Cysmotif Searcher Pipeline for Antimicrobial Peptide Identification in Plant Transcriptomes
A. A. Shelenkov1,2,a*, A. A. Slavokhotova1, and T. I. Odintsova1
1Vavilov Institute of General Genetics, Russian Academy of Sciences, 119333 Moscow, Russia2Central Research Institute of Epidemiology, Rospotrebnadzor, 111123 Moscow, Russia
* To whom correspondence should be addressed.
Received June 24, 2018; Revision received August 23, 2018
In this paper, we present the new Cysmotif searcher pipeline for identification of various antimicrobial peptides (AMPs), the most important components of innate immunity, in plant transcriptomes. Cysmotif searcher reveals and classifies short cysteine-rich amino acid sequences containing an open reading frame and a signal peptide cleavage site. Due to the combination of various search methods, Cysmotif searcher allows to obtain the most complete repertoire of AMPs for one or more transcriptomes in a short amount of time. The pipeline performance is estimated on the model plant Arabidopsis thaliana and nine other plants, including cultivated and wild species. The obtained results are compared to the existing annotation (A. thaliana) and results of conventional homology search (other plants). The comparison is carried out for known families of plant AMPs and newly discovered peptides that could not be assigned to existing families. The applicability of Cysmotif searcher in detecting new AMPs is discussed, and some practical recommendations on the pipeline usage for end users are given. The Cysmotif searcher pipeline is free for academic use and can be downloaded from Github (http://github.com/fallandar/cysmotifsearcher).
KEY WORDS: antimicrobial peptides, protein annotation, motif searching, computational pipelineDOI: 10.1134/S0006297918110135
Abbreviations: AMP, antimicrobial peptide; LTP, lipid transfer protein; ORF, open reading frame.
Antimicrobial peptides (AMPs) are key components of innate immunity in
various organisms, including bacteria, fungi, plants, invertebrates,
and vertebrates [1-3].
Expression of these peptides, either constitutive or induced by
specific pathogens, provides fast and energy-efficient first line of
defense of the host organism. Since the mode of action of AMPs is based
on damaging the membranes of pathogen cells, this dramatically
decreases the probability of resistance development in pathogenic
organisms. This makes AMPs promising agents in the development of
antimicrobial and antifungal drugs [4]. In
addition, AMPs are intriguing objects in the studies of
plant–pathogen interactions [5-8].
Currently, modern techniques, such as second generation sequencing and peptidomic analysis, are used for analysis of AMP repertoire in insects [9-11] and other invertebrates [12]; however, these methods have been very rarely used in plants [13, 14].
It should be mentioned that plant AMP repertoire is extremely divergent and species-specific [13-15]. Some of the discovered AMPs are of great interest for fundamental investigations and practical applications [13, 14] since they are highly efficient and possess unique structural features and mode of action [16-19]. Most plant AMPs are cysteine-rich peptides that can be classified into families according to the so-called cysteine motifs (the arrangement of cysteine residues in the polypeptide chain) [20]. Only cysteine motif itself is relatively conserved, while other parts of the peptide amino acid sequence could display very low degree of homology between the family members. Morevover, the similarity between AMP families is completely absent. Hence, it is evident that prediction of novel peptides based on homology search only (e.g., using BLAST or other sequence alignment programs) is possible yet not comprehensive for AMPs belonging to known families and can hardly be applied to the discovery of novel AMP families.
The development of reliable algorithms for prediction of such potential AMPs is vitally important task considering the growing volume of transcriptomic data available for an increasing number of plants [21]. The most effective strategy seems to be a bioinformatics-based approach that uses known structural motifs and is able to reveal all possible new motifs. However, this task is particularly difficult for plants since their AMPs, unlike animal AMPs, usually possess “more flexible” motifs with a variable number of amino acid residues between cysteines.
In this paper, we present a novel pipeline that can be used for plant AMP identification in transcriptomic data and provide a case study comparing the performance of various algorithms in the identification of AMPs from wild-growing and domestic plants, including the model species Arabidopsis thaliana (Thale cress).
MATERIALS AND METHODS
General description of AMPs. A brief description of AMP families and their cysteine motifs is given below in Table 1. We compared our pipeline to other available programs based on this classification, namely, on the number of AMPs that the programs assign to various families.
