REVIEW: Cytokines as Mediators of Neuroinflammation in Experimental Autoimmune Encephalomyelitis
V. S. Gogoleva1,2,a*, K.-S. N. Atretkhany1,2, M. S. Drutskaya1,2, I. A. Mufazalov3, A. A. Kruglov4, and S. A. Nedospasov1,2,4,b*
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia2Lomonosov Moscow State University, Biological Faculty, 119234 Moscow, Russia
3University of California, San Francisco, CA 94143, USA
4Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119234 Moscow, Russia
* To whom correspondence should be addressed.
Received May 12, 2018; Revision received June 10, 2018
Cytokines play a pivotal role in maintaining homeostasis of the immune system and in regulation of the immune response. Cytokine dysregulation is often associated with development of various pathological conditions, including autoimmunity. Recent studies have provided insights into the cytokine signaling pathways that are involved not only in pathogenesis of autoimmune neuroinflammatory disorders, such as multiple sclerosis, but also in neurodegenerative states, for example, Alzheimer’s disease. Understanding the exact molecular mechanisms of disease pathogenesis and evaluation of relevant experimental animal models are necessary for development of effective therapeutic approaches.
KEY WORDS: experimental autoimmune encephalomyelitis, inflammation, neurodegeneration, proinflammatory cytokines, TNF, IL-6, IL-17ADOI: 10.1134/S0006297918090110
Abbreviations: AD, Alzheimer’s disease; BBB, blood-brain barrier; CCR, chemokine receptor; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; GM-CSF, granulocyte-macrophage colony-stimulating factor; gp130, glycoprotein 130; IFN, interferon; IL, interleukin; IL-1R1, IL-1β receptor subunit; IL-6R, interleukin-6 receptor; LT, lymphotoxin; MOG, myelin oligodendrocyte protein; MS, multiple sclerosis; NK-cells, natural killer cells; TGFβ, transforming growth factor beta; Th, T helper; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; Tregs, regulatory T cells.
In developed countries, aging of the population is a risk factor for
development of central nervous system (CNS) disorders. Despite the
steadily increasing number of patients with these serious diseases, up
to now no effective therapy for such neuropathologies has been
developed. For a long time, it was believed that the CNS does not
interact with the immune system because of its immune privilege being
maintained, in particular, by the blood-brain barrier (BBB). The BBB is
a physical border between the CNS and immune cells circulating in the
blood and soluble factors produced by them. This barrier is maintained
by tight junctions between the brain endothelial cells, their basal
membrane, pericytes and astrocytes. However, recent studies in
neuroimmunology have shown that an interaction of the immune system
components with the CNS is required for maintaining homeostasis and
that homeostatic imbalance caused by inflammatory processes may lead to
various pathologies. Thus, immune dysregulation and development of
pathological processes trigger the effector immune response to
CNS-specific antigens that is mediated by infiltration of the
circulating immune cells into the CNS and by activation of the
CNS-resident cells. In the inflammatory microenvironment the
infiltrating and resident cells of the CNS secrete cytokines which in
steady state regulate the immune response and homeostasis.
Proinflammatory cytokines play a pivotal role in the pathogenesis of
autoimmune diseases, in particular, of those associated with pathology
of the nervous system. For instance, increased levels of many
proinflammatory cytokines, such as interleukins 6, 1, 17 (IL-6, IL-1,
IL-17) and tumor necrosis factor (TNF), were found in the cerebrospinal
fluid and in demyelinating plaques of patients with multiple sclerosis
(MS) suggesting their possible involvement in the pathogenesis of MS
[1, 2]. The roles of various
cytokines in neuroinflammation and neurodegeneration have been
investigated in the mouse experimental models using reverse genetics
approach.
NEURODEGENERATIVE DISEASES AND NEUROINFLAMMATION
Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) are the most frequent and best studied neurodegenerative diseases. It is thought that initiation of neurodegenerative diseases is associated with the accumulation of misfolded proteins in the brain causing neuronal damage in different brain regions [3, 4]. The role of the immune system in the pathogenesis of these diseases is intensively studied, particularly, in mouse models. Thus, systemic inflammation in mice may promote the accumulation of a β-amyloid precursor that further results in development of pathological processes specific for AD: the accumulation of β-amyloid plaques and activation of microglia [5]. Genetic studies have revealed the association between predisposition to AD and mutations in the gene encoding the triggering receptor expressed on myeloid cells 2 (TREM2) which is present on the microglia cell surface [6, 7]. It has been shown that in a mouse model of AD microglia acquires a protective phenotype in a two-step process due to increased expression of genes responsible for phagocytosis of the accumulated β-amyloid plaques [8]. Another study found that the microglia phenotype depends on activation of TREM2–APOE signaling pathway which is associated with the pathogenic role of the microglia in the neurodegeneration model [9]. Recent data have identified that the inflammation, mainly initiated by activated myeloid cells, plays an important role in the pathogenesis of AD. Thus, the immune system is believed to play an important role in the pathogenesis of this neurodegenerative disorder [10].
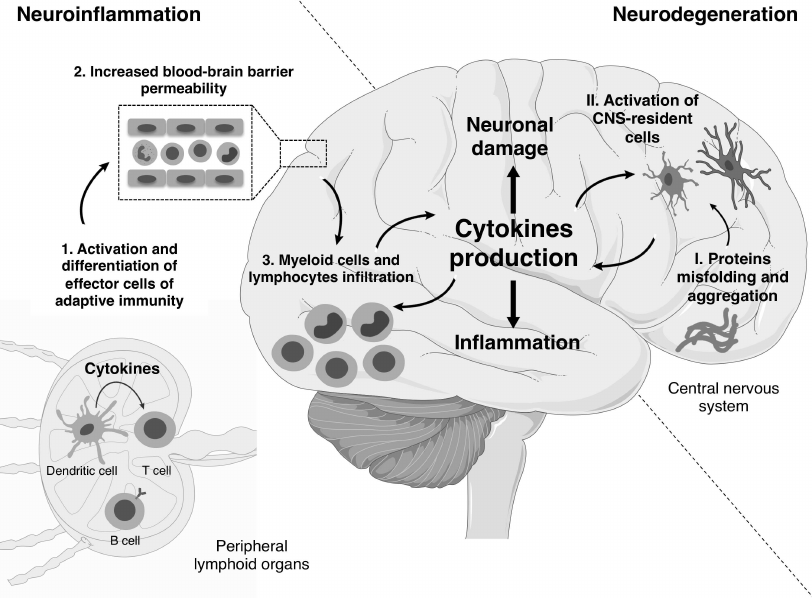
Mechanisms of development and pathogenesis of neurodegenerative and neuroinflammatory diseases are different. First, the main causes of neurodegenerative diseases are protein aggregates of β-amyloid, α-synuclein and a mutant form of superoxide dismutase 1 (SOD1) in the case of AD, PD, and the hereditary form of ALS, respectively [11]. On the other hand, in the classical neuroinflammatory disease, i.e., multiple sclerosis, pathological processes are mainly mediated by cells of the immune system. Second, in proteinopathies, proinflammatory cytokines are mainly produced by myeloid cells, predominantly, CNS-resident cells, whereas in neuroinflammation cytokines are produced by both T cells and myeloid cells [12, 13]. Third, in neuroinflammation the BBB is destroyed during the early stages of disease onset leading to infiltration of the immune cells that later will support the inflammation in the CNS (figure).
Role of cytokines in the development of neuroinflammatory and neurodegenerative diseases. During the neuroinflammation, priming occurs in peripheral lymphoid organs with subsequent differentiation of effector T cells due to cytokines being released by antigen-presenting cells (1). Then the increased BBB permeability promotes the infiltration of lymphoid and myeloid cells into the CNS (2, 3). The infiltrating cells recruit other immune cells from the periphery through the production of cytokines and also activate CNS-resident cells driving inflammation in the CNS. The development of neurodegenerative diseases is characterized by accumulation of protein aggregates (I). This process results in the activation of stromal cells and phagocytes of the CNS, i.e., astrocytes and microglia, respectively (II). Subsequently, astrocytes and microglia secrete cytokines, and this chronic CNS-intrinsic cytokine production is associated with loss of tissue function. The effector phase of neuroinflammatory and neurodegenerative diseases is characterized by neuronal damage and local inflammation in the CNS. (Materials of the Servier Medical Art were used for the figure)
MECHANISMS OF MULTIPLE SCLEROSIS DEVELOPMENT
Multiple sclerosis (MS) is a complex autoimmune disease characterized by damage of the myelin sheath in numerous inflammatory lesions. To date, MS affects approximately 2.5 million people worldwide, and the average age of MS diagnosis is 30. In the majority of patients, clinical manifestations include disorders of motor, cognitive, and visual functions, as well as the loss of sensitivity [14]. The development of MS is contributed by both genetic and environmental factors (e.g., the Epstein–Barr virus infection) that results further in development of the autoimmune response directed against the myelin sheath of nerve fibers [15].
Fundamental mechanisms of the disease pathophysiology were studied in a mouse model of multiple sclerosis – experimental autoimmune encephalomyelitis (EAE) [16, 17]. It should be noted that EAE induction can be both “active” or “passive”. “Active” EAE is induced by immunization with a CNS-specific antigen in complete Freund’s adjuvant with the subsequent injection of pertussis toxin. “Passive” EAE requires adoptive transfer of neuroantigen-specific T cells into recipient mice leading to development of the immune pathology [18]. Depending on the genetic strain of mice, myelin basic protein (MBP), myelin oligodendrocyte protein (MOG) or proteolipid protein (PLP) are used as antigens for the disease induction. Moreover, a model of spontaneous EAE development was established using transgenic mice with overexpression of the antigen-specific T- or B-cell receptor [19]. However, it should be noted that EAE is not an ideal model of multiple sclerosis. First, mice on C57BL/6 background, a commonly used mouse strain in immunological studies, show a monophasic disease course, whereas the most frequent MS type is relapsing-remitting MS characterized by periods of exacerbation and recovery. Second, the composition of key cell populations involved in the adaptive arm of immune response during EAE can vary, depending on the chosen antigen or on the mouse strain. Thus, when the disease is induced with the MOG35-55-peptide, the T cell response in C57BL/6 mice is characterized mainly by CD4+ T cells, whereas in humans CD8+ T cells and B cells are also involved [20, 21]. In clinical studies, the efficiency of B-cell depletion has been already approved, whereas the role of B cells in EAE requires further investigation. It has been demonstrated that B cells can modulate the severity of the disease by their antigen-presenting function, as well as via secretion of anti-inflammatory cytokines IL-10 and IL-35 [22, 23]. Moreover, EAE in C57BL/6 mice is characterized by more severe tissue damage of spinal cord than the brain. Nevertheless, despite the obvious limitations, EAE remains the most frequent experimental model of MS, also used in preclinical trials.
