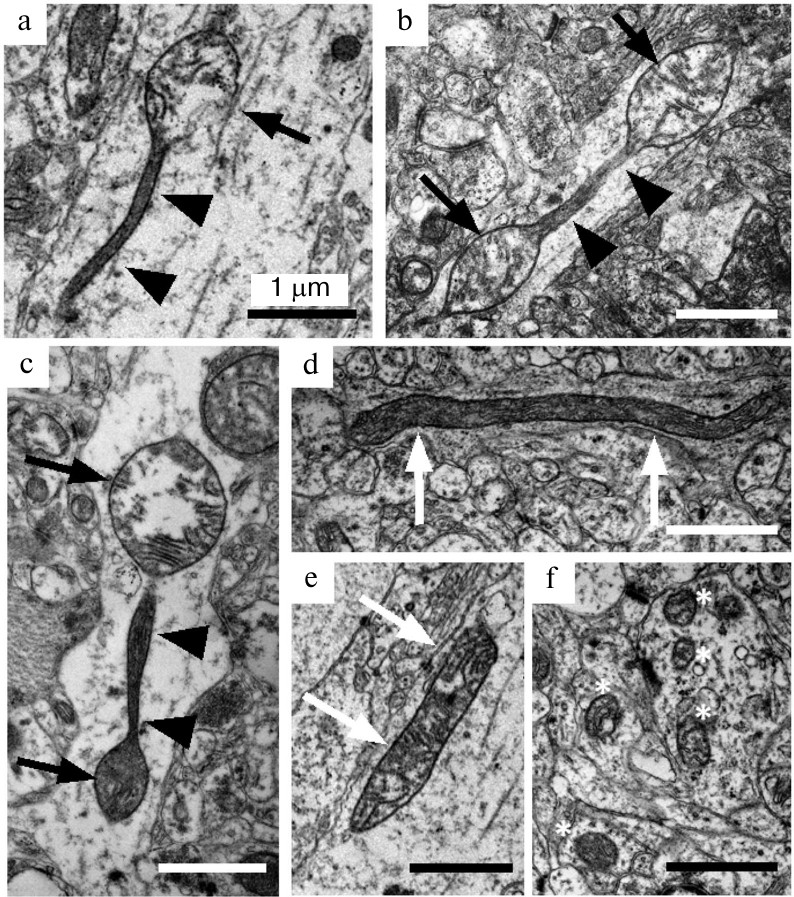
Preparation of samples and electron microscopy examination. Rats were sedated with carbon dioxide and decapitated. The isolated prefrontal cortex was fixed with 2.5% glutaraldehyde, postfixed with osmium tetroxide, and embedded into an Epon 812 resin. A 1 × 1 mm region of the resin block that corresponds to layers III-V of the cortex was used for preparation of ultrathin slices on a LEICA ULTRACUT EM UC6 ultramicrotome (Leica Microsystems GmbH, Germany). Ultrathin slices were examined with a JEM 1400 electron microscope (JEOL Ltd., Japan) in the Center for Collective Use for Microscopic Analysis of Biological Objects (Siberian Branch of the Russian Academy of Sciences) under 12,000-30,000× magnification. Using the ImageJ program, the number of longitudinal cross-sections of axons (n ≥ 24 per group, total n = 414) on the produced images as well as mitochondria contained in them were counted. The imaged mitochondria were classified into three morphological types: mitochondria with spindle-like segments alternating with thin long membrane segments (Fig. 1, a-c), elongated mitochondria with constant cross-section diameter (Fig. 1, d and e), and ovoid mitochondria (Fig. 1f).
Fig. 1. Ultrastructural characteristics of morphological types of mitochondria in axons from Wistar and OXYS rat brain. Mitochondria-on-a-string (MOAS) in axons from OXYS rats (a-c) are characterized by spindle-like segments (black arrows) alternating with narrow segments of approximately 1 μm length (short black arrows). Elongated mitochondria (white arrows) represent another type of large non-ovoid mitochondria found in the axons from Wistar (d) and OXYS (e) rats. Separate ovoid mitochondria (white stars) comprise the majority of mitochondria in brain neuropil (f). Scale – 1 μm.
Statistical processing. In order to reveal statistically significant differences in the investigated parameters, the data were processed using the STATISTICA 10 program. A factorial dispersion analysis (ANOVA) with post-hoc-comparison of group means (Newman–Keuls test) and non-parametric analysis (Mann–Whitney criterion) with post-hoc-analysis according to the Fisher criterion were used. Genotype and age were considered as independent factors. The data are presented as means ± S.E.M. The results were considered statistically significant at p < 0.05.