Table 1. Description and cysteine motifs of
main AMP families
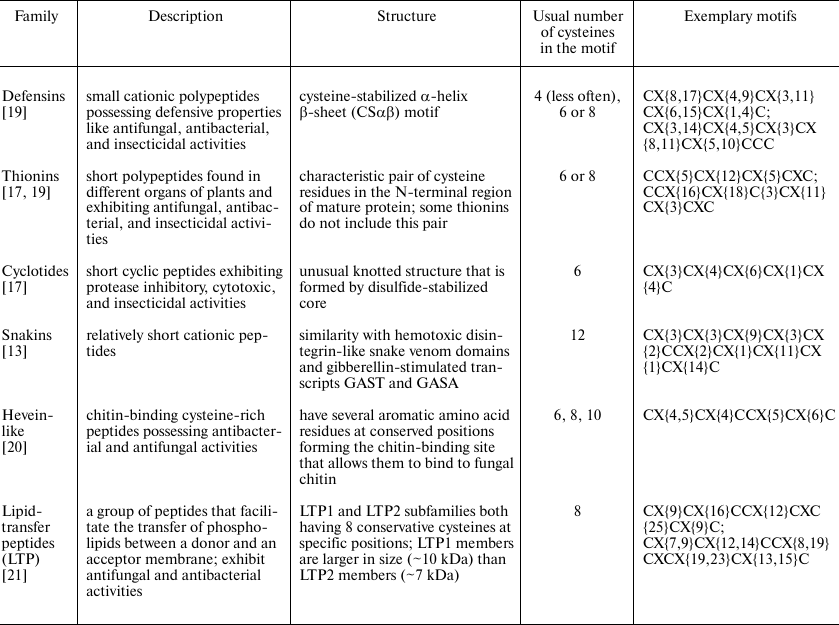
Each AMP family has its own cysteine motif denoted as: CX{i1, j1}CX{i2, j2}… X{iN–1, jN–1}C, where C is cysteine residue; X is any residue except cysteine; numbers in curly braces indicate the range for the number of variable residues present (ik < jk, k = 1 … (N – 1)); and N is the total number of cysteines in the motif.
That is, e.g., both sequences, C{ESQSHRFKGT}C{RSKWL}C{AML}C{MTEGFPGVA}C{RGF}C and C{WGTFKREW}C{ISLTRDRS}C{STWDV}C{KFGEGH}C{K}C, possess the motif CX{8,17}CX{4,9}CX{3,11}CX{6,15}CX{1,4}C, although they do not have significant similarity with each other.
Main pipeline. Earlier [13, 14], we have developed software for searching potential AMPs in transcriptomic data. Although these algorithms and programs allowed us to make reliable predictions, they were rather slow and too organism-specific to be used in comparative analysis of many transcriptomes simultaneously. To solve this problem, we have combined our software modules into single computational pipeline that allows seamless integration of third-party tools and is fast enough to get AMP prediction for 10 plant transcriptomes in less than 1 h on an average desktop machine. In addition, we have revised and extended the list of AMP motifs based on the current data.
In our pipeline, the search for AMPs is based on detection of several cysteine residues arranged in a rather strict order (motif) in amino acid sequences obtained by translation of original transcriptome sequencing data. As follows from the Table 1, the members of AMP families possess cysteine motifs specific for their families, and such motifs can be used in searching and classification of AMPs, e.g., in newly sequenced transcriptomes.
The flow charts of our algorithm (Cysmotif searcher) and the third-party SPADA software are shown in Figs. 1a and 1b, respectively.
Fig. 1. Comparison of Cysmotif searcher algorithm (a) and third-party SPADA software (b).
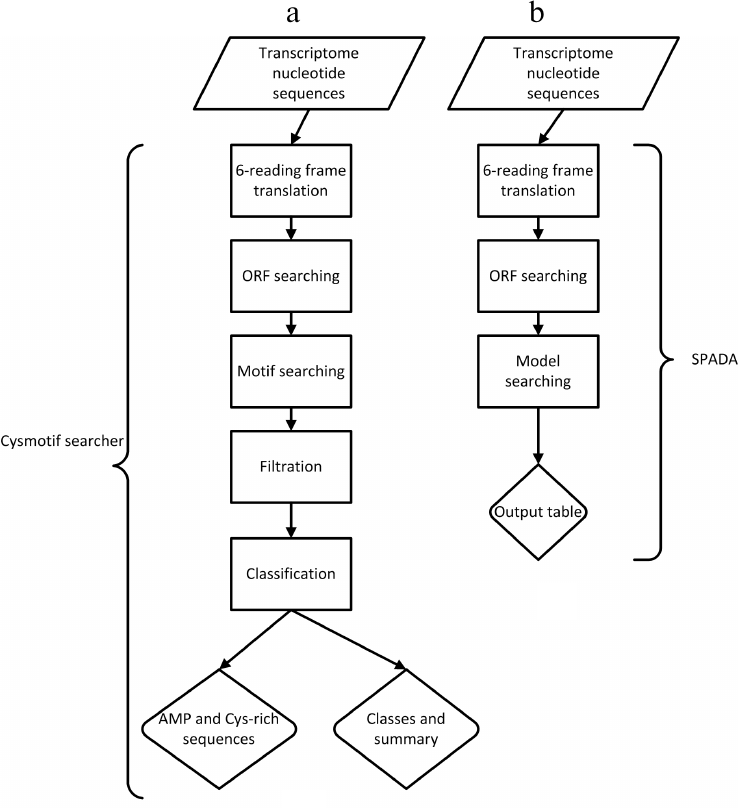
The first step of the pipeline includes 6-reading frame translation of the input transcripts and detection of open reading frames (ORFs) starting from methionine residues. This gives us additional evidence that such sequence could really be a translated protein sequence. The transcripts are not subjected to any filtration process, e.g., based on the available homology-based annotation or experimentally obtained data. This allows to avoid algorithm overfitting and relying on untested third-party data. In addition, as it was mentioned above, the use of homology-based data does not allow to identify novel AMP families.