It was shown in mouse models that the escape of autoreactive T cells from thymic deletion to the periphery and the loss of immune tolerance can lead to the development of T-cell mediated immune response directed against CNS-specific autoantigens [24]. For many years, the CNS was thought to be an immunologically privileged organ, but more recently lymphatic vessels responsible for drainage of the CNS and cerebrospinal fluid were found in the brain meninges [25]. This led to the revision of the existing hypotheses about the mechanisms of neuroinflammation. It is now understood that neuroinflammation can be initiated by antigens from the CNS entering the draining lymph nodes in a soluble form or as a result of the transfer by antigen-presenting cells. Moreover, viral antigens mimicking CNS-specific autoantigens also can induce an immune response in the periphery [26].
The next stage of the disease development is associated with T-cell priming and is accompanied by differentiation of CD4+ T cells into various effector populations in the draining lymph nodes. Thus, in the EAE model main pathogenic CD4+ T cell subpopulations are Th1 and Th17, and their adoptive transfer can induce the disease in naïve mice [27]. In this case, FoxP3+ regulatory T cells play a protective role, and their deletion leads to exacerbation of the disease [28]. The pathogenic and protective properties of different T cell subsets are considerably determined by production of certain cytokines. In particular, skewing of T cell differentiation towards protective Th2 subset is the mechanism of action of existing drugs approved for MS treatment [29, 30]. For example, one of the immunomodulatory effects of interferon-β (IFNβ) is the inhibition of IL-12 production by T cells, a key factor of Th1 differentiation [31]. Besides CD4+ T cells, which are the main regulators in EAE, activation and maturation of CD8+ T cells and B cells occur in the periphery [32, 33].
After induction of the adaptive immune response, these cells migrate into the CNS. For this, activation of the vascular endothelium is required through the induction of the signaling cascade α4β1-integrin/VCAM-1, as well as the increased BBB permeability for the subsequent cell infiltration [34]. The interaction of α4β1-integrin on the surface of immune cells with the protein on endothelial cells – vascular cell adhesion molecule 1 (VCAM-1) – facilitates the accumulation of lymphocytes in inflammatory lesions in the brain and spinal cord. Therefore, α4β1-integrin is one of the approved targets for the MS therapy [35]. The activation of the CCR6/CCL20 (chemokine receptor 6/chemokine ligand 20) signaling axis is another mechanism of migration of autoreactive and regulatory T cells into the CNS. Epithelial cells in the CNS constitutively express CCL20 that allows CCR6+ Th17 cells to migrate into the CNS [36].
Infiltrated CD4+ T cells, as well as the activated CNS-resident cells, microglia and astrocytes, produce many cytokines and chemokines, which, in turn, promote the influx of effector cells and drive the demyelination, axonal damage and the persistent inflammation.
Thus, the pathogenesis of MS is a multi-stage process and involves various populations of T cells, as well as B cells and myeloid cells, both in the periphery and CNS. EAE is the most suitable and well-studied mouse model of MS not only for understanding the fundamental mechanisms of induction and development of the disease but also for clinical studies.
ROLE OF CYTOKINES IN EAE DEVELOPMENT
Cytokines play an important role on all stages of EAE development. The pathogenic or protective function of each cytokine depends on the cellular source, the target cell population, as well as on the phase of the disease when this cytokine has been secreted. Biology of cytokines in MS was mainly studied in the model of MOG-induced EAE. In this model CD4+ T cells are key cells implicated into the disease pathogenesis, and, therefore, in this review we conventionally classify the cytokines considering their role in the differentiation of pathogenic T cells. Direct experimental evidences of contribution of different cytokines in EAE development were obtained using reverse genetics approach or pharmacological inhibition and are presented in the table.
Specific features of experimental autoimmune encephalomyelitis in mice
with genetic or pharmacological inhibition of cytokines or their
receptors
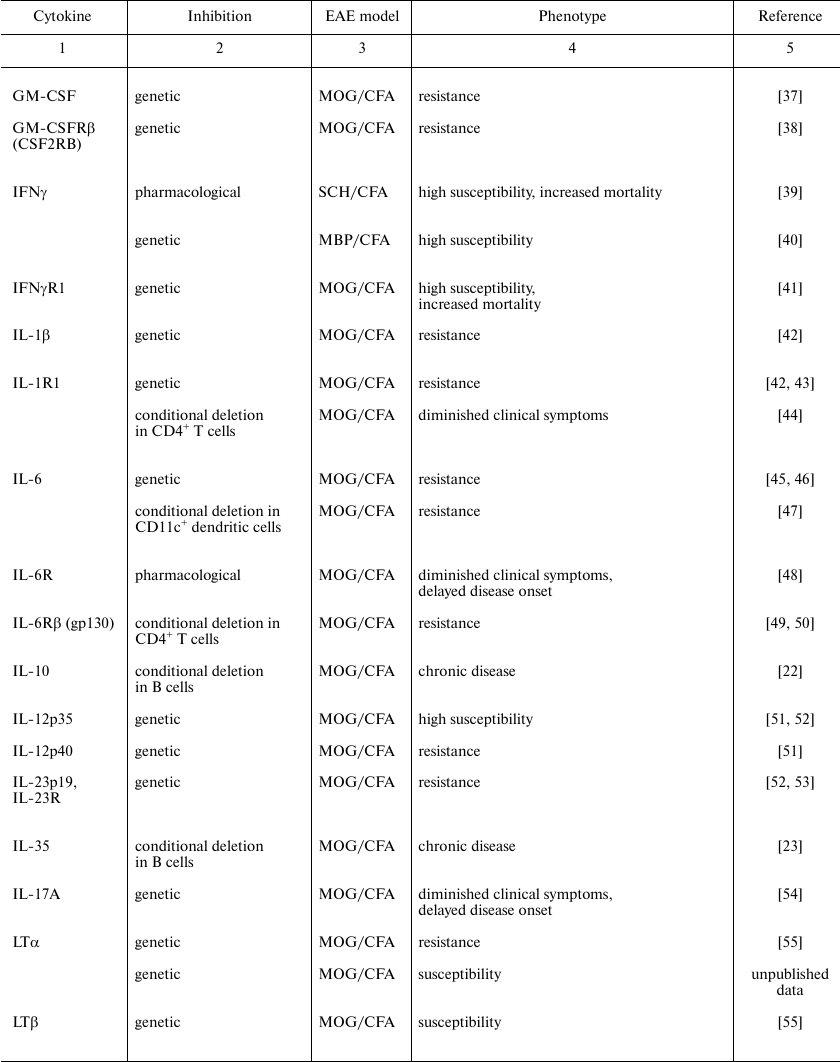
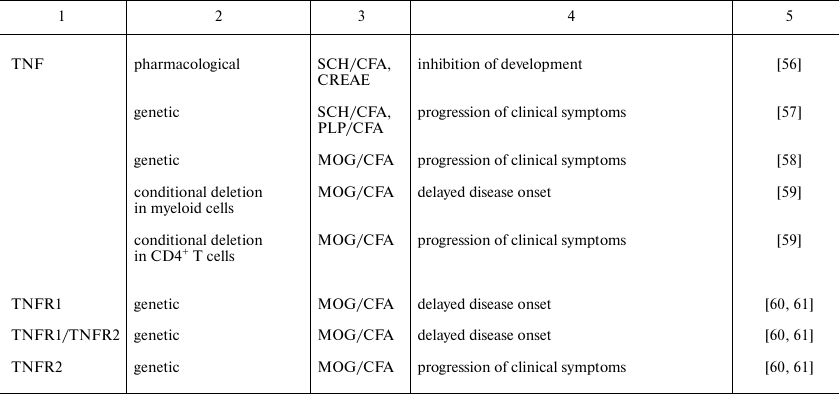
Note: CFA, complete Freund adjuvant; CREAE, chronic relapsing EAE; MBP,
myelin basic protein; MOG, myelin oligodendrocyte protein; PLP,
proteolipid protein; SCH, spinal cord homogenate.
Cytokines determining the differentiation of naïve T cells. IL-6 is a cytokine of the IL-6 family of cytokines characterized by gp130, a transmembrane protein, present as a subunit of the receptor complex and responsible for signal transduction [62]. IL-6 is produced by various cell types of both hematopoietic and stromal origin, i.e., fibroblasts, neurons and endothelial cells. On the contrary, IL-6 receptor (IL-6R) expression is restricted to certain cell types, namely, leukocytes, hepatocytes and microglia. IL-6R exists not only in membrane-bound, but also in soluble form (sIL-6R) as a result of proteolytic cleavage by the enzyme ADAM17. This determines the existence of two distinct mechanisms of signal transduction, the classical and trans-signaling cascades, respectively [63].
The “classical” IL-6 receptor complex consists of the cytokine IL-6, α-subunit (IL-6R) that binds IL-6, and β-subunit (gp130), which is responsible for the intracellular signal transduction. In the case of trans-signaling, sIL-6R binds to inactive dimers of the ubiquitously expressed gp130, and thus noticeably widens the spectrum of the IL-6 signaling target cells. Both the classic and trans-signaling activate the Jak–STAT signaling pathway, which predominantly leads to activation of the transcription factor STAT3. The signaling pathway Jak–STAT is tightly controlled, in particular, it is negatively regulated by a suppressor of cytokine signaling 3 (SOCS3) and by phosphatase SHP-2, which inhibit activities of Jak-kinases and activation of STAT3 [64].
It is interesting that IL-6 is one of only a few cytokines which are absolutely crucial for the EAE development. Il6-deficient mice are completely resistant to EAE induction [45, 46]. IL-6 in different combinations with IL-1β, IL-21, IL-23 and the transforming growth factor β (TGFβ) directs the differentiation of naïve T cells into pathogenic Th17 cells [65-68]. On the other hand, studies on reporter FoxP3-GFP mice and mice with the conventional knockout of gp130 in T cells demonstrated that IL-6 inhibited the differentiation of T cells into FoxP3+ regulatory T cells [49, 69]. CD11c+ dendritic cells can induce the differentiation of pathogenic Th17 cells via trans-presentation mechanism shown for the EAE model [47]. Besides that, the positive feedback loop between IL-17A and IL-6 is formed. Thus, injection of MOG-specific Th17 cells into healthy wild type mice leads to the increased production of IL-6 in serum. Adoptive transfer of Th17 cells from the wild type mice with EAE into the IL-6-deficient mice leads to induction of an attenuated disease suggesting a pathogenic role of the IL-17A/IL-6 signaling axis in EAE [70]. Moreover, IL-6 can maintain inflammation in the CNS where astrocytes are the major source of IL-6 [71, 72].
