Mobile Loop in the Active Site of Metallocarboxypeptidases as an Underestimated Determinant of Substrate Specificity
V. Kh. Akparov1,a*, V. I. Timofeev2,3,b, I. G. Khaliullin4,c, G. E. Konstantinova1, I. P. Kuranova2,3,b, T. V. Rakitina3,5,d, and V. K. Švedas6,e
1State Research Institute for Genetics and Selection of Industrial Microorganisms, 117545 Moscow, Russia2Shubnikov Institute of Crystallography, Crystallography and Photonics Federal Scientific Research Center, Russian Academy of Sciences, 119333 Moscow, Russia
3Kurchatov Institute National Research Center, 123098 Moscow, Russia
4Laboratory of Ion and Molecular Physics, Moscow Institute of Physics and Technology (State University), 141700 Dolgoprudny, Moscow Region, Russia
5Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Laboratory of Hormonal Regulation Proteins, Russian Academy of Sciences, 117997 Moscow, Russia
6Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received June 5, 2018; Revised September 19, 2018; Accepted September 19, 2018
It is generally accepted that the primary specificity of metallocarboxypeptidases is mainly determined by the structure of the so-called primary specificity pocket. However, the G215S/A251G/T257A/D260G/T262D mutant of carboxypeptidase T from Thermoactinomyces vulgaris (CPT) with the primary specificity pocket fully reproducing the one in pancreatic carboxypeptidase B (CPB) retained the broad, mainly hydrophobic substrate specificity of the wild-type enzyme. In order to elucidate factors affecting substrate specificity of metallocarboxypeptidases and the reasons for the discrepancy with the established views, we have solved the structure of the complex of the CPT G215S/A251G/T257A/D260G/T262D mutant with the transition state analogue N-sulfamoyl-L-phenylalanine at a resolution of 1.35 Å and compared it with the structure of similar complex formed by CPB. The comparative study revealed a previously underestimated structural determinant of the substrate specificity of metallocarboxypeptidases and showed that even if substitution of five amino acid residues in the primary specificity pocket results in its almost complete structural correspondence to the analogous pocket in CPB, this does not lead to fundamental changes in the substrate specificity of the mutant enzyme due to the differences in the structure of the mobile loop located at the active site entrance that affects the substrate-induced conformational rearrangements of the active site.
KEY WORDS: metallocarboxypeptidase T from Thermoactinomyces vulgaris, metallocarboxypeptidase B, S1′-subsite, substrate selectivity, N-sulfamoyl-L-phenylalanine, X-ray analysisDOI: 10.1134/S0006297918120167
Abbreviations: BSA, buried surface area; CPB, carboxypeptidase B from porcine pancreas; CPT, carboxypeptidase T from Thermoactinomyces vulgaris; CPT5, CPT mutant with A243G, T250A, A253G, D260G, T262D substitutions; CPTwt, wild-type carboxypeptidase T; CPU, carboxypeptidase U; CPO, carboxypeptidase O; SPhe, N-sulfamoyl-L-phenylalanine; ZAAL, N-benzyloxycarbonyl-L-alanyl-L-alanyl-L-leucine.
Human genome contains almost 600 genes encoding proteases, including
approximately 150 genes for zinc-dependent metalloenzymes essential for
various physiological and pathological processes, such as immune
response, inflammation, cell death, proliferation, physiological
homeostasis, oncological changes, cardiovascular and neurodegenerative
pathologies, and infection diseases [1-3]. Metallocarboxypeptidases that cleave off
C-terminal amino acids are well known exopeptidases that, as has
been thought previously, are mostly involved in catabolic processes
and, in the case of carboxypeptidases A and B, digestion. However,
analysis of genomic data and biochemical studies revealed that the
involvement of metallocarboxypeptidases in metabolism is significantly
broader [4, 5]. The discovery
of new functional roles of these enzymes in living systems has
attracted considerable interest in the fine mechanisms of catalysis,
substrate specificity, and regulation of catalytic activity of
metallocarboxypeptidases.
Carboxypeptidase T from Thermoactinomyces vulgaris (CPT, EC 3.4.17.18) belongs to zinc-dependent microbial carboxypeptidases. It displays distant homology to pancreatic carboxypeptidases A and B (CPA and CPB) having fewer than 30% identical residues [6]. The polypeptide chain of mature CPT consists of 326 residues. The folding pattern of CPT is similar to that of mammalian carboxypeptidase and typical for the α/β-domain hydrolases. The CPT molecule consists of β-sheets formed by eight β-strands surrounded by six α-helices [7].