Then, the software searches for 138 cysteine motifs in the amino acid sequences obtained. The motifs were obtained from literature data (e.g., [22]) or derived empirically by us based on the previous experience of AMP searching [13, 14].
After that, the pipeline checks for the presence of signal peptide in a sequence using the SignalP program [25]. In addition, the sequences are filtered based on their length (≤150 amino acids in mature peptide). All these criteria are mandatory for AMPs according to the currently available data. Output sequences are divided into families (defensins, thionins, etc.; see Table 1) based on their corresponding motifs. In addition, we filtered out the sequences that had passed all the criteria, but had additional cysteines not belonging to the motif revealed in their mature peptide. Such sequences were assigned to a special artificial family called “cysteine-rich peptides” or CYSRICH in short.
The sequences that have passed all the steps described above are output to FASTA files with an indication of the family they have been assigned to. The output also includes tabular data showing sequence, motif, and family for each discovered AMP and a summary text file containing general statistics regarding the motifs revealed. The pipeline also allows integration of a third-party software called SPADA.
SPADA. SPADA [26] stands for “small peptide alignment discovery application”. It represents a computational pipeline including, in turn, various third-party programs (to be downloaded separately), that can identify members of a protein family in a target sequence based on multiple sequence alignment for this gene or protein family. SPADA includes pre-built manually curated protein sequence alignments for all cysteine-rich peptide families in plant genomes (mostly based on A. thaliana and Oryza sativa genomes). The user can select which programs to use in a pipeline by changing the configuration file.
SPADA is a homology-based gene annotation program that allows using protein or gene family profiles instead of individual sequences to perform similarity searches. The quality of results is further increased by providing automated access to third-party annotation tools such as, among others, similarity search tool HMMER [27], the ab initio gene predictor Augustus [28], and SignalP program [25] that predicts the presence and location of signal peptide cleavage sites, etc. In brief, SPADA performs 6-reading frame translation of the input nucleotide sequence and ORF search, and then applies its built-in models to ORF sequences revealed.
However, the sequences of found peptides are not presented in well-known formats like FASTA in the output data, which complicates further processing. The main disadvantage of SPADA is that it is not suitable for discovering new protein families (e.g., AMPs) due to inherent searching strategies. In addition, it does not provide classification of the found peptides, making further analysis (e.g., using BLAST) necessary.
When SPADA is called within our pipeline, its results are converted to FASTA and compared to the output data from the main AMP motif searching algorithms (Fig. 2). Results are combined, and only unique sequences are included in joint output files. Since SPADA does not provide any classification data, its results are not assigned to protein families and provided as they are. It should be noted that the sequences revealed only by SPADA do not contain our cysteine motifs or do not meet other criteria, or both, since otherwise our algorithm would have revealed them.
Fig. 2. Main pipeline workflow.
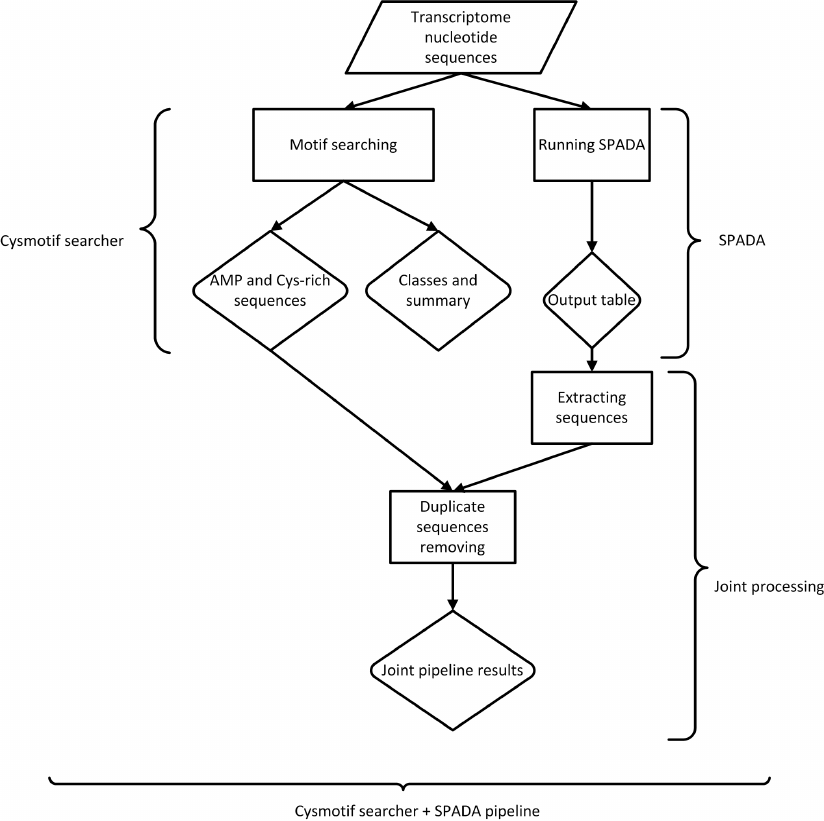
Thus, the output of our pipeline can include the results of both our motif-based algorithm and SPADA, in which non-unique sequences are excluded.