Thus, during the disease onset, IL-6 regulates the balance between pathogenic Th17 cells and protective Tregs in the periphery. Moreover, IL-6 produced by the CNS-resident cells can be pathogenic due to maintenance of local inflammation through activation of the IL-6 trans-signaling, but at the late stages of the disease it can play the anti-inflammatory role.
IL-1β. IL-1β belongs to the IL-1 cytokine family and is produced by monocytes, dendritic cells, microglia, and also by B cells and natural killers (NK cells) [73]. IL-1β is synthesized in the cell in the form of inactive precursor. For the secretion of the active form of IL-1β produced via proteolytic cleavage by caspase-1 or caspase-11, an assembly of a special molecular platform, inflammasome, is required. Recent studies unraveled inflammasome-independent IL-1β activation pathways [74-76]. IL-1β receptor consists of two subunits, IL-1R1 and IL-1RAP (IL-1R3), which are expressed on all innate immune cells and also on T cells [77]. The IL-1 receptor family is characterized by the presence of a TIR-domain (Toll/IL-1 receptor) in the cytoplasmic part. Formation of a heterodimeric receptor complex facilitates recruitment of the adaptor protein MyD88 and various components of the signaling cascade leading to activation of the transcription factor NFκB [78].
Il1b gene-deficient mice are resistant to EAE induction confirming the important role of IL-1β in EAE development [42]. IL-1R1-deficient mice develop significantly attenuated disease in comparison with the wild type mice due to absence of the antigen-specific Th17 cells [43]. In EAE model IL-1β is required for polarization of T cells into pathogenic Th17 cells and for their further proliferation [44, 79]. During the early stages of the disease, the IL-1 signaling pathway supports the differentiation into Th17 cells by increasing expression of the transcription factors IRF4 and RORγt [65]. IL-1β in the CNS is mainly produced by neutrophils and monocytes infiltrating into the spinal cord as it was shown on the reporter pro-IL-1β mice. In this case, the activation of the IL-1R1-signaling in the endothelial cells led to secretion of proinflammatory cytokines and chemokines recruiting myeloid cells into the CNS [42]. It also has been revealed that IL-1β was synthesized by myeloid cells in the draining lymph nodes after injection of the pertussis toxin, a component needed for the EAE induction. In this case, tissue-specific deletion of IL-1R1 in T cells resulted in a decreased amount of CD4+ Th17 cells in the draining lymph nodes, but it only partially influenced the resistance of the mice to the disease induction. It appeared that IL-1β was necessary for supporting the pathogenicity of Th17 cells producing the granulocyte-macrophage colony-stimulating factor (GM-CSF) [44, 80].
Unlike the IL-1β, IL-1α plays insignificant role in EAE [42, 81]. Thus, of the family of IL-1 cytokines, it is the IL-1β that is the most important for EAE development, and its main pathogenic role is the expansion of GM-CSF+ Th17 cells in the periphery.
IL-12 and IL-23. IL-12 and IL-23 are members of the IL-12 cytokine family with the IL-12Rβ subunit in heterodimeric receptors. Active IL-12 is an IL-12p70 heterodimer consisting of subunits p35 and p40. IL-23 contains the same subunit p40, but unlike IL-12, it is combined with the subunit p19. Both cytokines are secreted mainly by antigen-presenting cells. In accordance with the presence of the common subunit p40, IL-12 and IL-23 have the common receptor subunit IL-12Rβ1, but specific subunits are different – IL-12Rβ2 and IL-23R, respectively [82]. IL-12R and IL-23R are expressed on activated T cells, dendritic cells and NK cells. Moreover, IL-23R is also expressed on Th17 cells and γδ T cells [83]. As in the case of IL-6, triggering of receptors of the IL-12 family of cytokines leads to activation of the Jak–STAT signaling pathway, but the consequences of the cytokine binding to the receptor are distinct. Thus, IL-12 binding to the receptor leads to activation of the transcription factor STAT4, whereas IL-23 mediates the STAT3- and STAT4-signaling pathways [84].
IL-12 is one of the cytokines which determines Th1 cell differentiation. Initially it was thought that Th1 cells present the main pathogenic subpopulation of T cells in the EAE model. This concept was partially associated with complete resistance to EAE following ablation of Il12p40 (a subunit of the heterodimeric complex of IL-12) [51]. However, it was surprising that deletion of the gene encoding the second component of IL-12, Il12p35, led to progression of the disease. Later, it was established that the resistance to EAE was due to the absence of IL-23, but not of IL-12, because p40 is the common subunit for both IL-12 and IL-23. This conclusion was experimentally confirmed by the resistant phenotype of the Il23p19-deficient mice, similarly to that of Il23r knockout mice, characterized by a decreased number of IL-17+ cells as compared to wild type mice [52, 53]. It was shown that IL-23 was necessary for stabilizing the phenotype of pathogenic Th17 cells, and in the absence of IL-23/IL-23R-signaling Th17 cells were unable for the terminal differentiation and were more prone to cell death [68, 85]. Thus, IL-12 and IL-23 are required for the development and supporting the phenotype of pathogenic Th1 and Th17 cells.
Cytokines produced by effector cells. IL-17 is the most studied member of the IL-17 cytokine family. Th17 cells are a classic producer of IL-17A, however, IL-17A also can be secreted by CD8+ T cells and by the group 3 innate lymphoid cells 3 (ILC3), γδ T cells and invariant NK cells (NKT cells) [86]. IL-17A functions as a homodimer or heterodimer in the complex with IL-17F. The receptor to IL-17A consists of the IL-17RA subunit, which is common for all receptors of the IL-17R family, and the IL-17RC subunit. The IL-17RA subunit is expressed on all hemopoietic cells which defines the large spectrum of IL-17 target cell populations [87]. All IL-17R receptors have a conservative domain SEFIR (similar expression of fibroblast growth factor and IL-17R), which is required for the intracellular signal transduction through the adaptor protein Act1. The IL-17A/IL-17R interaction results in activation of NFκB transcription factor [88]. Normally, IL-17A-producing cells participate in the control of bacterial infections, but dysregulation of this cytokine is associated with various autoimmune diseases, such as psoriasis, rheumatoid arthritis and multiple sclerosis [89].
The role of IL-17 in EAE is insufficiently understood. Selective deletion of the IL-17A subunit leads to a slight improvement in the course of the disease, whereas the deletion of the IL-17F subunit does not affect EAE development [54]. Although T cells are important source of IL-17A in EAE, induced overexpression of IL-17A in CD4+ T cells does not lead to exacerbation of the disease [54]. Moreover, IL-17A-producing Th17 cells show plasticity, so that IL-17A produced by Th17 cells can play a pathogenic role during the early stages of EAE, but with disease progression the so-called ex-Th17 cells in the CNS secrete interferon-γ (IFNγ), TNF, and GM-CSF but not IL-17A [90]. As it has been demonstrated by the transcriptome analysis, the population of Th17 cells is heterogenous, and Th1-like cells, which are present in the CNS at the peak of the disease, are thought to be the most pathogenic [91]. Although IL-17A is a pivotal effector cytokine of Th17 cells, the contribution of IL-17A to the pathogenicity of Th17 subset is not clear, as well as the role of IL-17A from different sources and at different stages of the disease.
The binding of IL-17A to IL-17R induces production of many cytokines and expression of chemokines by CNS-resident cells leading to inflammation and increased immune cell recruitment to the CNS [92]. It was shown that activation of IL-17R on the brain endothelial cells facilitated their activation and the subsequent disruption of the BBB [93]. Further studies elucidating the cellular sources of this cytokine and the role of various ligand and receptor subunits are required for understanding IL-17 mediated neuroinflammation and possible clinical application.
IFNγ. IFNγ is the type II interferon which can be produced by activated T cells, NK and NKT cells [94]. IFNγ is an effector cytokine of Th1 cells and an activator of macrophages. The IFNγ dimer binds to the receptor consisting of two subunits – the α-subunit IFNγR1, which is ubiquitously expressed and interacts with the ligand, and the β-subunit IFNγR2, whose expression is inducible and is necessary for the signal transduction. The classical IFNγ signaling cascade acts through phosphorylation and activation of the receptor-bound Jak kinases, and that results in phosphorylation and activation of the transcription factor STAT1 [95].
The role of IFNγ in EAE is contradictory [96]. It was assumed that neutralization of the pivotal Th1 effector cytokine should protect mice against EAE development. In contrast, neutralization of IFNγ with monoclonal antibodies resulted in EAE exacerbation, and genetic inactivation of Ifng in mice caused a disease with massive immune cell infiltration into the CNS [39, 40]. Perhaps, such effects were due to the fact that systemic IFNγ ablation affected IFNγ-producing cells, which played a protective role [97]. The deletion of Ifngr1 in 129/Sv mice, which are resistant to EAE induction, led to development of more severe disease and increased mortality than in the wild type mice [41]. Naves et al. [98] have revealed that injection of IFNγ at the initiation stage of EAE promoted the disease pathogenesis, whereas IFNγ injection at the effector stage had protective effect [98]. Thus, the suppressive role of IFNγ during the effector stage can be a possible mechanism of exacerbated disease in Ifng-deficient mice. In the absence of IFNγ, activated CD4+ T cells can be expanded in the periphery and in the CNS, and this expansion can be accompanied by reduced expression of the transcription factor FoxP3 in regulatory T cells [99]. In this case, the adoptive transfer of regulatory T cells, differentiated in the presence of IFNγ and anti-CD3 in vitro, can promote suppressive functions of regulatory T cells and the further EAE inhibition [100]. Thus, the protective effect of IFNγ is a result of its action on the suppressor activity of Tregs, while at the initiation stage of EAE IFNγ plays the pathogenic role.
GM-CSF. GM-CSF is a member of the superfamily of colony-stimulating factors (CSF), however, recent studies have implicated that its major function is the immune regulation in vivo. In fact, GM-CSF directs the differentiation of hematopoietic precursors into granulocytes and monocytes, but, unlike the other members of the CSF superfamily, GM-CSF participates in a crosstalk between myeloid cells and lymphocytes during inflammation [101, 102]. GM-CSF is secreted by various cell types, i.e. cells of myeloid and lymphoid origin as well as by stromal cells [103]. The heterodimeric GM-CSF receptor consists of the ligand-binding α-subunit and the signal-transducing β-subunit, which is common for GM-CSF, IL-3 and IL-5 [104]. GM-CSFR signaling triggers activation of the Jak–STAT pathway due to phosphorylation of the Jak2 kinase and the transcription factor STAT5. In addition to the Jak–STAT-signaling, other signaling pathways can also be activated, including the MAP-kinase and PI3K–Akt-signaling cascades, as well as NFκB [105].