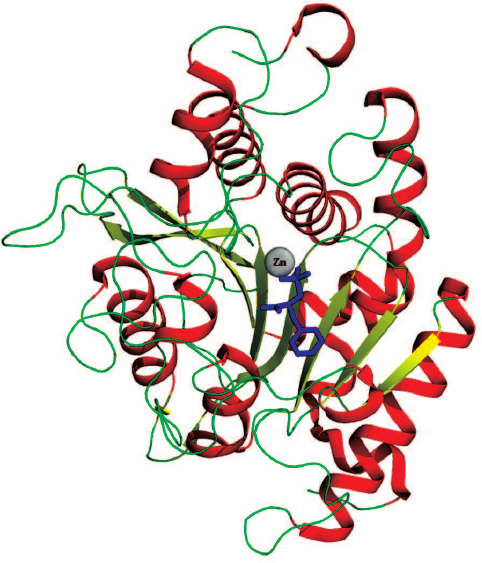
The CPT active site is located in the central part of the β-sheets (Fig. 1) and contains the catalytic residues and the binding sites for the side groups of the amino acid substrates. The binding site for the C-terminal carboxyl group and the side group of the cleaved amino acid is commonly known as the primary specificity pocket, or the S1′ subsite according to the nomenclature of Schechter and Berger [8]. By the beginning of 2000s, the notion has been formed based on the studies reported by the Vallee [9], Lipscomb [10], Gardell [11], Stepanov [12], and Osterman [7, 13] research groups that the substrate specificity of metallocarboxypeptidases toward the cleaved amino acid is determined by the nature of seven residues forming the S1′ subsite (numeration is given for CPB/CPT): Leu203/Leu211, Ser207/Gly215, Gly243/Ala251, Ala250/Thr257, Gly253/Asp260, Asp255/Thr262, and Thr268/275, with the major role played by the residue at the bottom of the primary specificity pocket [11, 13, 14]. This position in CPA is occupied by Ile255 with the hydrophobic side chain; hence, CPA cleaves hydrophobic C-terminal amino acids. CPB and carboxypeptidase U (CPU) contain aspartic acid at position 255 [15], and these enzymes hydrolyze with high selectivity substrates containing C-terminal residues with positively charged side radicals. In carboxypeptidase O (CPO), position 255 is occupied by arginine [16], and this enzyme cleaves negatively charged C-terminal residues.
Fig. 1. 3D structure of CPT5. SPhe is shown as a skeletal formula, zinc ion is shown as a sphere.
Substitution of Asp255 with arginine in CPB results in the generation of the CPO-like enzyme [17], which finally validated the hypothesis on the essential role of this residue in the substrate recognition by metallocarboxypeptidases. Nevertheless, the studies of a large number of metallocarboxypeptidases revealed other factors affecting the primary specificity of this family of enzymes. For example, the spectrum of cleaved substrates depends on the nature of residue at the position adjacent to 255. Thus, the wild-type CPT (CPTwt) with the aspartic acid residue at position 260 (253 in CPA) is capable of cleaving both hydrophobic and positively charged C-terminal residues, although with a 10-fold lower efficiency [12]. Not only structural elements in direct contact with the cleaved residue participate in the substrate recognition by metallocarboxypeptidase, but also remote fragments of the enzyme. For example, earlier we found that structural calcium ions affect the substrate specificity of CPT [18].
The primary specificity pocket in CPTwt is spatially similar to those in CPB and CPA [7] and differs from the primary specificity pocket of CPB by five substitutions. It was suggested that introduction of G215S, A251G, T257A, D260G, and T262D mutations to reconstruct the CPB primary specificity pocket in CPT should convert the hydrophobicity-selective CPT into the CPB-like enzyme or, at least, increase its ability to cleave positively charged residues at the C-terminus of the substrate. This was not the case, and CPT5 containing the reconstructed S1′ subsite of CPB not only preserved the specificity towards hydrophobic substrates, but also demonstrated a decreased ability to transform substrates with positively charged C-terminal residues [19]. These results indicated that the specificity of metallocarboxypeptidases toward the cleaved amino acids is determined not only by the residue complementarity to the S1′ subsite, but also by other structural factors that might be even more influential.
High-resolution three-dimensional structure of the complex of the G215S/A251G/T257A/D260G/T262D CPT mutant (CPT5) with the transition state analogue N-sulfamoyl-L-phenylalanine (SPhe) was solved with the objective to find structural determinants essential for the metallocarboxypeptidase substrate specificity, as well as to reveal the reasons for the discrepancies with the commonly accepted notion on the role of amino acid residues in the primary specificity pocket of this enzyme family by comparing the generated structure with the structures of similar complexes formed by CPB and CPA. This comparison allowed identification of previously underestimated determinant of the substrate specificity of metallocarboxypeptidases.
MATERIALS AND METHODS
Materials. Glass capillaries from Confocal Science Inc. (Japan) (length, 60 mm; inner diameter, 0.5 mm) were used for CPT crystallization. Deionized water from a MilliQ water purification system (Millipore, USA) with a resistivity of 19 MOhm-cm, YM10 ultrafiltration membranes and ultrafiltration devices from Amicon (USA), and Centripack microfiltration cartridges (Millipore) were used. 2-Methyl-2,4-pentanediol (MPD), L-cystine and L-cysteine, diisopropyl fluorophosphate, isopropyl-β-d-thiogalactopyranoside (IPTG), 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and 4-morpholinoethanesulfonic acid monohydrate (MES) were purchased from Sigma (USA). Subtilisin 72 was isolated from Protosubtilin G10x manufactured by the Ladyzhensky Plant of Biological and Enzymatic Preparations (Ukraine) according to the technique described in [20].
Chromogenic substrates carbobenzoxy-L-alanyl-L-alanyl-L-leucine p-nitroanilide (ZAALpNA) for subtilisin activity assay and carbobenzoxy-L-alanyl-L-alanyl-L-leucine (ZAAL) for CPT activity assay were synthesized at the Laboratory of Protein Chemistry, State Research Institute of Genetics [21]. [N-(ε-aminocaproyl)-p-aminobenzyl]succinyl-Sepharose 4B (CABS-Sepharose) was synthesized as described in [22]. SPhe was acquired from the Solyaris Innovation Research Center (Russia).