RESULTS AND DISCUSSION
The relative content of MOAS (the number of MOAS per one section of the axon of cortical neuron) (Fig. 2a) depended only on the genotype of animals, it was higher for OXYS rats (F1,407 = 6.51, p < 0.02) and was age-independent (F3,406 = 1.36, p = 0.26). It is essential that the most significant differences between the rat lines were observed at the age of 20 days, when the content of MOAS in axons from the OXYS rats was 8-fold higher than that in axons from the Wistar rats (0.139 ± 0.058 versus 0.017 ± 0.017, p < 0.019). Thus, unlike the results of Morozov et al. [9] who reported the age-dependent accumulation of MOAS in the dendrites of the prefrontal cortex of rhesus monkeys, we did not find any increase in the MOAS content in axons from the brain of either Wistar or OXYS rats with aging. At the same time, the fraction of axons containing MOAS in Wistar rats increased from 1.7% at the age of 20 days to 8.3% at the age of 18 months, while it did not change significantly in OXYS rats (13.9% at the age of 20 days and 16% at the age of 18 months, which was twice as much as in the Wistar rats of the same age).
Fig. 2. Frequency of occurrence of mitochondria-on-a-string (MOAS) in axons from the brain of Wistar and OXYS rats. Dispersion analysis revealed elevated content of MOAS (F1,407 = 6.51, p < 0.02) in axons from the cortex of OXYS rats (a). Data presented as a mean value ± S.E.M; * – significant difference. The increase of MOAS fraction in the total number of mitochondria in axons from cortex of OXYS rats occurs on the background of the decrease of the fraction of elongated mitochondria (b).
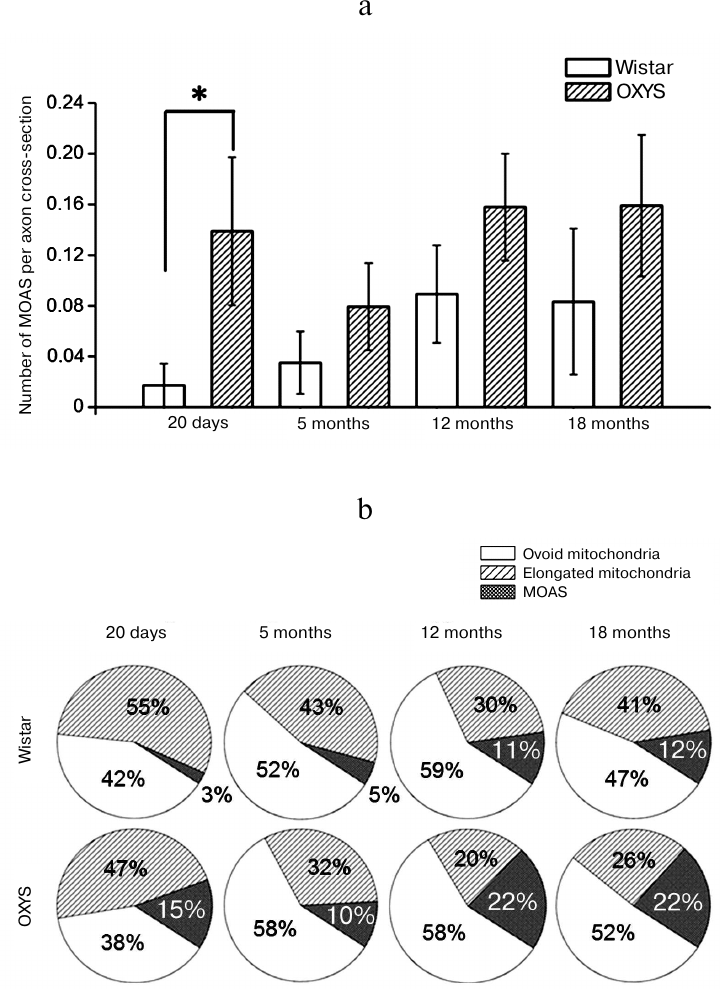
The increase in the number of MOAS in the axons from OXYS rats can be explained by the impaired fission of elongated mitochondria into separate ovoid organelles [9], which hinders their further transport. The number of elongated mitochondria per one axon did not change with the age in Wistar rats (p = 0.47), while it decreased in OXYS rats and was 1.8-fold lower (p < 0.036) at the age of 5 months, and 3- and 2.4-fold lower at the age of 12 and 18 months (p < 0.005 and p < 0.011, respectively) than at the age of 20 days. Thus, the fraction of MOAS (percentage of the total amount of mitochondria in neuropil) increased in OXYS rats on the background of the decrease of the fraction of elongated mitochondria (Fig. 2b). In particular, their fraction decreased more than twice from the age of 20 days to 12 months (from 47 to 20%, p < 0.002), while the fraction of ovoid mitochondria did not change. It must be noted that the total amount of mitochondria of all morphological types – ovoid mitochondria, elongated mitochondria, and MOAS – in the axons from cortex of both Wistar and OXYS rats did not differ and did not change significantly with aging.