GM-CSF is required for EAE development. Mice lacking GM-CSF are resistant to EAE induction with decreased cellular infiltration of the CNS [37]. At least partially, GM-CSF is involved in regulation of the balance between effector T cells and Treg cells, because neutralization of Tregs using monoclonal antibodies to CD25 makes the GM-CSF-deficient mice susceptible to the EAE development [106]. Following BBB disruption and leukocyte infiltration into the CNS, GM-CSF produced by T cells affects cells of the myeloid origin, i.e. activates microglia and recruits circulating proinflammatory Ly6C+ monocytes into the CNS [107, 108]. Codarri et al. have shown that the secretion of GM-CSF by Th1 and Th17 cells is sufficient for EAE initiation, and that the GM-CSF production depends on the expression of IL-23/IL-23R and transcription factor RORγt [109]. Recent evidence also suggests that IL-1β and IL-23, but not IL-6 and TGFβ, can induce the expression of GM-CSF in Th17 cells [80]. Complete GM-CSF receptor deletion provides the resistance to EAE. This phenotype is also reproduced in mice with tissue-specific deletion of Csfr2b in Ly6ChiCCR2+ monocytes but not in dendritic cells or neutrophils. It is important that GM-CSF is not required for the development of proinflammatory Ly6Chi monocytes but is needed for their effector functions [38]. Conditional overexpression of GM-CSF in CD4+ T cells facilitates CNS infiltration by myeloid cells, which produce such proinflammatory mediators as IL-1β and reactive oxygen species [110]. Taken together, GM-CSF in the EAE model is produced by T cells and promotes the recruitment of different myeloid cells into the CNS, which further support local inflammation by production of various proinflammatory cytokines.
TNF and lymphotoxin (LT). TNF is a member of TNF superfamily, which is characterized by signaling of homo- or heterotrimeric ligands through homotrimeric receptors. TNF is produced mainly by myeloid cells, but also by B cells, activated T cells, as wells as CNS-resident cells – astrocytes and microglia. TNF is a transmembrane protein (tmTNF), which is released in a soluble form (sTNF) following proteolytic cleavage by TACE (ADAM17). TNF binds to two receptors expressed on the cell membrane or to their soluble forms produced via the same mechanism as sTNF. The TNFR1 receptor (p55, CD120a) is present nearly on all cell types, and TNFR2 (p75, CD120b) is expressed on the cells of the hematopoietic origin, especially, leukocytes and B cells [111]. TNF binding to its receptors leads to recruitment of intracellular adaptor proteins activating different signaling pathways. The intracellular part of TNFR1 contains “death domain”, therefore, the functional outcomes of TNF-TNFR1 signaling are cell survival or cell death. TNFR2 does not contain this domain, therefore, the signal transduction from TNFR2 leads to activation of the transcription factor NFκB which stimulates the cell survival and proliferation [112, 113].
According to published works, the role of TNF in EAE development is contradictory. On the one hand, the pharmacological inhibition of TNF improved clinical symptoms of EAE, because TNF may play the pathogenic role by inducing chemokine expression in the CNS [56, 114]. On the other hand, data obtained using reverse genetics approach revealed a more complicated picture [115]. Mice lacking Tnf develop more severe disease than the wild type mice (i), mice with Tnfr1(p55) deletion showed delayed disease onset and diminished disease progression (ii). In contrast, genetic inactivation of Tnfr2(p75) led to exacerbated EAE with the massive infiltration of CD4+ T cells into the CNS and increased production of Th1 effector cytokines (iii) [57, 58, 60]. Double knockout TNFRp55/p75–/– mice have been shown to develop attenuated disease, suggesting a pathogenic role of TNFR1 and a protective role of TNFR2 in EAE [61]. The recent studies have demonstrated that selective inhibition of TNFR1 protected mice against EAE development. Moreover, a protective function of TNFR2 was also shown in the neurodegeneration model in vivo [116, 117].
Understanding TNF biology in the context of EAE is difficult not only because of its interaction with two receptors and the presence of two related ligands (see below), but also because role of TNF depends on its cellular source. Mice with genetic ablation of TNF in myeloid cells, despite delayed disease onset, had no improvement during the late stages of EAE, indicating that the role of TNF from this source was mainly pathogenic [59]. Moreover, in experiments with the inducible TNF deletion in microglia, it was convincingly shown that TNF from monocytes, but not from the CX3CR1+ microglia, is crucial for EAE development [118]. It is interesting that functions of TNFR2 are different depending on its expression on microglia or on monocytes/macrophages. Deletion of TNFR2 on CX3CR1+ microglia increased the leukocyte infiltration into the CNS, whereas in the LysMCre:Tnfrsf1bfl/fl mice with inactivated TNFR2 in monocytes/macrophages the disease severity was reduced [119]. TNFR2 is expressed on various suppressor cells, including protective regulatory T cells, and TNFR2 ablation in FoxP3+ Tregs resulted in progression of clinical symptoms of the disease with decreased suppressor properties of Tregs (Atretkhany et al., in print). These results emphasize that protective role of TNFR2 in the EAE model, at least partially, is associated with the signaling in Tregs.
Another member of TNF superfamily, lymphotoxin (LΤα3), can also bind to TNFR1 and TNFR2. The lymphotoxin exists in the soluble and transmembrane forms (LTα3 and LTα1β2, respectively), and each form interacts with different receptors. The homotrimer LTα3 binds to TNFR1 and TNFR2, while the heterotrimer LTα1β2 can bind only to LTβR [120]. Unlike TNF, the lymphotoxin is produced by a limited number of cell types, namely, by activated T cells, B cells, and ILC [121]. It is difficult to interpret data about the role of lymphotoxin in EAE, because the genes Lta, Ltb and Tnf are closely linked in the TNF/LT locus, limiting the possibilities of independent editing of these genes. Another reason is their overlapping signaling through the corresponding receptors. Moreover, Lta–/– and LtbR–/– mice do not have lymph nodes, making it difficult to interpret the data when dissecting the molecular mechanisms of the disease pathogenesis [122]. Earlier studies of pharmacological inhibition of TNF and LTα3 concluded that these cytokines play a pathogenic role, because injection of the antibodies prevented the development of the passively induced EAE [114]. The genetic deletion of Lta leading to the absence of both the soluble and membrane-bound forms of lymphotoxin resulted in the complete resistance of mice to EAE. Nonetheless, it was found that the ablation only of the Ltb gene did not reproduce the phenotype associated with the resistance. Therefore, it was concluded that just the soluble form of lymphotoxin was required for EAE [55]. Note that in the cited work the mice with the conventional knockout of Lta were used, and later it was shown that these mice have defects in TNF production by some cell types, thus making doubtful the unambiguous interpretation of the reported phenotype [122]. According to our observations, Lta knockout mice with normal TNF production were fully susceptible to EAE (unpublished data). Moreover, injection of antibodies specific to LTα caused the depletion of LTα-expressing cells, i.e., the cells with the membrane form of the lymphotoxin, and also weakened clinical symptoms of the disease in these mice[123]. However, the heterotrimeric structure of the membrane-bound lymphotoxin makes it impossible to separate effects of the soluble and membrane-bound forms using the pharmacological inhibition. It was also shown that Th17 cells facilitated the development of tertiary lymphoid organs in the CNS, and the interaction between LTα1β2 on Th17 cells and LTβR on stromal cells was necessary for neuroinflammation [124].
Taken together, the role of TNF in EAE can be both pathogenic and protective, depending on the cellular source of TNF, as well as the target population and the type of receptor involved, and lastly, on the stage of the disease. On the other hand, the role of lymphotoxin has to be revised questioning some earlier studies, in which pharmacological and genetic inhibition of lymphotoxin in EAE, may have also affected TNF.
CYTOKINES AND THERAPY OF MULTIPLE SCLEROSIS
Cytokines play an important role in the development of autoimmune diseases, including multiple sclerosis (MS). The detection of cytokines in the cerebrospinal fluid or in demyelinated lesions of MS patients suggested that anti-cytokine therapy could be one of the main approaches for the treatment of MS, like in the case of rheumatoid arthritis. However, it should be pointed out that such therapy does not lead to clearance of autoreactive T cells, which are the causative agent of the disease. Clinical trials of TNF and IL-12/23 inhibitors were not successful in MS therapy, and at present, there are no antagonist or agonist of the cytokine signaling among the FDA-approved drugs for MS [125, 126]. On the other hand, neutralization of IL-17A with the secukinumab has been effective during the second phase of clinical trials and remains promising for further evaluation of the effect of this blocker in the therapy of MS [127].
The failure of existing anti-cytokine therapies can be partially attributed to the fact that systemic inhibition of an individual cytokine will result in blocking the pathogenic functions, but inevitably in blocking important protective functions. Therefore, more specific therapy should be considered, including inhibition of the cytokine from a defined pathogenic cell type, inhibition (or stimulation) of a specific receptor or signaling cascade in case if this cytokine is characterized by activation of signaling pathways through several receptor complexes [128, 129]. Another possible explanation of the inefficiency of anti-TNF therapy may be due to existing polymorphism of the gene encoding TNFR1, which is shown to be associated with the development of MS [130, 131]. More accurate evaluation of precise molecular mechanisms of cytokine regulation and their signaling pathways can drive the development of new therapeutics. On the other hand, it should be noted that immunotherapy with specific antibodies remains a promising trend in the therapy of MS, and α4β1-integrin, CD52 and CD20 can be used as targets [35, 132, 133].
Funding
The work was supported by the Russian Science Foundation (project no. 14-25-00160).
REFERENCES
1.Hauser, S. L., Doolittle, T. H., Lincoln, R.,
Brown, R. H., and Dinarello, C. A. (1990) Cytokine accumulations in CSF
of multiple sclerosis patients: frequent detection of interleukin-1 and
tumor necrosis factor but not interleukin-6, Neurology,
40, 1735-1739.
2.Lock, C., Hermans, G., Pedotti, R., Brendolan, A.,
Schadt, E., Garren, H., Langer-Gould, A., Strober, S., Cannella, B.,
Allard, J., Klonowski, P., Austin, A., Lad, N., Kaminski, N., Galli, S.
J., Oksenberg, J. R., Raine, C. S., Heller, R., and Steinman, L. (2002)
Gene-microarray analysis of multiple sclerosis lesions yields new
targets validated in autoimmune encephalomyelitis, Nat. Med.,
8, 500-508.
3.Jucker, M., and Walker, L. C. (2013)
Self-propagation of pathogenic protein aggregates in neurodegenerative
diseases, Nature, 501, 45-51.
4.Blokhuis, A. M., Groen, E. J., Koppers, M., van den
Berg, L. H., and Pasterkamp, R. J. (2013) Protein aggregation in
amyotrophic lateral sclerosis, Acta Neuropathol., 125,
777-794.