Mutant variant of the pro-cpT5 gene was obtained from Evrogen (Russia); the presence of mutations was confirmed by sequencing.
Purification of recombinant CPT. Expression of the pro-cpT5 gene was carried out Escherichia coli BL21(DE3) pLysS cells (Novagen, USA) according to the manufacturer’s instructions [23]. The cells were disrupted by sonication after induction of the protein expression with IPTG.
proCPT5 was purified from inclusion bodies as described previously [24]. To obtained mature enzyme, subtilisin 72 was added to the proCPT5 solution at a CPT5/subtilisin ratio of 200 : 1 (w/w), and the mixture was incubated for 4 h at 37°C. Subtilisin was then inactivated by adding diisopropyl fluorophosphate. Activated CPT5 was further concentrated by ultrafiltration to 2 ml and centrifuged to remove the precipitate.
Following concentration, the pH of the enzyme solution was adjusted to pH 6.0 by adding 100 mM MES/NaOH (pH 5.8), and the solution was applied onto a CABS-Sepharose column (column volume, 20 ml) equilibrated with 10 mM MES/NaOH buffer, pH 6.0, containing 0.5 M NaCl, 10 mM CaCl2, and 0.1 mM ZnSO4. The column was washed with the same buffer, and CPT5 was eluted with 10 mM Tris-HCl buffer, pH 9.0. Fractions containing the active protein were combined and concentrated to 1 ml. Next, the buffer was exchanged for the crystallization buffer (0.01 M MES/NaOH, pH 6.0, containing 1 mM CaCl2, 0.1 mM ZnSO4, and 0.25 M NaCl) by triple 10-fold dilution/concentration in an Amicon device followed by ultrafiltration to the protein concentration of 10 mg/ml according to Bradford [25]. The resulting solution was cold-sterilized by filtering in Centripack centrifuge cartridges and used for further protein crystallization. The absence of subtilisin activity was confirmed using the specific chromogenic substrate ZAALpNA. SDS-PAGE was carried out according to Laemmli’s protocol [26].
Kinetic studies. Kinetic experiments were conducted at 25°C in a thermostated 1-ml quartz cuvette with a 1-cm optical path. The substrate solution (0.5 ml) in 0.25 M Tris-HCl buffer, pH 7.5, containing 0.01 M CaCl2, was incubated at 25°C for 5 min and the reaction was initiated by adding 2-5 μl of the enzyme solution. The reaction was monitored with a Shimadzu UV-1800 (Japan) spectrophotometer at 225 nm.
Determination of inhibition constants. Integral kinetic curves of the ZAAL hydrolysis catalyzed by CPT5 in the presence and absence of the inhibitor SPhe were analyzed in coordinates:
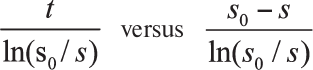 [27],
[27],
where t – time, s and s0 – running and initial substrate concentrations. This allowed the calculation of the apparent (KM/Vmax)app value at each used concentration of the inhibitor (I). To determine the inhibition constant (Ki), the dependence of the calculated (KM/Vmax)app parameter on the inhibitor concentration was examined in accordance with the linear equation:
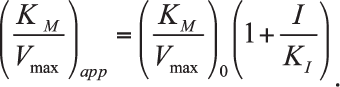
Buried surface area. Calculation of the buried surface area (BSA) of amino acids in the enzyme active site was conducted using the Pisa program (European Institute of Bioinformatics; http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) [28].
Protein crystallization. Crystals of the CPT5 complex with SPhe were formed under the microgravity conditions by the method of counter-diffusion through a gel layer in a capillary (Table 1) using instrumentation and technology developed by the Japan Aerospace Exploration Agency (JAXA) [29, 30]. An aliquot (8 μl) of the protein solution produced as described above was placed into a capillary with a diameter of 0.5 mm. The free end of the capillary was hermetically sealed with clay, and a silicon tube filled with 1% agarose gel presoaked in the precipitation solution for 24 h was attached to the other end of the capillary. The tube with the gel was cut to the length of 10 mm. The capillary with the tube were placed into a cylinder with the precipitation solution (1.6 M ammonium sulfate in 10 mM MES/NaOH buffer, pH 6.0, 0.5 M NaCl, 10 mM CaCl2, 0.1 mM ZnSO4) containing 100 mM SPhe. Several such devices were delivered to the International Space Station in a special packaging, where crystallization was carried out at 20°C. Crystals for X-ray diffraction analysis were taken out of the capillary into the precipitation solution, transferred to the cryosolution (20% glycerol (w/w) in the precipitation solution), and frozen in liquid nitrogen vapor.
Table 1. Statistical characteristics of the
X-ray diffraction data set and refinement of the CPT5 structure in a
complex with SPhe

* Numbers in parenthesis represent the highest resolution shell.
Collection and processing of X-ray diffraction data from CPT5 crystals. X-ray diffraction data were collected from the obtained crystals at the resolution of 1.35 Å at 100 K on a SPring-8 synchrotron (Japan, station BL41XU) using a MAR Mosaic 225 mm CCD detector. The diffraction data were generated by rotating a single crystal with the distance between the crystal and the detector of 100 mm and the wavelength of 0.8 Å; oscillation and rotation angles were 0.5 and 180°, respectively. The data were processed with the iMosflm program. The crystals were assigned to the P6(3)22 space group; one asymmetric unit contained one enzyme molecule.