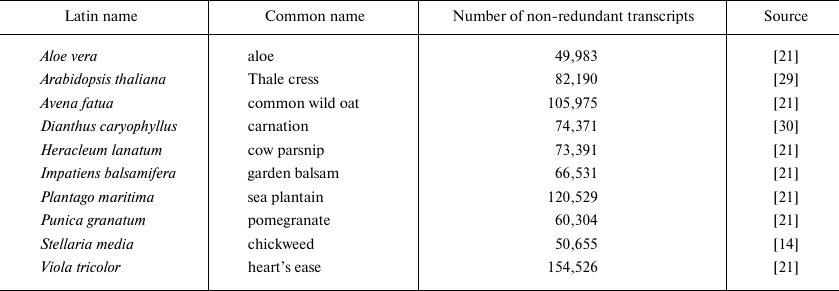
Plant transcriptomes. Ten plant transcriptomes were used for evaluating the performance of our pipeline. Some of the represented species were domestic plants, and the other were wild-growing plants, since the latter are expected to have more diverse AMP repertoire [13, 14]. The well-known A. thaliana was used as a model plant. Seven out of ten transcriptomes were taken from the 1000 Plants (1KP) project [21]. Complete genome sequences are currently available only for A. thaliana and Dianthus caryophyllus. The description of transcriptomes is given below in Table 2.
Table 2. Plant transcriptomes used for
evaluating the pipeline performance
Pipeline testing and comparison. To test our pipeline and to demonstrate its advantages, we have run two versions of it (with and without SPADA) on 10 plant transcriptomes described above. We also ran SPADA alone and CS-AMPPred [31] program on the same dataset to compare the performance. These programs use clearly defined criteria for identification of AMPs (or cysteine-rich peptides) and do not require additional data for training their algorithms, so they allow to perform a fruitful comparison. We have chosen for comparison only the programs that allow complete offline analysis of entire newly sequenced transcriptome at once. Apparently, existing databases (e.g., PhytAMP [32]) or web-servers (e.g., CAMP [33]) do not satisfy this requirement, so they and other similar programs were not included in the comparison.
CS-AMPPred [31] stands for “cysteine-stabilized antimicrobial peptides predictor”. This software uses the support vector machine (SVM) model for antimicrobial activity prediction in cysteine-stabilized peptides. The properties included in the model consist of indexes of α-helix and loop formation, as well as average values of peptide net charge, hydrophobicity, and flexibility. The model was derived based on 310 cysteine-stabilized AMPs.
The programs were executed using their default parameters. However, since all of them use some pre-calculated data like peptide models, family alignments, and sequence motifs, we can say that they are slightly biased towards the discovery of peptides that have been used to “train” them. In addition, it is evident that AMP discovery heavily depends on RNA sequencing quality. Hence, we can expect that many more potential AMPs could be revealed in such well-studied and extensively sequenced organism as A. thaliana. In addition, SPADA program has a set of pre-calculated alignments based on this very plant.
We performed BLAST annotation using NCBI “nr” database for all peptides predicted by the pipeline and other programs involved in comparison to validate the assignment of revealed AMPs to specific families.
RESULTS
The total running time on Linux machine with Intel(R) Xeon(R) CPU E7-8880 v3 2.30 GHz processor using one thread is shown in the Table 3. Note that CS-AMPPred requires amino acid sequences as input, while SPADA and Cysmotif searcher can perform translation themselves. Data are presented only for three exemplary plant transcriptomes to not obscure the explanation.