5.Krstic, D., Madhusudan, A., Doehner, J., Vogel, P.,
Notter, T., Imhof, C., Manalastas, A., Hilfiker, M., Pfister, S.,
Schwerdel, C., Riether, C., Meyer, U., and Knuesel, I. (2012) Systemic
immune challenges trigger and drive Alzheimer-like neuropathology in
mice, J. Neuroinflamm., 9, 151.
6.Jonsson, T., Stefansson, H., Steinberg, S.,
Jonsdottir, I., Jonsson, P. V., Snaedal, J., Bjornsson, S.,
Huttenlocher, J., Levey, A. I., Lah, J. J., Rujescu, D., Hampel, H.,
Giegling, I., Andreassen, O. A., Engedal, K., Ulstein, I., Djurovic,
S., Ibrahim-Verbaas, C., Hofman, A., Ikram, M. A., van Duijn, C. M.,
Thorsteinsdottir, U., Kong, A., and Stefansson, K. (2013) Variant of
TREM2 associated with the risk of Alzheimer’s disease, N.
Engl. J. Med., 368, 107-116.
7.Ulland, T. K., Song, W. M., Huang, S. C., Ulrich,
J. D., Sergushichev, A., Beatty, W. L., Loboda, A. A., Zhou, Y.,
Cairns, N. J., Kambal, A., Loginicheva, E., Gilfillan, S., Cella, M.,
Virgin, H. W., Unanue, E. R., Wang, Y., Artyomov, M. N., Holtzman, D.
M., and Colonna, M. (2017) TREM2 maintains microglial metabolic fitness
in Alzheimer’s disease, Cell, 170, 649-663.e13.
8.Keren-Shaul, H., Spinrad, A., Weiner, A.,
Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., David, E.,
Baruch, K., Lara-Astaiso, D., Toth, B., Itzkovitz, S., Colonna, M.,
Schwartz, M., and Amit, I. (2017) A unique microglia type associated
with restricting development of Alzheimer’s disease, Cell,
169, 1276-1290.
9.Krasemann, S., Madore, C., Cialic, R., Baufeld, C.,
Calcagno, N., El Fatimy, R., Beckers, L., O’Loughlin, E., Xu, Y.,
Fanek, Z., Greco, D. J., Smith, S. T., Tweet, G., Humulock, Z., Zrzavy,
T., Conde-Sanroman, P., Gacias, M., Weng, Z., Chen, H., Tjon, E.,
Mazaheri, F., Hartmann, K., Madi, A., Ulrich, J. D., Glatzel, M.,
Worthmann, A., Heeren, J., Budnik, B., Lemere, C., Ikezu, T., Heppner,
F. L., Litvak, V., Holtzman, D. M., Lassmann, H., Weiner, H. L.,
Ochando, J., Haass, C., and Butovsky, O. (2017) The TREM2–APOE
pathway drives the transcriptional phenotype of dysfunctional microglia
in neurodegenerative diseases, Immunity, 47, 566-581.
10.Heppner, F. L., Ransohoff, R. M., and Becher, B.
(2015) Immune attack: the role of inflammation in Alzheimer disease,
Nat. Rev. Neurosci., 16, 358-372.
11.Glass, C. K., Saijo, K., Winner, B., Marchetto,
M. C., and Gage, F. H. (2010) Mechanisms underlying inflammation in
neurodegeneration, Cell, 140, 918-934.
12.Herz, J., Filiano, A. J., Smith, A., Yogev, N.,
and Kipnis, J. (2017) Myeloid cells in the central nervous system,
Immunity, 46, 943-956.
13.Raj, T., Rothamel, K., Mostafavi, S., Ye, C.,
Lee, M. N., Replogle, J. M., Feng, T., Lee, M., Asinovski, N.,
Frohlich, I., Imboywa, S., Von Korff, A., Okada, Y., Patsopoulos, N.
A., Davis, S., McCabe, C., Paik, H. I., Srivastava, G. P.,
Raychaudhuri, S., Hafler, D. A., Koller, D., Regev, A., Hacohen, N.,
Mathis, D., Benoist, C., Stranger, B. E., and De Jager, P. L. (2014)
Polarization of the effects of autoimmune and neurodegenerative risk
alleles in leukocytes, Science, 344, 519-523.
14.Compston, A., and Coles, A. (2008) Multiple
sclerosis, Lancet, 372, 1502-1517.
15.Ascherio, A., and Munger, K. L. (2010)
Epstein–Barr virus infection and multiple sclerosis: a review,
J. Neuroimmune Pharmacol., 5, 271-277.
16.Constantinescu, C. S., Farooqi, N.,
O’Brien, K., and Gran, B. (2011) Experimental autoimmune
encephalomyelitis (EAE) as a model for multiple sclerosis (MS), Br.
J. Pharmacol., 164, 1079-1106.
17.Croxford, A. L., Kurschus, F. C., and Waisman, A.
(2011) Mouse models for multiple sclerosis: historical facts and future
implications, Biochim. Biophys. Acta, 1812, 177-183.
18.Miller, S. D., and Karpus, W. J. (2007)
Experimental autoimmune encephalomyelitis in the mouse, Curr.
Protoc. Immunol., Chap. 15, Unit 15, doi:
10.1002/0471142735.im1501s77.
19.Ben-Nun, A., Kaushansky, N., Kawakami, N.,
Krishnamoorthy, G., Berer, K., Liblau, R., Hohlfeld, R., and Wekerle,
H. (2014) From classic to spontaneous and humanized models of multiple
sclerosis: impact on understanding pathogenesis and drug development,
J. Autoimmun., 54, 33-50.
20.Dendrou, C. A., Fugger, L., and Friese, M. A.
(2015) Immunopathology of multiple sclerosis, Nat. Rev.
Immunol., 15, 545-558.
21.Babbe, H., Roers, A., Waisman, A., Lassmann, H.,
Goebels, N., Hohlfeld, R., Friese, M., Schroder, R., Deckert, M.,
Schmidt, S., Ravid, R., and Rajewsky, K. (2000) Clonal expansions of
CD8(+) T cells dominate the T cell infiltrate in active multiple
sclerosis lesions as shown by micromanipulation and single cell
polymerase chain reaction, J. Exp. Med., 192,
393-404.
22.Fillatreau, S., Sweenie, C. H., McGeachy, M. J.,
Gray, D., and Anderton, S. M. (2002) B cells regulate autoimmunity by
provision of IL-10, Nat. Immunol., 3, 944-950.
23.Shen, P., Roch, T., Lampropoulou, V., O'Connor,
R. A., Stervbo, U., Hilgenberg, E., Ries, S., Dang, V. D., Jaimes, Y.,
Daridon, C., Li, R., Jouneau, L., Boudinot, P., Wilantri, S., Sakwa,
I., Miyazaki, Y., Leech, M. D., McPherson, R. C., Wirtz, S., Neurath,
M., Hoehlig, K., Meinl, E., Grutzkau, A., Grun, J. R., Horn, K., Kuhl,
A. A., Dorner, T., Bar-Or, A., Kaufmann, S. H. E., Anderton, S. M., and
Fillatreau, S. (2014) IL-35-producing B cells are critical regulators
of immunity during autoimmune and infectious diseases, Nature,
507, 366-370.
24.Goverman, J. M. (2011) Immune tolerance in
multiple sclerosis, Immunol. Rev., 241, 228-240.
25.Louveau, A., Smirnov, I., Keyes, T. J., Eccles,
J. D., Rouhani, S. J., Peske, J. D., Derecki, N. C., Castle, D.,
Mandell, J. W., Lee, K. S., Harris, T. H., and Kipnis, J. (2015)
Structural and functional features of central nervous system lymphatic
vessels, Nature, 523, 337-341.
26.Korn, T., and Kallies, A. (2017) T cell responses
in the central nervous system, Nat. Rev. Immunol., 17,
179-194.
27.Jager, A., Dardalhon, V., Sobel, R. A., Bettelli,
E., and Kuchroo, V. K. (2009) Th1, Th17, and Th9 effector cells induce
experimental autoimmune encephalomyelitis with different pathological
phenotypes, J. Immunol., 183, 7169-7177.
28.McGeachy, M. J., Stephens, L. A., and Anderton,
S. M. (2005) Natural recovery and protection from autoimmune
encephalomyelitis: contribution of CD4+CD25+
regulatory cells within the central nervous system, J. Immunol.,
175, 3025-3032.
29.Dhib-Jalbut, S., and Marks, S. (2010)
Interferon-beta mechanisms of action in multiple sclerosis,
Neurology, 74, S17-24.
30.Vieira, P. L., Heystek, H. C., Wormmeester, J.,
Wierenga, E. A., and Kapsenberg, M. L. (2003) Glatiramer acetate
(copolymer-1, copaxone) promotes Th2 cell development and increased
IL-10 production through modulation of dendritic cells, J.
Immunol., 170, 4483-4488.
31.Wang, X., Chen, M., Wandinger, K. P., Williams,
G., and Dhib-Jalbut, S. (2000) IFN-beta-1b inhibits IL-12 production in
peripheral blood mononuclear cells in an IL-10-dependent mechanism:
relevance to IFN-beta-1b therapeutic effects in multiple sclerosis,
J. Immunol., 165, 548-557.
32.Stern, J. N., Yaari, G., Vander Heiden, J. A.,
Church, G., Donahue, W. F., Hintzen, R. Q., Huttner, A. J., Laman, J.
D., Nagra, R. M., Nylander, A., Pitt, D., Ramanan, S., Siddiqui, B. A.,
Vigneault, F., Kleinstein, S. H., Hafler, D. A., and O’Connor, K.
C. (2014) B cells populating the multiple sclerosis brain mature in the
draining cervical lymph nodes, Sci. Transl. Med., 6,
248ra107.
33.Saxena, A., Martin-Blondel, G., Mars, L. T., and
Liblau, R. S. (2011) Role of CD8 T cell subsets in the pathogenesis of
multiple sclerosis, FEBS Lett., 585, 3758-3763.
34.Ransohoff, R. M., and Engelhardt, B. (2012) The
anatomical and cellular basis of immune surveillance in the central
nervous system, Nat. Rev. Immunol., 12, 623-635.
35.Polman, C. H., O’Connor, P. W., Havrdova,
E., Hutchinson, M., Kappos, L., Miller, D. H., Phillips, J. T., Lublin,
F. D., Giovannoni, G., Wajgt, A., Toal, M., Lynn, F., Panzara, M. A.,
Sandrock, A. W., and AFFIRM Investigators (2006) A randomized,
placebo-controlled trial of natalizumab for relapsing multiple
sclerosis, N. Engl. J. Med., 354, 899-910.