Structure solution and refinement. The structure of the CPT5 complex with SPhe was solved by the molecular replacement method using the Phaser program [31] and coordinates of CPT5 in a free state (PDB ID: 4IHM) as a start model. The Refmac program was used for the structure refinement [32]. Manual model correction was carried out with the Coot program [33] using electron density maps calculated with the coefficients 2|Fo| – |Fc| and |Fo| – |Fc|. Electron density maps were also used in the Coot program to localize water molecules, zinc ion, sulfate ions, and molecules of glycerol and SPhe. The structure with a 1.35 Å resolution was refined to Rcryst = 13.9%; Rfree = 15.4%. The model coordinates were deposited to the Protein Data Bank (PDB ID: 4IAV). Statistical parameters of the refinement are presented in Table 1. For the structure comparison, superposition of Cα-atoms with the Lsqkab program from the CCP4 library was used.
RESULTS AND DISCUSSION
Crystal structure of the CPT5 complex with SPhe was elucidated at a high resolution (1.35 Å) due to the efficient purification of the enzyme by affinity chromatography, high quality crystals produced under microgravity conditions, the use of the world’s most powerful synchrotron SPring-8 (Japan, station BL41XU) under umbrella of the Program of Russia–Japan collaboration in space (space experiment “crystallizer”). The electron density found in the center of the obtained structure was identified as a molecule of SPhe (Fig. 2).
Fig. 2. Electron density of SPhe calculated with the help of |Fo| – |Fc| coefficient at level 2.0. The ligand was excluded from the electron density map.
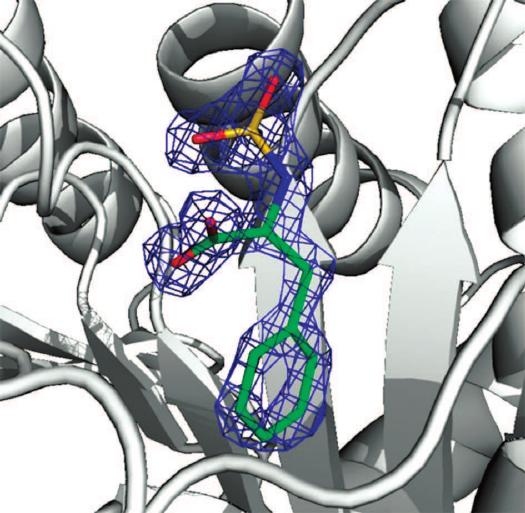
The sulfamoyl residue is located close to the zinc ion and it is in the asymmetric tetrahedral configuration with the bond between the nitrogen and zinc atoms typical for sulfamoyl analogues of the transition state [34, 35]. The Tyr255 residue (similar to Tyr248 in the CPB complex with SPhe) involved in the substrate-induced conformational changes of the enzyme active site is located at the so-called “lower” position preventing the access of water to the catalytic site and forms a hydrogen bond with the C-terminal carboxyl group of the ligand. The “upper” position of Tyr255 residue is characteristic for the carboxypeptidase with free active site; in this case, the tyrosine hydroxyl group is shifted by 12 Å from the “lower” position. The presence of sulfate or acetate ions in the binding site of the C-terminal carboxyl group is sufficient for the initiation of Tyr255 transition from the “upper” position to the “lower” one [36]. No significant differences in the positions of α-carbon atoms in the active sites of CPT5 and CPTwt were observed, which confirmed the suggestion on the absence of protein structure disruptions upon introduction of mutations (as judged from the catalytic activity of the mutant).
Comparison of the structure of the CPT5–SPhe complex with the previously elucidated structure of CPB in a similar complex (5J1Q) [37] demonstrated that the standard deviation (SD) of zinc atoms and α-carbon atoms in the CPT5 catalytic site (His69, Glu72, His204, Arg129, Glu277, Asn146, Arg147, and Tyr255) and the respective atoms in CPB was 0.41 Å. In the case of elimination of Tyr255 located in the mobile loop at the entrance to the catalytic site, the deviation was 0.24 Å. Deviation of atoms in the S1′ subsites (211/203, 215/207, 251/243, 257/250, 260/253, 262/255, and 275/268; numbering for CPT/CPB) was 0.30 Å. Hence, the spatial structure of the S1′ subsite in the CPT5 mutant did not change significantly and remained similar to the structure of the S1′ subsite in CPB. The highest deviation (0.41 Å) was detected for the Asp262 residue due to its dual position (positions A and B; Fig. 3); moreover, occupation of position A, which coincided with the position Asp255 in CPB, was 90%. This is related to the fact that according to the Pisa program, the side chain of Asp262 in position A forms two hydrogen bonds (with Asp263 and Thr275) and contacts with Ser215, Gly260, and Thr275, while in position B, it forms only one hydrogen bond with Thr275 (Fig. 3).
Fig. 3. Position of Asp262 and adjacent amino acid residues in the enzyme conformations A and B. Numeration is given for CPT (except Tyr192).