Table 3. Total running time (in minutes) for
the investigated programs for three plant transcriptomes
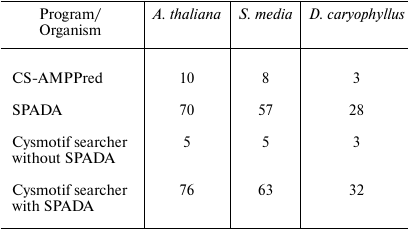
Brief results and family classification are presented in Tables 4 and 5. The comparison includes the results obtained by SPADA alone, Cysmotif searcher pipeline alone (without SPADA), and Cysmotif searcher pipeline within which SPADA is called (in this case results are combined and processed to remove sequence duplication). Since CS-AMPPred demonstrated very low specificity, as will be discussed below, its results were obtained by running it on a set of sequences revealed by SPADA and Cysmotif searcher.
Table 4. The number of unique potential
AMPs, including cysteine-rich peptides, discovered by the investigated
programs in 10 plant transcriptomes
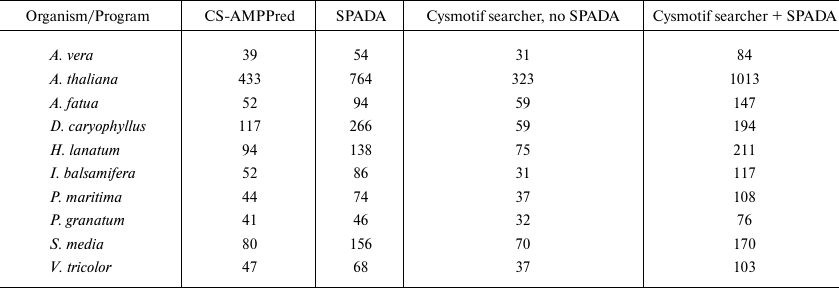
Table 5. Distribution of potential AMPs
among peptide families
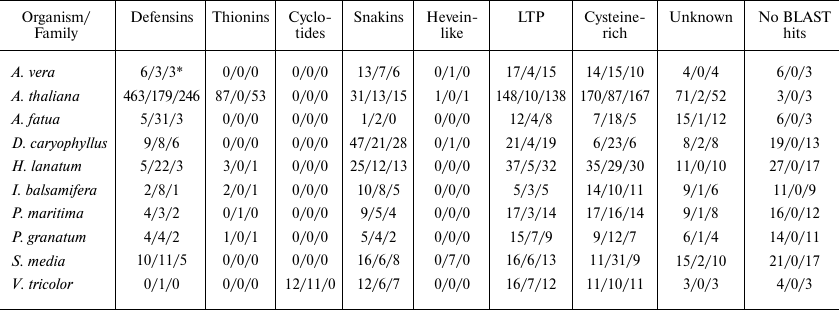
* N1/N2/N3 represents results for the number of peptides revealed by
BLAST annotation only (without using our pipeline)/Cysmotif searcher
classification/SPADA + BLAST, respectively.
In order to additionally verify the consistency of pipeline performance and to exclude false positives, motif searching was conducted in the amino acid sequences built with a random number generator. ORFs obtained for A. thaliana were taken as initial data, since the largest number of motifs has been revealed in them. Each of the translated ORF sequences was randomly shuffled (keeping its amino acid composition) and then subjected to motif searching. Two motifs corresponding to defensins and five cysteine-rich peptides were revealed, which makes less than 2% of the corresponding numbers for real sequences. Such a great specificity was achieved due to strict result filtration and substitution of some “fuzzy” motifs with more rigorous ones during previous practical application of our pipeline.
Table 5 shows classification of cysteine-rich peptides revealed in plant transcriptomes by BLAST (i.e., without the use of out pipeline) (first value in each cell), revealed by SPADA and then annotated by BLAST (second value), and found by Cysmotif searcher (based on its own motif classification) (third value).
For example, we can see that our pipeline allows to annotate up to 80% of known cysteine-rich AMP of A. thaliana, while providing high specificity, that is, virtually complete exclusion of false positive results.
Note that Cysmotif searcher’s classification is compared to BLAST annotation, while only the latter is given for SPADA since it does not assign peptides to classes or families.
DISCUSSION
The first thing that should me mentioned is dramatically low specificity of CS-AMPPred. When executed on a set of all translated sequences from a transcriptome, it assigns up to 90% transcripts or ORFs from these transcripts to AMPs, which obviously represent false positives. Therefore, to make comparison sensible, we ran CS-AMPPred again on the results of Cysmotif searcher and SPADA.
Second, we see that the unprocessed results of SPADA alone and Cysmotif searcher without SPADA complement each other, that is, they just slightly overlap (Tables 4 and 5).