36.Reboldi, A., Coisne, C., Baumjohann, D.,
Benvenuto, F., Bottinelli, D., Lira, S., Uccelli, A., Lanzavecchia, A.,
Engelhardt, B., and Sallusto, F. (2009) C-C chemokine receptor
6-regulated entry of TH-17 cells into the CNS through the choroid
plexus is required for the initiation of EAE, Nat. Immunol.,
10, 514-523.
37.McQualter, J. L., Darwiche, R., Ewing, C., Onuki,
M., Kay, T. W., Hamilton, J. A., Reid, H. H., and Bernard, C. C. (2001)
Granulocyte macrophage colony-stimulating factor: a new putative
therapeutic target in multiple sclerosis, J. Exp. Med.,
194, 873-882.
38.Croxford, A. L., Lanzinger, M., Hartmann, F. J.,
Schreiner, B., Mair, F., Pelczar, P., Clausen, B. E., Jung, S., Greter,
M., and Becher, B. (2015) The cytokine GM-CSF drives the inflammatory
signature of CCR2+ monocytes and licenses autoimmunity,
Immunity, 43, 502-514.
39.Billiau, A., Heremans, H., Vandekerckhove, F.,
Dijkmans, R., Sobis, H., Meulepas, E., and Carton, H. (1988)
Enhancement of experimental allergic encephalomyelitis in mice by
antibodies against IFN-gamma, J. Immunol., 140,
1506-1510.
40.Ferber, I. A., Brocke, S., Taylor-Edwards, C.,
Ridgway, W., Dinisco, C., Steinman, L., Dalton, D., and Fathman, C. G.
(1996) Mice with a disrupted IFN-gamma gene are susceptible to the
induction of experimental autoimmune encephalomyelitis (EAE), J.
Immunol., 156, 5-7.
41.Willenborg, D. O., Fordham, S., Bernard, C. C.,
Cowden, W. B., and Ramshaw, I. A. (1996) IFN-gamma plays a critical
down-regulatory role in the induction and effector phase of myelin
oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis,
J. Immunol., 157, 3223-3227.
42.Levesque, S. A., Pare, A., Mailhot, B.,
Bellver-Landete, V., Kebir, H., Lecuyer, M. A., Alvarez, J. I., Prat,
A., de Rivero Vaccari, J. P., Keane, R. W., and Lacroix, S. (2016)
Myeloid cell transmigration across the CNS vasculature triggers
IL-1-beta-driven neuroinflammation during autoimmune encephalomyelitis
in mice, J. Exp. Med., 213, 929-949.
43.Sutton, C., Brereton, C., Keogh, B., Mills, K.
H., and Lavelle, E. C. (2006) A crucial role for interleukin (IL)-1 in
the induction of IL-17-producing T cells that mediate autoimmune
encephalomyelitis, J. Exp. Med., 203, 1685-1691.
44.Mufazalov, I. A., Schelmbauer, C., Regen, T.,
Kuschmann, J., Wanke, F., Gabriel, L. A., Hauptmann, J., Muller, W.,
Pinteaux, E., Kurschus, F. C., and Waisman, A. (2017) IL-1 signaling is
critical for expansion but not generation of autoreactive
GM-CSF+ Th17 cells, EMBO J., 36, 102-115.
45.Samoilova, E. B., Horton, J. L., Hilliard, B.,
Liu, T. S., and Chen, Y. (1998) IL-6-deficient mice are resistant to
experimental autoimmune encephalomyelitis: roles of IL-6 in the
activation and differentiation of autoreactive T cells, J.
Immunol., 161, 6480-6486.
46.Okuda, Y., Sakoda, S., Bernard, C. C., Fujimura,
H., Saeki, Y., Kishimoto, T., and Yanagihara, T. (1998) IL-6-deficient
mice are resistant to the induction of experimental autoimmune
encephalomyelitis provoked by myelin oligodendrocyte glycoprotein,
Int. Immunol., 10, 703-708.
47.Heink, S., Yogev, N., Garbers, C., Herwerth, M.,
Aly, L., Gasperi, C., Husterer, V., Croxford, A. L., Moller-Hackbarth,
K., Bartsch, H. S., Sotlar, K., Krebs, S., Regen, T., Blum, H., Hemmer,
B., Misgeld, T., Wunderlich, T. F., Hidalgo, J., Oukka, M., Rose-John,
S., Schmidt-Supprian, M., Waisman, A., and Korn, T. (2017)
Trans-presentation of IL-6 by dendritic cells is required for the
priming of pathogenic TH17 cells, Nat. Immunol., 18,
74-85.
48.Serada, S., Fujimoto, M., Mihara, M., Koike, N.,
Ohsugi, Y., Nomura, S., Yoshida, H., Nishikawa, T., Terabe, F.,
Ohkawara, T., Takahashi, T., Ripley, B., Kimura, A., Kishimoto, T., and
Naka, T. (2008) IL-6 blockade inhibits the induction of myelin
antigen-specific Th17 cells and Th1 cells in experimental autoimmune
encephalomyelitis, Proc. Natl. Acad. Sci. USA, 105,
9041-9046.
49.Korn, T., Mitsdoerffer, M., Croxford, A. L.,
Awasthi, A., Dardalhon, V. A., Galileos, G., Vollmar, P., Stritesky, G.
L., Kaplan, M. H., Waisman, A., Kuchroo, V. K., and Oukka, M. (2008)
IL-6 controls Th17 immunity in vivo by inhibiting the conversion
of conventional T cells into Foxp3+ regulatory T cells,
Proc. Natl. Acad. Sci. USA, 105, 18460-18465.
50.Holz, K., Prinz, M., Brendecke, S. M., Holscher,
A., Deng, F., Mitrucker, H.-W., Rose-John, S., and Holscher, C. (2018)
Differing outcome of experimental autoimmune encephalitis in
macrophage/neutrophil- and T cell-specific gp130-deficient mice,
Front. Immunol., 9, 836.
51.Becher, B., Durell, B. G., and Noelle, R. J.
(2002) Experimental autoimmune encephalitis and inflammation in the
absence of interleukin-12, J. Clin. Invest., 110,
493-497.
52.Cua, D. J., Sherlock, J., Chen, Y., Murphy, C.
A., Joyce, B., Seymour, B., Lucian, L., To, W., Kwan, S., Churakova,
T., Zurawski, S., Wiekowski, M., Lira, S. A., Gorman, D., Kastelein, R.
A., and Sedgwick, J. D. (2003) Interleukin-23 rather than
interleukin-12 is the critical cytokine for autoimmune inflammation of
the brain, Nature, 421, 744-748.
53.Awasthi, A., Riol-Blanco, L., Jager, A., Korn,
T., Pot, C., Galileos, G., Bettelli, E., Kuchroo, V. K., and Oukka, M.
(2009) Cutting edge: IL-23 receptor gfp reporter mice reveal distinct
populations of IL-17-producing cells, J. Immunol., 182,
5904-5908.
54.Haak, S., Croxford, A. L., Kreymborg, K.,
Heppner, F. L., Pouly, S., Becher, B., and Waisman, A. (2009) IL-17A
and IL-17F do not contribute vitally to autoimmune neuro-inflammation
in mice, J. Clin. Invest., 119, 61-69.
55.Suen, W. E., Bergman, C. M., Hjelmstrom, P., and
Ruddle, N. H. (1997) A critical role for lymphotoxin in experimental
allergic encephalomyelitis, J. Exp. Med., 186,
1233-1240.
56.Baker, D., Butler, D., Scallon, B. J.,
O’Neill, J. K., Turk, J. L., and Feldmann, M. (1994) Control of
established experimental allergic encephalomyelitis by inhibition of
tumor necrosis factor (TNF) activity within the central nervous system
using monoclonal antibodies and TNF receptor-immunoglobulin fusion
proteins, Eur. J. Immunol., 24, 2040-2048.
57.Frei, K., Eugster, H. P., Bopst, M.,
Constantinescu, C. S., Lavi, E., and Fontana, A. (1997) Tumor necrosis
factor alpha and lymphotoxin alpha are not required for induction of
acute experimental autoimmune encephalomyelitis, J. Exp. Med.,
185, 2177-2182.
58.Liu, J., Marino, M. W., Wong, G., Grail, D.,
Dunn, A., Bettadapura, J., Slavin, A. J., Old, L., and Bernard, C. C.
(1998) TNF is a potent anti-inflammatory cytokine in
autoimmune-mediated demyelination, Nat. Med., 4,
78-83.
59.Kruglov, A. A., Lampropoulou, V., Fillatreau, S.,
and Nedospasov, S. A. (2011) Pathogenic and protective functions of TNF
in neuroinflammation are defined by its expression in T lymphocytes and
myeloid cells, J. Immunol., 187, 5660-5670.
60.Suvannavejh, G. C., Lee, H. O., Padilla, J., Dal
Canto, M. C., Barrett, T. A., and Miller, S. D. (2000) Divergent roles
for p55 and p75 tumor necrosis factor receptors in the pathogenesis of
MOG(35-55)-induced experimental autoimmune encephalomyelitis, Cell.
Immunol., 205, 24-33.
61.Eugster, H. P., Frei, K., Bachmann, R.,
Bluethmann, H., Lassmann, H., and Fontana, A. (1999) Severity of
symptoms and demyelination in MOG-induced EAE depends on TNFR1, Eur.
J. Immunol., 29, 626-632.
62.Rose-John, S. (2018) Interleukin-6 family
cytokines, Cold Spring Harb. Perspect. Biol., 10,
a028415.
63.Drutskaya, M. S., Nosenko, M. A., Atretkhany, K.
S., Efimov, G. A., and Nedospasov, S. A. (2015) Interleukin-6 from
molecular mechanisms of signal transduction to physiological properties
and therapeutic targeting, Mol. Biol. (Moscow), 49,
937-943.
64.Babon, J. J., Kershaw, N. J., Murphy, J. M.,
Varghese, L. N., Laktyushin, A., Young, S. N., Lucet, I. S., Norton, R.
S., and Nicola, N. A. (2012) Suppression of cytokine signaling by
SOCS3: characterization of the mode of inhibition and the basis of its
specificity, Immunity, 36, 239-250.
65.Chung, Y., Chang, S. H., Martinez, G. J., Yang,
X. O., Nurieva, R., Kang, H. S., Ma, L., Watowich, S. S., Jetten, A.
M., Tian, Q., and Dong, C. (2009) Critical regulation of early Th17
cell differentiation by interleukin-1 signaling, Immunity,
30, 576-587.