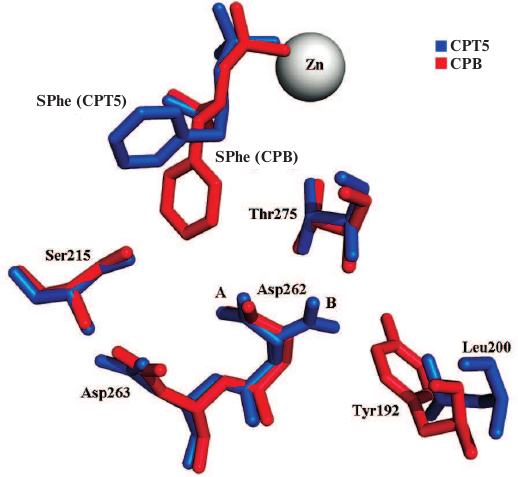
Note that the Asp262 group in CPB in conformation B experiences steric hindrances (distance 2.78 Å) from the benzene ring of Tyr192 residue absent in CPT; that is why conformation B is not realized in this case. It seems unlikely that these differences could be the reason for better binding of hydrophobic ligands by CPT5 in comparison with CPB.
The sulfur atom in SPhe (corresponding to the sp3-hybridized carbon atom of the transition state) in a complex with CPT5 was shifted by 0.3 Å relative to the sulfur atom in CPB. The phenolic oxygen in Tyr255 linked to the carboxyl groups of the ligands by a hydrogen bond was shifted by 0.6 Å, and zinc atoms were shifted by 0.1 Å. The difference in the distances between the sulfur atoms and zinc ions could suggest different stability of the respective transition complexes and, hence, different catalytic activity of CPT5 and CPB toward hydrophobic substrates.
The superposition of the S1′ subsites of CPT5 and CPB demonstrated that the ligand side chain binds to these two enzymes in different conformations. Therefore, its interactions with the enzymes are different. In the case of CPT5, the aromatic ring of SPhe is positioned in parallel to the aromatic ring of Tyr255 and, it is likely that they participate in the π–π interactions. Moreover, the side radical of SPhe forms hydrophobic contacts with Leu211, Gly251, Leu254, Ala257, and Thr275 (Fig. 4).
Fig. 4. Position of SPhe in active centers of CPT5 and CPB. In addition to ligands and zinc ions, the following residues are shown: L211/203, S215/207, G251/243, L254/247, Y255/248, G260/253, D262/255, A257/250, and T275/268 (numeration is given for CPT/CPB).
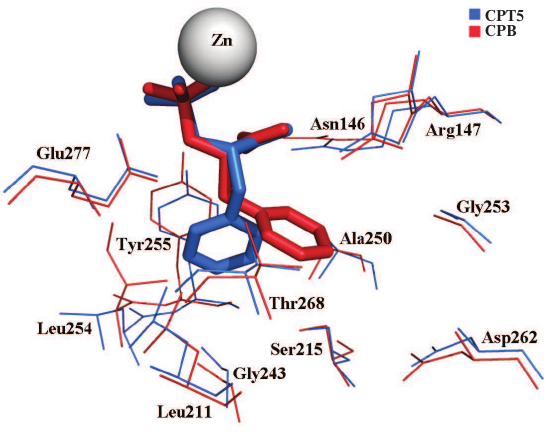
The hydrophobic environment of the ligand side group enables high efficiency of the hydrophobic substrate transformation and binding of the hydrophobic inhibitor by CPT5 (Table 2). At the same time, the aromatic ring of the SPhe side group in a complex with CPB is directed towards Asp255 and localizes in the hydrophilic environment of the Asp255 carboxylic group and hydroxyl groups of Ser207 and Thr268, as well as of the carbonyl group of the Ala250 main chain (Fig. 4). Only the methylene group of Gly253/260 and the side group of Ile247/254 have larger areas of hydrophobic contacts with the ligand in CPB in comparison with CPT5 (Fig. 5). This environment is unfavorable for binding of the hydrophobic transition state analogues, which determines low transformation efficiency of hydrophobic substrates with bulky side chains by CPB (Table 2).
Table 2. Kinetic parameters of hydrolysis of
the hydrophobic substrate ZAAL and inhibition constant by the reaction
transition state analogue SPhe
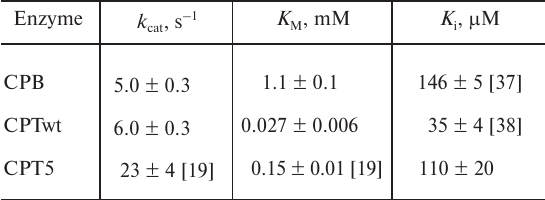
Fig. 5. Buried surface area of residues in the active sites of CPT5 and CPB in a complex with SPhe. Numeration is given for CPT.