Third, in view of the “training” mentioned above, it is not surprising that all programs revealed at least 4 times more AMPs in the transcriptome of A. thaliana (Tables 4 and 5) than in other plants. This does not make sense from a biological point of view, since both Thale cress and, for example, chickweed are wild-growing plants (namely, weeds) and not expected to have significant differences in their AMP repertoires. As we revealed in our earlier studies (data not presented), the same is true for other well-sequenced organisms like rice (O. sativa). Therefore, such a large number of AMPs is likely to be a consequence of better transcriptome sequencing for model plants and large amount of data available for building AMP models.
However, the most interesting part is comparison of the number of predicted peptides assigned to various families. As described above, Cysmotif searcher was developed specifically for revealing AMPs possessing rather rigid motifs and low amino acid sequence similarity. Thus, it is not a surprise that Cysmotif searcher usually predicts, in particular, defensins in some poorly annotated transcriptomes better than SPADA or BLAST (e.g., 22/3/5 defensins, respectively, for H. lanatum, and 31/3/5 defensins, respectively, for A. fatua). Some of the sequences that were assigned to defensins by Cysmotif searcher were also revealed by BLAST, but they were annotated only generally as cysteine-rich or unknown proteins. The same holds for snakins, that were sometimes annotated by BLAST as gibberellin-regulated proteins (snakins represent a sub-class of these proteins). At the same time, SPADA found more lipid transfer proteins (LTPs) (e.g., 138/10 for A. thaliana). These proteins have less stringent motifs and higher sequence similarity than, e.g., defensins, so this is a consequence of increased specificity in Cysmotif searcher. This is also true for the thionin family. In addition, SPADA and Cysmotif searcher annotated rather large number of peptides that did not have BLAST hits or were annotated as unknown peptides (e.g., 73 for A. thaliana and 25 for P. maritima), which confirms the pipeline’s ability to annotate new cysteine-rich peptides.
Therefore, we can see that SPADA and Cysmotif searcher complement each other by making better predictions for different protein families. However, SPADA does not classify the peptides it reveals and does not present the results in FASTA or similar format, which complicates further investigations for a researcher with no programming background. When SPADA is executed within Cysmotif searcher pipeline, its results are given separately in a FASTA file with the found peptides while keeping all its usual output files unchanged. The final output files contain a combination of unique sequences revealed by either Cysmotif searcher or SPADA programs. It is useful because SPADA does not involve filtering of identical sequences obtained from different ORFs. Thus, a user is able to easily choose the results suitable for specific investigations, e.g., only SPADA results can be taken, or only the results for one peptide family like defensins. In addition, Cysmotif searcher is much faster than SPADA, so it can be used for fast screening of a transcriptome to check if additional processing and/or investigations are needed. In the latter case, SPADA is not called within the pipeline.
Here, we propose the new Cysmotif searcher pipeline for fast screening of one or more plant transcriptomes to predict AMP repertoire. Cysmotif searcher performs translation of the input transcriptomic data and selects short sequences containing ORF and signal peptide cleavage site. Then, the search for potential AMPs can be conducted using only the algorithm developed by us, in which case all the sequences possessing family-specific cysteine motifs will be revealed and classified. Also, Cysmotif searcher allows seamless integration of the third-party software SPADA that uses special models to reveal cysteine-rich peptides, into its workflow. Therefore, Cysmotif searcher without SPADA seems to be the most suitable tool for fast screening of potential AMPs, while joint output of these two programs should be considered when looking for full repertoire of cysteine-rich peptides.
For the 10 plants selected, we have shown that Cysmotif searcher reveals large number of predicted AMPs both alone and with SPADA module. Both SPADA and Cysmotif searcher made rather good annotation of potential AMPs, which was confirmed by BLAST search in “nr” database, and both of these algorithms have given much better results for the well-sequenced transcriptome of A. thaliana than for other plants.
In addition, our pipeline allows to draw attention to some cysteine-rich peptides, whose motifs differ from the “classical” motifs of the known families and that could therefore contain the sequences representing some novel AMP family. All the output data of our pipeline are given as files and tables that can be easily inspected visually.
In view of the data shown above, and since Cysmotif searcher allows to easily integrate SPADA into its pipeline and to present results in a more user-friendly format, we believe that it is a very useful tool for initial AMP screening in the transcriptomes of plants and other eukaryotes. However, to extend the field of its application, more specific motifs are required. Those can be obtained when more AMP sequences become available. Adding new motifs to the pipeline is as easy as adding new line to a text file, therefore, our pipeline could be upgraded without substantial efforts.
Funding
This work was supported by the Russian Science Foundation (project 16-16-00032).
Conflict of Interests
Authors declare no conflict of interests.
REFERENCES
1.Bahar, A. A., and Ren, D. (2013) Antimicrobial
peptides, Pharmaceuticals (Basel), 6, 1543-1575.
2.Pasupuleti, M., Schmidtchen, A., and Malmsten, M.