66.Lee, Y., Awasthi, A., Yosef, N., Quintana, F. J.,
Xiao, S., Peters, A., Wu, C., Kleinewietfeld, M., Kunder, S., Hafler,
D. A., Sobel, R. A., Regev, A., and Kuchroo, V. K. (2012) Induction and
molecular signature of pathogenic TH17 cells, Nat. Immunol.,
13, 991-999.
67.Ghoreschi, K., Laurence, A., Yang, X. P., Tato,
C. M., McGeachy, M. J., Konkel, J. E., Ramos, H. L., Wei, L., Davidson,
T. S., Bouladoux, N., Grainger, J. R., Chen, Q., Kanno, Y., Watford, W.
T., Sun, H. W., Eberl, G., Shevach, E. M., Belkaid, Y., Cua, D. J.,
Chen, W., and O’Shea, J. J. (2010) Generation of pathogenic
T(H)17 cells in the absence of TGF-beta signalling, Nature,
467, 967-971.
68.Zhou, L., Ivanov, I. I., Spolski, R., Min, R.,
Shenderov, K., Egawa, T., Levy, D. E., Leonard, W. J., and Littman, D.
R. (2007) IL-6 programs T(H)-17 cell differentiation by promoting
sequential engagement of the IL-21 and IL-23 pathways, Nat.
Immunol., 8, 967-974.
69.Bettelli, E., Carrier, Y., Gao, W., Korn, T.,
Strom, T. B., Oukka, M., Weiner, H. L., and Kuchroo, V. K. (2006)
Reciprocal developmental pathways for the generation of pathogenic
effector TH17 and regulatory T cells, Nature, 441,
235-238.
70.Ogura, H., Murakami, M., Okuyama, Y., Tsuruoka,
M., Kitabayashi, C., Kanamoto, M., Nishihara, M., Iwakura, Y., and
Hirano, T. (2008) Interleukin-17 promotes autoimmunity by triggering a
positive-feedback loop via interleukin-6 induction, Immunity,
29, 628-636.
71.Erta, M., Quintana, A., and Hidalgo, J. (2012)
Interleukin-6, a major cytokine in the central nervous system, Int.
J. Biol. Sci., 8, 1254-1266.
72.Giralt, M., Ramos, R., Quintana, A., Ferrer, B.,
Erta, M., Castro-Freire, M., Comes, G., Sanz, E., Unzeta, M., Pifarre,
P., Garcia, A., Campbell, I. L., and Hidalgo, J. (2013) Induction of
atypical EAE mediated by transgenic production of IL-6 in astrocytes in
the absence of systemic IL-6, Glia, 61, 587-600.
73.Garlanda, C., Dinarello, C. A., and Mantovani, A.
(2013) The interleukin-1 family: back to the future, Immunity,
39, 1003-1018.
74.Kayagaki, N., Warming, S., Lamkanfi, M., Vande
Walle, L., Louie, S., Dong, J., Newton, K., Qu, Y., Liu, J., Heldens,
S., Zhang, J., Lee, W. P., Roose-Girma, M., and Dixit, V. M. (2011)
Non-canonical inflammasome activation targets caspase-11,
Nature, 479, 117-121.
75.Afonina, I. S., Muller, C., Martin, S. J., and
Beyaert, R. (2015) Proteolytic processing of interleukin-1 family
cytokines: variations on a common theme, Immunity, 42,
991-1004.
76.Netea, M. G., van de Veerdonk, F. L., van der
Meer, J. W., Dinarello, C. A., and Joosten, L. A. (2015)
Inflammasome-independent regulation of IL-1-family cytokines, Annu.
Rev. Immunol., 33, 49-77.
77.Sims, J. E., and Smith, D. E. (2010) The IL-1
family: regulators of immunity, Nat. Rev. Immunol., 10,
89-102.
78.Dinarello, C. A. (2009) Immunological and
inflammatory functions of the interleukin-1 family, Annu. Rev.
Immunol., 27, 519-550.
79.Manel, N., Unutmaz, D., and Littman, D. R. (2008)
The differentiation of human T(H)-17 cells requires transforming growth
factor-beta and induction of the nuclear receptor RORgammat, Nat.
Immunol., 9, 641-649.
80.El-Behi, M., Ciric, B., Dai, H., Yan, Y.,
Cullimore, M., Safavi, F., Zhang, G. X., Dittel, B. N., and Rostami, A.
(2011) The encephalitogenicity of T(H)17 cells is dependent on IL-1-
and IL-23-induced production of the cytokine GM-CSF, Nat.
Immunol., 12, 568-575.
81.Matsuki, T., Nakae, S., Sudo, K., Horai, R., and
Iwakura, Y. (2006) Abnormal T cell activation caused by the imbalance
of the IL-1/IL-1R antagonist system is responsible for the development
of experimental autoimmune encephalomyelitis, Int. Immunol.,
18, 399-407.
82.Sun, L., He, C., Nair, L., Yeung, J., and
Egwuagu, C. E. (2015) Interleukin 12 (IL-12) family cytokines: role in
immune pathogenesis and treatment of CNS autoimmune disease,
Cytokine, 75, 249-255.
83.Gaffen, S. L., Jain, R., Garg, A. V., and Cua, D.
J. (2014) The IL-23–IL-17 immune axis: from mechanisms to
therapeutic testing, Nat. Rev. Immunol., 14, 585-600.
84.Vignali, D. A., and Kuchroo, V. K. (2012) IL-12
family cytokines: immunological playmakers, Nat. Immunol.,
13, 722-728.
85.McGeachy, M. J., Chen, Y., Tato, C. M., Laurence,
A., Joyce-Shaikh, B., Blumenschein, W. M., McClanahan, T. K.,
O’Shea, J. J., and Cua, D. J. (2009) The interleukin 23 receptor
is essential for the terminal differentiation of interleukin
17-producing effector T helper cells in vivo, Nat.
Immunol., 10, 314-324.
86.Gaffen, S. L. (2009) Structure and signalling in
the IL-17 receptor family, Nat. Rev. Immunol., 9,
556-567.
87.Ishigame, H., Kakuta, S., Nagai, T., Kadoki, M.,
Nambu, A., Komiyama, Y., Fujikado, N., Tanahashi, Y., Akitsu, A.,
Kotaki, H., Sudo, K., Nakae, S., Sasakawa, C., and Iwakura, Y. (2009)
Differential roles of interleukin-17A and -17F in host defense against
mucoepithelial bacterial infection and allergic responses,
Immunity, 30, 108-119.
88.Amatya, N., Garg, A. V., and Gaffen, S. L. (2017)
IL-17 signaling: the Yin and the Yang, Trends Immunol.,
38, 310-322.
89.Stadhouders, R., Lubberts, E., and Hendriks, R.
W. (2018) A cellular and molecular view of T helper 17 cell plasticity
in autoimmunity, J. Autoimmun., 87, 1-15.
90.Hirota, K., Duarte, J. H., Veldhoen, M., Hornsby,
E., Li, Y., Cua, D. J., Ahlfors, H., Wilhelm, C., Tolaini, M., Menzel,
U., Garefalaki, A., Potocnik, A. J., and Stockinger, B. (2011) Fate
mapping of IL-17-producing T cells in inflammatory responses, Nat.
Immunol., 12, 255-263.
91.Gaublomme, J. T., Yosef, N., Lee, Y., Gertner, R.
S., Yang, L. V., Wu, C., Pandolfi, P. P., Mak, T., Satija, R., Shalek,
A. K., Kuchroo, V. K., Park, H., and Regev, A. (2015) Single-cell
genomics unveils critical regulators of Th17 cell pathogenicity,
Cell, 163, 1400-1412.
92.Waisman, A., Hauptmann, J., and Regen, T. (2015)
The role of IL-17 in CNS diseases, Acta Neuropathol.,
129, 625-637.
93.Kebir, H., Kreymborg, K., Ifergan, I.,
Dodelet-Devillers, A., Cayrol, R., Bernard, M., Giuliani, F., Arbour,
N., Becher, B., and Prat, A. (2007) Human TH17 lymphocytes promote
blood-brain barrier disruption and central nervous system inflammation,
Nat. Med., 13, 1173-1175.
94.Petermann, F., and Korn, T. (2011) Cytokines and
effector T cell subsets causing autoimmune CNS disease, FEBS
Lett., 585, 3747-3757.
95.Green, D. S., Young, H. A., and Valencia, J. C.
(2017) Current prospects of type II interferon gamma signaling and
autoimmunity, J. Biol. Chem., 292, 13925-13933.
96.Arellano, G., Ottum, P. A., Reyes, L. I., Burgos,
P. I., and Naves, R. (2015) Stage-specific role of interferon-gamma in
experimental autoimmune encephalomyelitis and multiple sclerosis,
Front. Immunol., 6, 492.
97.Chong, W. P., van Panhuys, N., Chen, J., Silver,
P. B., Jittayasothorn, Y., Mattapallil, M. J., Germain, R. N., and
Caspi, R. R. (2015) NK-DC crosstalk controls the autopathogenic Th17
response through an innate IFN-gamma-IL-27 axis, J. Exp. Med.,
212, 1739-1752.
98.Naves, R., Singh, S. P., Cashman, K. S., Rowse,
A. L., Axtell, R. C., Steinman, L., Mountz, J. D., Steele, C., De
Sarno, P., and Raman, C. (2013) The interdependent, overlapping, and
differential roles of type I and II IFNs in the pathogenesis of
experimental autoimmune encephalomyelitis, J. Immunol.,
191, 2967-2977.
99.Chu, C. Q., Wittmer, S., and Dalton, D. K. (2000)
Failure to suppress the expansion of the activated CD4 T cell
population in interferon gamma-deficient mice leads to exacerbation of
experimental autoimmune encephalomyelitis, J. Exp. Med.,
192, 123-128.
100.Wang, Z., Hong, J., Sun, W., Xu, G., Li, N.,
Chen, X., Liu, A., Xu, L., Sun, B., and Zhang, J. Z. (2006) Role of
IFN-gamma in induction of Foxp3 and conversion of CD4+
CD25– T cells to CD4+ Tregs, J. Clin.
Invest., 116, 2434-2441.
101.Metcalf, D. (2008) Hematopoietic cytokines,
Blood, 111, 485-491.
102.Becher, B., Tugues, S., and Greter, M. (2016)
GM-CSF: from growth factor to central mediator of tissue inflammation,
Immunity, 45, 963-973.
103.Wicks, I. P., and Roberts, A. W. (2016)
Targeting GM-CSF in inflammatory diseases, Nat. Rev. Rheumatol.,
12, 37-48.
104.Broughton, S. E., Nero, T. L., Dhagat, U., Kan,
W. L., Hercus, T. R., Tvorogov, D., Lopez, A. F., and Parker, M. W.
(2015) The betac receptor family – structural insights and
their functional implications, Cytokine, 74, 247-258.