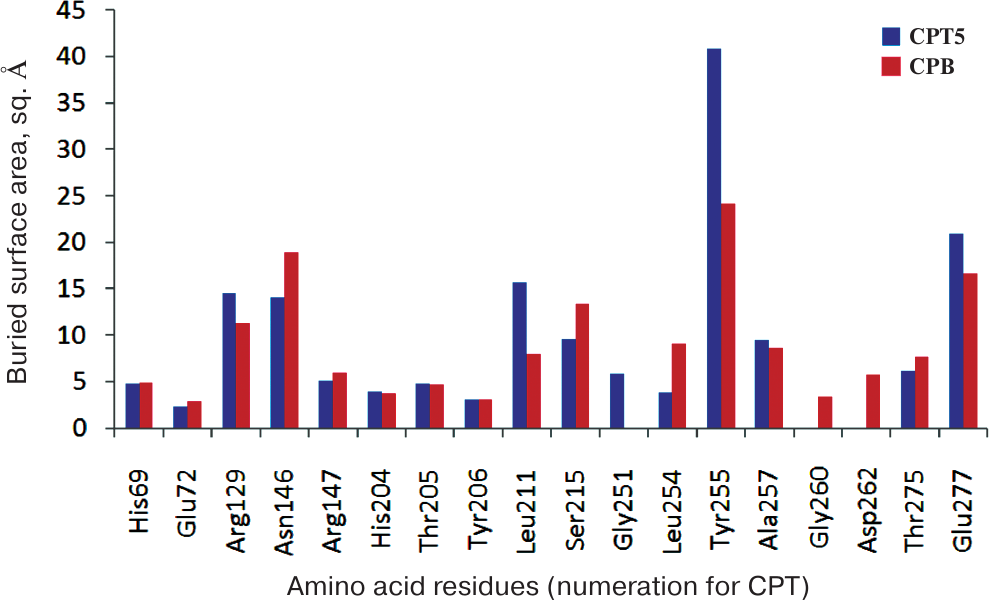
Hence, the conformation of SPhe and its interactions in a complex with CPTwt [38] are different than those in CPT5 and CPB, which is related to the five substitutions differing CPTwt from CPT5. This brings up the question of why the side chain of SPhe in the complex with CPT5 can be located in the hydrophobic subsite formed by Tyr255, Leu211, Leu254, and Ala257, while the side chain of the same ligand in the complex with CPB must be in a hydrophilic environment of the biding site for the arginine side chain if the corresponding residues forming the S1′ subsites demonstrate good superposition? The comparison of surfaces of the S1′ subsites in CPB and CPT5 revealed that unlike the S1′ subsite in CPB, the S1′ subsite in CPT5 has an additional cavity in the region adjacent to the active site mobile loop (Fig. 6). This site in CPT5 is occupied by the ligand, and in CPB – by the Ile247 residue that prevents the side group of the ligand from moving to this area from its hydrophilic environment. Ile247 plays an important and previously underestimated role in the substrate specificity of metallocarboxypeptidases. By moving together with Tyr248 to the “lower” position in CPB, it protects the S1′ subsite from water, which decreases the permittivity and promotes ionic interaction of Asp255 with the positively charged substrate. In the case of CPA, Ile247 in the “lower” position increases the hydrophobicity of the S1′ subsite and hydrophobic selectivity of CPA containing both Ile255 and Ile243. The inability of Leu254 in the “lower” position to isolate the S1′ subsite in CPT from water and to fix the ligand in the narrowed binding zone results in the possibility for the substrate to occupy either the position close to Thr252 (polar substrate) or the hydrophobic site (hydrophobic substrate), which provides the structural basis for the wide substrate specificity of CPT. The “lower” position of Leu254 in CPT depends on the nature of the substrate, thereby representing an example of the effect of induced complementarity on the enzyme selectivity.
Fig. 6. Position of SPhe molecule in the S1′ subsites of CPB and CPT. Ligands and Ile247 (CPB) are presented as stick-figure molecules, remaining amino acid residues are shown with lines; zinc ion is shown as a sphere. The cavity in the S1′ subsite of CPT is marked with an arrow; in CPB, this volume is occupied by the Ile247 side chain.
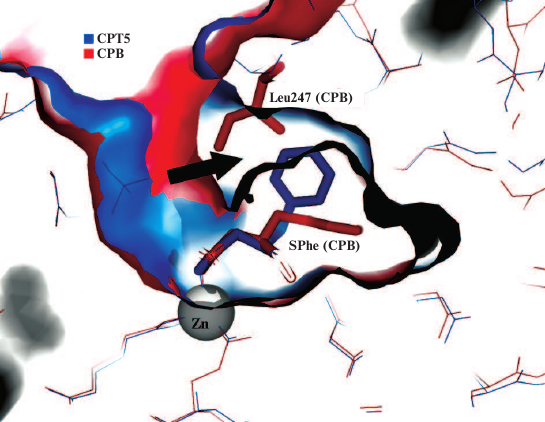
Different positions of residue 254 in CPT5 and CPB (247 in CPB) are due to different structures of the mobile loop in which they are located. This loop consists of the α-helix in CPB and 310 helix in CPT5 resulting from the deletion at position 253 (246 in CPA). No other obstructions preventing the ligand from occupying in CPB the same favorable position as in CPT5 has been observed. Note that the mobile loop in carboxypeptidase D, which exhibits the specificity similar to CPB, is also a 310 helix, and the substrate position “under the loop” is the major one for this carboxypeptidase (PDB ID: 1H8L) [39], while the substrate position similar to that in CPB is not realized due to different location of the negative charge and other groups in the active site.
In conclusion, elucidation of the high-resolution structure of the CPT mutant with the G215S, A251G, T257A, D260G, and T262D substitutions in a complex with the transition state analogue SPhe and comparison of this structure with the structures of similar complexes formed by CPB revealed previously underestimated structural determinant defining the substrate specificity of metallocarboxypeptidases. Comparative investigation of the structures demonstrated that substitution of five amino acid residues in the primary specificity pocket of CPT results in the structure closely matching the respective pocket in CPB; however, this does not lead to fundamental changes in the substrate specificity of the mutant due to the differences in the structures of the mobile loops located at the entrance of the active sites in CPB and CPTwt and controlling the substrate-induced conformational rearrangements of these sites.
Funding
This work was supported by the Russian Science Foundation (grant no. 17-14-01256; protein purification, optimization of crystallization conditions, solving, refining, and analyzing the structures), Federal Space Program 2016-2025 (ISS Nauka Research and Development Program; crystallization under conditions of zero gravity), and by the Ministry of Science and Higher Education within the State assignment FSRC “Crystallography and Photonics” RAS in part of collection and processing of X-ray diffraction datasets.