(2012) Antimicrobial peptides: key components of the innate immune
system, Crit. Rev. Biotechnol., 32, 143-171.
3.Pushpanathan, M., Gunasekaran, P., and Rajendhran,
J. (2013) Antimicrobial peptides: versatile biological properties,
Int. J. Pept., 2013, 675391.
4.Maccari, G., Di Luca, M., Nifosi, R., Cardarelli,
F., Signore, G., Boccardi, C., and Bifone, A. (2013) Antimicrobial
peptides design by evolutionary multiobjective optimization, PLoS
Comput. Biol., 9, e1003212.
5.Jochumsen, N., Marvig, R. L., Damkiaer, S., Jensen,
R. L., Paulander, W., Molin, S., Jelsbak, L., and Folkesson, A. (2016)
The evolution of antimicrobial peptide resistance in Pseudomonas
aeruginosa is shaped by strong epistatic interactions, Nat.
Commun., 7, 13002.
6.Pasupuleti, M. (2009) Structural, Functional and
Evolutionary Studies of Antimicrobial Peptides: Doctoral
Dissertation, Department of Clinical Sciences, Lund University.
7.Tennessen, J. A. (2005) Molecular evolution of
animal antimicrobial peptides: widespread moderate positive selection,
J. Evol. Biol., 18, 1387-1394.
8.Hiemstra, P., and Zaat, S. (eds.) (2013)
Antimicrobial Peptides and Innate Immunity (part of the
Progress in Inflammation Research book series), Springer,
Basel.
9.Wang, M., and Hu, X. (2017) Antimicrobial peptide
repertoire of Thitarodes armoricanus, a host species of
Ophiocordyceps sinensis, predicted based on de novo
transcriptome sequencing and analysis, Infect. Genet. Evol.,
54, 238-244.
10.Kim, I. W., Markkandan, K., Lee, J. H.,
Subramaniyam, S., Yoo, S., Park, J., and Hwang, J. S. (2016)
Transcriptome profiling and in silico analysis of the
antimicrobial peptides of the grasshopper Oxya chinensis
sinuosa, J. Microbiol. Biotechnol., 26,
1863-1870.
11.Gupta, S. K., Kupper, M., Ratzka, C., Feldhaar,
H., Vilcinskas, A., Gross, R., Dandekar, T., and Forster, F. (2015)
Scrutinizing the immune defence inventory of Camponotus
floridanus applying total transcriptome sequencing, BMC
Genomics, 16, 540.
12.Pujol, N., Zugasti, O., Wong, D., Couillault, C.,
Kurz, C. L., Schulenburg, H., and Ewbank, J. J. (2008) Anti-fungal
innate immunity in C. elegans is enhanced by evolutionary
diversification of antimicrobial peptides, PLoS Pathog.,
4, e1000105.
13.Slavokhotova, A. A., Shelenkov, A. A., and
Odintsova, T. I. (2015) Prediction of Leymus arenarius (L.)
antimicrobial peptides based on de novo transcriptome assembly,
Plant. Mol. Biol., 89, 203-214.
14.Slavokhotova, A. A., Shelenkov, A. A.,
Korostyleva, T. V., Rogozhin, E. A., Melnikova, N. V., Kudryavtseva, A.
V., and Odintsova, T. I. (2017) Defense peptide repertoire of
Stellaria media predicted by high throughput next generation
sequencing, Biochimie, 135, 15-27.
15.Utkina, L. L., Andreev, Y. A., Rogozhin, E. A.,
Korostyleva, T. V., Slavokhotova, A. A., Oparin, P. B., Vassilevski, A.
A., Grishin, E. V., Egorov, T. A., and Odintsova, T. I. (2013) Genes
encoding 4-Cys antimicrobial peptides in wheat Triticum kiharae
Dorof. et Migush.: multimodular structural organization, instraspecific
variability, distribution and role in defence, FEBS J.,
280, 3594-3608.
16.Fujimura, M., Minami, Y., Watanabe, K., and
Tadera, K. (2003) Purification, characterization, and sequencing of a
novel type of antimicrobial peptides, Fa-AMP1 and Fa-AMP2, from seeds
of buckwheat (Fagopyrum esculentum Moench.), Biosci.
Biotechnol. Biochem., 67, 1636-1642.
17.Rogozhin, E. A., Slezina, M. P., Slavokhotova, A.
A., Istomina, E. A., Korostyleva, T. V., Smirnov, A. N., Grishin, E.
V., Egorov, T. A., and Odintsova, T. I. (2015) A novel antifungal
peptide from leaves of the weed Stellaria media L,
Biochimie, 116, 125-132.
18.Astafieva, A. A., Enyenihi, A. A., Rogozhin, E.