The authors who were the first to describe the phenomenon of MOAS formation [8, 9] associated their emergence not with the shift of balance between fission and fusion of mitochondria but with the impaired functioning of the protein complex responsible for formation of membrane constriction and for its cleavage during mitochondria fission, where a major role belongs to the DRP1 protein. At the same time, according to their data, MOAS are formed on the background of constant content of the DRP1 protein, a classic marker of mitochondrial dynamics [8]. As was mentioned above, the disruption of balance between fusion and fission of mitochondria in neurons from hippocampus from OXYS rats occurs on the background of the changing ratio of the protein markers of fusion and fission (MFN1/DRP1) due to increase in DRP1 level with age. But, as we have shown previously [14], at the age of 20 days in the absence of AD signs, both the content of the MFN1 protein and the MFN1/DRP1 ratio and, correspondingly, formation of large mitochondria were increased in neurons of the hippocampus of OXYS rats. It is generally accepted that the increased content of large mitochondria formed as a result of fusion of small mitochondria during formation of the mitochondrial network is directed to the increase in energy supply under conditions of decreased activity of respiratory chain [5], which we observed in 20-day-old OXYS rats [14], and can be considered as a compensatory reaction. As shown in the present work, the enhanced formation of MOAS in OXYS rats is observed already at the age of 20 days, which, according to the hypothesis of Zhang et al. [8], also can be considered as an adaptive response to the bioenergetic stress. At the same time, it can be suggested that the long-term elevation of the number of MOAS can negatively affect the axonal transport – disrupt the traffic of organelles along the elements of cytoskeleton creating “jams” in neuronal axons and preventing further vesicular transport and transport of mitochondria [18]. As a result, the abnormal mitochondrial transport leads to the synaptic starvation and ineffective elimination of the damaged mitochondria, disrupts synaptic functions and causes neuron degeneration in AD [18].
Thus, MOAS recently detected in axons from cortex of AD patients and transgenic animals modeling early stage of the disease as well as of old rhesus monkeys [8, 9] are also present in insignificant amounts in Wistar rats, and the fraction of axons containing MOAS increases with age. The number of MOAS in the axons from the cortex of OXYS rats (model of sporadic form of AD) is significantly elevated, including the early pre-clinical stage of the disease. At present, the importance of this phenomenon is not quite clear, however, the produced results confirm the significance of the disruption of mitochondrial dynamics in the AD pathogenesis, as well as its association with the formation of mitochondria with the characteristic morphology – “mitochondria-on-a-string”.
Funding
This work was financially supported by the Russian Science Foundation, grant no. 16-15-10005.
Acknowledgments
Authors are grateful to the Center of Collective Use for Microscopic Analysis of Biological Objects, Siberian Branch of the Russian Academy of Sciences, for the help in conducting the study.
Conflict of Interests
Authors declare no conflict of interests neither in financial nor in any other area.
Ethical Approval
All procedures conducted in this study with participation of laboratory animals were in accordance with the ethical standards of the Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, and legal requirements adopted by the Russian Federation and international organizations.
REFERENCES
1.Morley, J. E., Armbrecht, H. J., Farr, S. A., and
Kumar, V. B. (2012) The senescence accelerated mouse (SAMP8) as a model
for oxidative stress and Alzheimer’s disease, Biochim.
Biophys. Acta, 1822, 650-656.
2.Swerdlow, R. H., Burns, J. M., and Khan, S. M.
(2014) The Alzheimer’s disease mitochondrial cascade hypothesis:
progress and perspectives, Biochim. Biophys. Acta, 1842,
1219-1231.
3.Payne, B. A. I., and Chinnery, P. F. (2015)
Mitochondrial dysfunction in aging: much progress but many unresolved
questions, Biochim. Biophys. Acta, 1847, 1347-1353.