Conflicts of Interest
Authors declare no conflicts of interest in financial or any other sphere.
REFERENCES
1.Perez-Silva, J. G., Espanol, Y., Velasco, G., and
Quesada, V. (2016) The Degradome database: expanding roles of mammalian
proteases in life and disease, Nucleic Acids Res., 44,
D351-D355.
2.Turk, B., Turk, D., and Turk, V. (2012) Protease
signalling: the cutting edge, EMBO J., 31, 1630-1643.
3.Turk, B. (2006) Targeting proteases: successes,
failures and future prospects, Nat. Rev. Drug Discov., 5,
785-799.
4.Tanco, S., Tort, O., Demol, H., Aviles, F. X.,
Gevaert, K., Van Damme, P., and Lorenzo, J. (2015) C-terminomics screen
for natural substrates of cytosolic carboxypeptidase 1 reveals
processing of acidic protein C- termini, Mol. Cell. Proteomics,
14, 177-190.
5.Sapio, M. R., and Fricker, L. D. (2014)
Carboxypeptidases in disease: insights from peptidomic studies,
Proteomics Clin. Appl., 8, 327-337.
6.Osterman, A. L., Stepanov, V. M., Rudenskaya, G.
N., Khodova, O. M., and Tsaplina, I. A. (1984) Carboxypeptidase T
– extracellular carboxypeptodase of Thermoactinomyces
– a distant analogue of animal carboxypeptidases,
Biokhimiya, 49, 292-301.
7.Teplyakov, A., Polyakov, K., Obmolova, G.,
Strokopytov, B., Kuranova, I., Osterman, A. L., Grishin, N.,
Smulevitch, S., Zagnitko, O., and Galperina, O. (1992) Crystal
structure of carboxypeptidase T from Thermoactinomyces vulgaris,
Eur. J. Biochem., 208, 281-288.
8.Schechter, I., and Berger, A. (1967) On the size of
the active site in proteases. I. Papain, Biochem. Biophys. Res.
Commun., 27, 157-162.
9.Auld, D. S., Galdes, A., Geoghegan, K. F.,
Holmquist, B., Martinelli, R., and Vallee, B. L. (1984)
Cryospectrokinetic characterization of intermediates in biochemical
reactions: carboxypeptidase A, Proc. Natl. Acad. Sci. USA,
81, 5041-5045.
10.Reeke, G. N., Hartsuck, J. A., Ludwig, M. L.,
Quiocho, F. A., Steitz, T. A., and Lipscomb, W. N. (1967) The structure
of carboxypeptidase A. VI. Some results at 2.0-A resolution, and the
complex with glycyl-tyrosine at 2.8-A resolution, Proc. Natl. Acad.
Sci. USA, 58, 2220-2226.
11.Gardell, S. J., Craik, C. S., Clauser, E.,
Goldsmith, E. J., Stewart, C. B., Graf, M., and Rutter, W. J. (1988) A
novel rat carboxypeptidase, CPA2: characterization, molecular cloning,
and evolutionary implications on substrate specificity in the
carboxypeptidase gene family, J. Biol. Chem., 263,
17828-17836.
12.Stepanov, V. M. (1995) Carboxypeptidase T,
Methods Enzymol., 248, 675-683.
13.Osterman, A. L., Grishin, N. V., Smulevitch, S.
V., Matz, M. V., Zagnitko, O. P., Revina, L. P., and Stepanov, V. M.
(1992) Primary structure of carboxypeptidase T: delineation of
functionally relevant features in Zn-carboxypeptidase family, J.
Protein Chem., 11, 561-570.
14.Reeck, G. R., Walsh, K. A., Hermodson, M. A., and
Neurath, H. (1971) New forms of bovine carboxypeptidase B and their
homologous relationships to carboxypeptidase A, Proc. Natl. Acad.
Sci. USA, 68, 1226-1230.
15.Bunnage, M. E., Blagg, J., Steele, J., Owen, D.
R., Allerton, C., McElroy, A. B., Miller, D., Ringer, T., Butcher, K.,
Beaumont, K., Evans, K., Gray, A. J., Holland, S. J., Feeder, N.,
Moore, R. S., and Brown, D. G. (2007) Discovery of potent and selective
inhibitors of activated thrombin-activatable fibrinolysis inhibitor for
the treatment of thrombosis, J. Med. Chem., 50,
6095-6103.
16.Bown, D. P., and Gatehouse, J. A. (2004)
Characterization of a digestive carboxypeptidase from the insect pest
corn earworm (Helicoverpa armigera) with novel specificity
towards C-terminal glutamate residues, Eur. J. Biochem.,
271, 2000-2011.
17.Edge, M., Forder, C., Hennam, J., Lee, I., Tonge,
D., Hardern, I., Fitton, J., Eckersley, K., East, S., Shufflebotham,
A., Blakey, D., and Slater, A. (1998) Engineered human carboxypeptidase
B enzymes that hydrolyse hippuryl-L-glutamic acid: reversed-polarity
mutants, Protein Eng., 11, 1229-1234.
18.Grishin, A. M., Akparov, V. Kh., and Chestykhina,
G. G. (2008) Leu254 residue and calcium ions as new structural
determinant of carboxypeptidase T substrate specificity,
Biochemistry (Moscow), 73, 1140-1145.