A., Kozlov, S. A., Grishin, E. V., Odintsova, T. I., Zubarev, R. A.,
and Egorov, T. A. (2015) Novel proline-hydroxyproline glycopeptides
from the dandelion (Taraxacum officinale Wigg.) flowers: de
novo sequencing and biological activity, Plant Sci.,
238, 323-329.
19.Slavokhotova, A. A., Naumann, T. A., Price, N.
P., Rogozhin, E. A., Andreev, Y. A., Vassilevski, A. A., and Odintsova,
T. I. (2014) Novel mode of action of plant defense peptides –
hevein-like antimicrobial peptides from wheat inhibit fungal
metalloproteases, FEBS J., 281, 4754-4764.
20.Tam, J. P., Wang, S., Wong, K. H., and Tan, W. L.
(2015) Antimicrobial peptides from plants, Pharmaceuticals
(Basel), 8, 711-757.
21.Matasci, N., Hung, L. H., Yan, Z., Carpenter, E.
J., Wickett, N. J., Mirarab, S., Nguyen, N., Warnow, T., Ayyampalayam,
S., Barker, M., Burleigh, J. G., Gitzendanner, M. A., Wafula, E., Der,
J. P., dePamphilis, C. W., Roure, B., Philippe, H., Ruhfel, B. R.,
Miles, N. W., Graham, S. W., Mathews, S., Surek, B., Melkonian, M.,
Soltis, D. E., Soltis, P. S., Rothfels, C., Pokorny, L., Shaw, J. A.,
DeGironimo, L., Stevenson, D. W., Villarreal, J. C., Chen, T., Kutchan,
T. M., Rolf, M., Baucom, R. S., Deyholos, M. K., Samudrala, R., Tian,
Z., Wu, X., Sun, X., Zhang, Y., Wang, J., Leebens-Mack, J., and Wong,
G. K. (2014) Data access for the 1000 Plants (1KP) project,
Gigascience, 3, 17.
22.Silverstein, K. A., Moskal, W. A., Jr., Wu, H.
C., Underwood, B. A., Graham, M. A., Town, C. D., and VandenBosch, K.
A. (2007) Small cysteine-rich peptides resembling antimicrobial
peptides have been under-predicted in plants, Plant J.,
51, 262-280.
23.Slavokhotova, A. A., Shelenkov, A. A., Andreev,
Y. A., and Odintsova, T. I. (2017) Hevein-like antimicrobial peptides
of plants, Biochemistry (Moscow), 82, 1659-1674.
24.Kader, J. C. (1996) Lipid-transfer proteins in
plants, Annu. Rev. Plant Physiol. Plant Mol. Biol., 47,
627-654.
25.Nielsen, H. (2017) Predicting secretory proteins
with SignalP, Methods Mol. Biol., 1611, 59-73.
26.Zhou, P., Silverstein, K. A., Gao, L., Walton, J.
D., Nallu, S., Guhlin, J., and Young, N. D. (2013) Detecting small
plant peptides using SPADA (Small Peptide Alignment Discovery
Application), BMC Bioinformatics, 14, 335.
27.Eddy, S. R. (1998) Profile hidden Markov models,
Bioinformatics, 14, 755-763.
28.Keller, O., Kollmar, M., Stanke, M., and Waack,
S. (2011) A novel hybrid gene prediction method employing protein
multiple sequence alignments, Bioinformatics, 27,
757-763.
29.Zhang, R., Calixto, C. P. G., Marquez, Y.,
Venhuizen, P., Tzioutziou, N. A., Guo, W., Spensley, M., Entizne, J.
C., Lewandowska, D., Ten Have, S., Frei Dit Frey, N., Hirt, H., James,
A. B., Nimmo, H. G., Barta, A., Kalyna, M., and Brown, J. W. S. (2017)
A high quality Arabidopsis transcriptome for accurate
transcript-level analysis of alternative splicing, Nucleic Acids
Res., 45, 5061-5073.
30.Tanase, K., Nishitani, C., Hirakawa, H., Isobe,
S., Tabata, S., Ohmiya, A., and Onozaki, T. (2012) Transcriptome
analysis of carnation (Dianthus caryophyllus L.) based on
next-generation sequencing technology, BMC Genomics, 13,
292.
31.Porto, W. F., Pires, A. S., and Franco, O. L.
(2012) CS-AMPPred: an updated SVM model for antimicrobial activity
prediction in cysteine-stabilized peptides, PLoS One, 7,
e51444.
32.Hammami, R., Ben Hamida, J., Vergoten, G., and
Fliss, I. (2009) PhytAMP: a database dedicated to antimicrobial plant
peptides, Nucleic Acids Res., 37, D963-968.
33.Waghu, F. H., Barai, R. S., Gurung, P., and
Idicula-Thomas, S. (2016) CAMPR3: a database on sequences, structures
and signatures of antimicrobial peptides, Nucleic Acids Res.,
44, D1094-1097.