4.Ziegler, D. V., Wiley, C. D., and Velarde, M. C.
(2015) Mitochondrial effectors of cellular senescence: beyond the free
radical theory of aging, Aging Cell, 14, 1-7.
5.Westermann, B. (2012) Bioenergetic role of
mitochondrial fusion and fission, Biochim. Biophys. Acta,
1817, 1833-1838.
6.Zhu, X., Perry, G., Smith, M. A., and Wang, X.
(2013) Abnormal mitochondrial dynamics in the pathogenesis of
Alzheimer’s disease, J. Alzheimers Dis., 33,
253-262.
7.Kim, D. I., Lee, K. H., Oh, J. Y., Kim, J. S., and
Han, H. J. (2017) Relationship between β-amyloid and mitochondrial
dynamics, Cell. Mol. Neurobiol., 37, 955-968.
8.Zhang, L., Trushin, S., Christensen, T. A.,
Bachmeier, B. V., Gateno, B., Schroeder, A., Yao, J., Itoh, K., Sesaki,
H., Poon, W. W., Gylys, K. H., Patterson, E. R., Parisi, J. E., Diaz
Brinton, R., Salisbury, J. L., and Trushina, E. (2016) Altered brain
energetics induces mitochondrial fission arrest in Alzheimer’s
disease, Sci. Rep., 6, 18725.
9.Morozov, Y. M., Datta, D., Paspalas, C. D., and
Arnsten, A. F. T. (2017) Ultrastructural evidence for impaired
mitochondrial fission in the aged rhesus monkey dorsolateral prefrontal
cortex, Neurobiol. Aging, 51, 9-18.
10.Youle, R. J., and van der Bliek, A. M. (2012)
Mitochondrial fission, fusion, and stress, Science, 337,
1062-1065.
11.Kolosova, N. G., Stefanova, N. A., Korbolina, E.
E., Fursova, A. Zh., and Kozhevnikova, O. S. (2014)
Senescence-accelerated OXYS rats – a genetic model of
premature aging and age-related diseases, Adv. Gerontol.,
4, 294-298.
12.Stefanova, N. A., Kozhevnikova, O. S., Vitovtov,
A. O., Maksimova, K. Y., Logvinov, S. V., Rudnitskaya, E. A.,
Korbolina, E. E., Muraleva, N. A., and Kolosova, N. G. (2014)
Senescence-accelerated OXYS rats: a model of age-related cognitive
decline with relevance to abnormalities in Alzheimer’s disease,
Cell Cycle, 13, 898-909.
13.Stefanova, N. A., Muraleva, N. A., Korbolina, E.
E., Kiseleva, E., Maksimova, K. Y., and Kolosova, N. G. (2015) Amyloid
accumulation is a late event in sporadic Alzheimer’s disease-like
pathology in nontransgenic rats, Oncotarget, 6,
1396-1413.
14.Tyumentsev, M. A., Stefanova, N. A., Muraleva, N.
A., Rumyantseva, Y. V., Kiseleva, E., Vavilin, V. A., and Kolosova, N.
G. (2018). Mitochondrial dysfunction as a predictor and driver of
Alzheimer’s disease-like pathology in OXYS rats, J. Alzheimers
Dis., 63, 1075-1088.
15.Stefanova, N. A., Muraleva, N. A., Skulachev, V.
P., and Kolosova, N. G. (2014) Alzheimer’s disease-like pathology
in senescence-accelerated OXYS rats can be partially retarded with
mitochondria-targeted antioxidant SkQ1, J. Alzheimers Dis.,
38, 681-694.
16.Stefanova, N. A., Maksimova, K. Y., Kiseleva, E.,
Rudnitskaya, E. A., Muraleva, N. A., and Kolosova, N. G. (2015)
Melatonin attenuates impairments of structural hippocampal
neuroplasticity in OXYS rats during active progression of
Alzheimer’s disease-like pathology, J. Pineal Res.,
59, 163-177.
17.Cai, Q., and Tammineni, P. (2017) Mitochondrial
aspects of synaptic dysfunction in Alzheimer’s disease, J.
Alzheimers Dis., 57, 1087-1103.
18.Correia, S. C., Perry, G., and Moreira, P. I.
(2016) Mitochondrial traffic jams in Alzheimer’s
disease – pinpointing the roadblocks, Biochim. Biophys.
Acta, 1862, 1909-1917.