105.Hercus, T. R., Dhagat, U., Kan, W. L.,
Broughton, S. E., Nero, T. L., Perugini, M., Sandow, J. J.,
D’Andrea, R. J., Ekert, P. G., Hughes, T., Parker, M. W., and
Lopez, A. F. (2013) Signalling by the betac family of cytokines,
Cytokine Growth Factor Rev., 24, 189-201.
106.Ghosh, D., Curtis, A. D., 2nd, Wilkinson, D.
S., and Mannie, M. D. (2016) Depletion of
CD4+CD25+ regulatory T cells confers
susceptibility to experimental autoimmune encephalomyelitis (EAE) in
GM-CSF-deficient Csf2–/– mice, J. Leukoc.
Biol., 100, 747-760.
107.Ponomarev, E. D., Shriver, L. P., Maresz, K.,
Pedras-Vasconcelos, J., Verthelyi, D., and Dittel, B. N. (2007) GM-CSF
production by autoreactive T cells is required for the activation of
microglial cells and the onset of experimental autoimmune
encephalomyelitis, J. Immunol., 178, 39-48.
108.King, I. L., Dickendesher, T. L., and Segal, B.
M. (2009) Circulating Ly-6C+ myeloid precursors migrate to
the CNS and play a pathogenic role during autoimmune demyelinating
disease, Blood, 113, 3190-3197.
109.Codarri, L., Gyulveszi, G., Tosevski, V.,
Hesske, L., Fontana, A., Magnenat, L., Suter, T., and Becher, B. (2011)
RORgammat drives production of the cytokine GM-CSF in helper T cells,
which is essential for the effector phase of autoimmune
neuroinflammation, Nat. Immunol., 12, 560-567.
110.Spath, S., Komuczki, J., Hermann, M., Pelczar,
P., Mair, F., Schreiner, B., and Becher, B. (2017) Dysregulation of the
cytokine GM-CSF induces spontaneous phagocyte invasion and
immunopathology in the central nervous system, Immunity,
46, 245-260.
111.Kalliolias, G. D., and Ivashkiv, L. B. (2016)
TNF biology, pathogenic mechanisms and emerging therapeutic strategies,
Nat. Rev. Rheumatol., 12, 49-62.
112.Brenner, D., Blaser, H., and Mak, T. W. (2015)
Regulation of tumour necrosis factor signalling: live or let die,
Nat. Rev. Immunol., 15, 362-374.
113.Varfolomeev, E., and Vucic, D. (2018)
Intracellular regulation of TNF activity in health and disease,
Cytokine, 101, 26-32.
114.Ruddle, N. H., Bergman, C. M., McGrath, K. M.,
Lingenheld, E. G., Grunnet, M. L., Padula, S. J., and Clark, R. B.
(1990) An antibody to lymphotoxin and tumor necrosis factor prevents
transfer of experimental allergic encephalomyelitis, J. Exp.
Med., 172, 1193-1200.
115.Murphy, C. A., Hoek, R. M., Wiekowski, M. T.,
Lira, S. A., and Sedgwick, J. D. (2002) Interactions between
hemopoietically derived TNF and central nervous system-resident glial
chemokines underlie initiation of autoimmune inflammation in the brain,
J. Immunol., 169, 7054-7062.
116.Steeland, S., Van Ryckeghem, S., Van Imschoot,
G., De Rycke, R., Toussaint, W., Vanhoutte, L., Vanhove, C., De Vos,
F., Vandenbroucke, R. E., and Libert, C. (2017) TNFR1 inhibition with a
Nanobody protects against EAE development in mice, Sci. Rep.,
7, 13646.
117.Dong, Y., Fischer, R., Naude, P. J., Maier, O.,
Nyakas, C., Duffey, M., Van der Zee, E. A., Dekens, D., Douwenga, W.,
Herrmann, A., Guenzi, E., Kontermann, R. E., Pfizenmaier, K., and
Eisel, U. L. (2016) Essential protective role of tumor necrosis factor
receptor 2 in neurodegeneration, Proc. Natl. Acad. Sci. USA,
113, 12304-12309.
118.Wolf, Y., Shemer, A., Polonsky, M., Gross, M.,
Mildner, A., Yona, S., David, E., Kim, K. W., Goldmann, T., Amit, I.,
Heikenwalder, M., Nedospasov, S., Prinz, M., Friedman, N., and Jung, S.
(2017) Autonomous TNF is critical for in vivo monocyte survival
in steady state and inflammation, J. Exp. Med., 214,
905-917.
119.Gao, H., Danzi, M. C., Choi, C. S., Taherian,
M., Dalby-Hansen, C., Ellman, D. G., Madsen, P. M., Bixby, J. L.,
Lemmon, V. P., Lambertsen, K. L., and Brambilla, R. (2017) Opposing
functions of microglial and macrophagic TNFR2 in the pathogenesis of
experimental autoimmune encephalomyelitis, Cell Rep., 18,
198-212.
120.Ruddle, N. H. (2014) Lymphotoxin and TNF: how
it all began – a tribute to the travelers, Cytokine Growth
Factor Rev., 25, 83-89.
121.Upadhyay, V., and Fu, Y. X. (2013) Lymphotoxin
signalling in immune homeostasis and the control of microorganisms,
Nat. Rev. Immunol., 13, 270-279.
122.Liepinsh, D. J., Grivennikov, S. I., Klarmann,
K. D., Lagarkova, M. A., Drutskaya, M. S., Lockett, S. J., Tessarollo,
L., McAuliffe, M., Keller, J. R., Kuprash, D. V., and Nedospasov, S. A.
(2006) Novel lymphotoxin alpha (LTalpha) knockout mice with unperturbed
tumor necrosis factor expression: reassessing LTalpha biological
functions, Mol. Cell. Biol., 26, 4214-4225.
123.Chiang, E. Y., Kolumam, G. A., Yu, X.,
Francesco, M., Ivelja, S., Peng, I., Gribling, P., Shu, J., Lee, W. P.,
Refino, C. J., Balazs, M., Paler-Martinez, A., Nguyen, A., Young, J.,
Barck, K. H., Carano, R. A., Ferrando, R., Diehl, L., Chatterjea, D.,
and Grogan, J. L. (2009) Targeted depletion of
lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune
disease, Nat. Med., 15, 766-773.
124.Pikor, N. B., Astarita, J. L., Summers-Deluca,
L., Galicia, G., Qu, J., Ward, L. A., Armstrong, S., Dominguez, C. X.,
Malhotra, D., Heiden, B., Kay, R., Castanov, V., Touil, H., Boon, L.,
O’Connor, P., Bar-Or, A., Prat, A., Ramaglia, V., Ludwin, S.,
Turley, S. J., and Gommerman, J. L. (2015) Integration of Th17- and
lymphotoxin-derived signals initiates meningeal-resident stromal cell
remodeling to propagate neuroinflammation, Immunity, 43,
1160-1173.
125.The Lenercept Multiple Sclerosis Study Group
and The University of British Columbia MS/MRI Analysis Group (1999) TNF
neutralization in MS: results of a randomized, placebo-controlled
multicenter study, Neurology, 53, 457-465.
126.Longbrake, E. E., and Racke, M. K. (2009) Why
did IL-12/IL-23 antibody therapy fail in multiple sclerosis? Expert.
Rev. Neurother., 9, 319-321.
127.Havrdova, E., Belova, A., Goloborodko, A.,
Tisserant, A., Wright, A., Wallstroem, E., Garren, H., Maguire, R. P.,
and Johns, D. R. (2016) Activity of secukinumab, an anti-IL-17A
antibody, on brain lesions in RRMS: results from a randomized,
proof-of-concept study, J. Neurol., 263, 1287-1295.
128.Efimov, G. A., Kruglov, A. A., Khlopchatnikova,
Z. V., Rozov, F. N., Mokhonov, V. V., Rose-John, S., Scheller, J.,
Gordon, S., Stacey, M., Drutskaya, M. S., Tillib, S. V., and
Nedospasov, S. A. (2016) Cell-type-restricted anti-cytokine therapy:
TNF inhibition from one pathogenic source, Proc. Natl. Acad. Sci.
USA, 113, 3006-3011.
129.Nosenko, M. A., Atretkhany, K. N., Mokhonov, V.
V., Efimov, G. A., Kruglov, A. A., Tillib, S. V., Drutskaya, M. S., and
Nedospasov, S. A. (2017) VHH-based bispecific antibodies targeting
cytokine production, Front. Immunol., 8, 1073.
130.De Jager, P. L., Jia, X., Wang, J., de Bakker,
P. I., Ottoboni, L., Aggarwal, N. T., Piccio, L., Raychaudhuri, S.,
Tran, D., Aubin, C., Briskin, R., Romano, S., International, M. S. G.
C., Baranzini, S. E., McCauley, J. L., Pericak-Vance, M. A., Haines, J.
L., Gibson, R. A., Naeglin, Y., Uitdehaag, B., Matthews, P. M., Kappos,
L., Polman, C., McArdle, W. L., Strachan, D. P., Evans, D., Cross, A.
H., Daly, M. J., Compston, A., Sawcer, S. J., Weiner, H. L., Hauser, S.
L., Hafler, D. A., and Oksenberg, J. R. (2009) Meta-analysis of genome
scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple
sclerosis susceptibility loci, Nat. Genet., 41,
776-782.
131.Gregory, A. P., Dendrou, C. A., Attfield, K.
E., Haghikia, A., Xifara, D. K., Butter, F., Poschmann, G., Kaur, G.,
Lambert, L., Leach, O. A., Promel, S., Punwani, D., Felce, J. H.,
Davis, S. J., Gold, R., Nielsen, F. C., Siegel, R. M., Mann, M., Bell,
J. I., McVean, G., and Fugger, L. (2012) TNF receptor 1 genetic risk
mirrors outcome of anti-TNF therapy in multiple sclerosis,
Nature, 488, 508-511.
132.Coles, A. J., Twyman, C. L., Arnold, D. L.,
Cohen, J. A., Confavreux, C., Fox, E. J., Hartung, H. P., Havrdova, E.,
Selmaj, K. W., Weiner, H. L., Miller, T., Fisher, E., Sandbrink, R.,
Lake, S. L., Margolin, D. H., Oyuela, P., Panzara, M. A., Compston, D.
A., and CARE-MS II investigators (2012) Alemtuzumab for patients with
relapsing multiple sclerosis after disease-modifying therapy: a
randomised controlled phase 3 trial, Lancet, 380,
1829-1839.
133.Hohlfeld, R., and Meinl, E. (2017) Ocrelizumab
in multiple sclerosis: markers and mechanisms, Lancet Neurol.,
16, 259-261.