19.Akparov, V. Kh., Grishin, A. M., Yusupova, M. P.,
Ivanova, N. M., and Chestukhina, G. G. (2007) Structural principles of
the wide substrate specificity of Thermoactinomyces vulgaris
carboxypeptidase T. Reconstruction of the carboxypeptidase B primary
specificity pocket, Biochemistry (Moscow), 72,
416-423.
20.Akparov, V. Kh., Belyanova, L. P., Baratova, L.
A., and Stepanov, V. M. (1979) Subtilisin 72 – a serine protease
from Bac. subtilus strain 72 – an enzyme similar to
subtilisin Carlsberg, Biokhmiya, 44, 886-891.
21.Lyublinskaya, L. A., Yakusheva, L. D., and
Stepanov, V. M. (1977) Synthesis of peptide substrates of subtilisin
and its analogues, Bioorg. Khim., 3, 273-279.
22.Cueni, L. B., Bazzone, T. J., Riordan, J. F., and
Vallee, B. L. (1980) Affinity chromatographic sorting of
carboxypeptidase A and its chemically modified derivatives, Anal.
Biochem., 107, 341-349.
23.Novagen pET System Manual TB055 (1997) 7th Edn.,
Novagen Madison, W.I.
24.Trachuk, L., Letarov, A., Kudelina, I. A.,
Yusupova, M. P., and Chestukhina, G. G. (2005) In vitro
refolding of carboxypeptidase T precursor from Thermoactinomyces
vulgaris obtained in Escherichia coli as cytoplasmic
inclusion bodies, Protein Expr. Purif., 40, 51-59.
25.Bradford, M. M. (1976) A rapid and sensitive
method for the quantitation of microgram quantities of protein
utilizing the principle of protein–dye binding, Anal.
Biochem., 72, 248-254.
26.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
27.Cornish-Bowden, A. (2013) Fundamentals of
Enzyme Kinetics, 4th Edn., Wiley-VCH, Weinheim.
28.Krissinel, E., and Henrick, K. (2007) Inference
of macromolecular assemblies from crystalline state, J. Mol.
Biol., 372, 774-797.
29.Takahashi, S., Tsurumura, T., Aritake, K.,
Furubayashi, N., Sato, M., Yamanaka, M., Hirota, E., Sano, S.,
Kobayashi, T., Tanaka, T., Inaka, K., Tanaka, H., and Urade, Y. (2010)
High-quality crystals of human haematopoietic prostaglandin D synthase
with novel inhibitors, Acta Crystallogr. Sect. F Struct. Biol.
Cryst. Commun., 66, 846-850.
30.Kuranova, I. P., Smirnova, E. A., Abramchik, Yu.
A., Chupova, L. A., Esipov, R. S., Akparov, V. Kh., Timofeev, V. I.,
and Kovalchuk, M. V. (2011) Crystal growth of phosphopantentein
adenylyltransferase, carboxypeptidase T, and thymidine phosphorylase on
the international space station by the capillary counter-diffusion
method, Crystallogr. Rep., 56, 884-891.
31.McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P.
D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007) Phaser
crystallographic software, J. Appl. Crystallogr., 40,
658-674.
32.Murshudov, G. N., Vagin, A. A., and Dodson, E. J.
(1997) Refinement of macromolecular structures by the
maximum-likelihood method, Acta Crystallogr. Sect. D Biol.
Crystallogr., 53, 240-255.
33.Emsley, P., and Cowtan, K. (2004) Coot:
model-building tools for molecular graphics, Acta Crystallogr. Sect.
D Biol. Crystallogr., 60, 2126-2132.
34.Park, J. D., Kim, D. H., Kim, S. J., Woo, J. R.,
and Ryu, S. E. (2002) Sulfamide-based inhibitors for carboxypeptidase
A. Novel type transition state analogue inhibitors for zinc proteases,
J. Med. Chem., 45, 5295-5302.
35.Akparov, V. K., Sokolenko, N., Timofeev, V., and
Kuranova, I. (2015) Structure of the complex of carboxypeptidase B and
N-sulfamoyl-L-arginine, Acta Crystallogr. Sect. F Struct. Biol.
Cryst. Commun., 71, 1335-1340.
36.Akparov, V. Kh., Timofeev, V. I., Maghsoudi, N.
N., and Kuranova, I. P. (2017) Three-dimensional structure of porcine
pancreatic carboxypeptidase B with an acetate ion and two zinc atoms in
the active center, Crystallogr. Rep., 62, 249-253.
37.Akparov, V., Timofeev, V., Khaliullin, I.,
Švedas, V., and Kuranova, I. (2018) Structure of the
carboxypeptidase B complex with N-sulfamoyl-L-phenylalanine – a
transition state analog of non-specific substrate, J. Biomol.
Struct. Dyn., 36, 956-965.
38.Akparov, V., Timofeev, V., Khaliullin, I.,
Švedas, V., Kuranova, I., and Rakitina, T. (2017) Crystal
structures of carboxypeptidase T complexes with transition-state
analogs, J. Biomol. Struct. Dyn., 35, 1-9.
39.Aloy, P., Companys, V., Vendrell, J., Aviles, F.
X., Fricker, L. D., Coll, M., and Gomis-Ruth, F. X. (2001) The crystal
structure of the inhibitor-complexed carboxypeptidase D domain II and
the modeling of regulatory carboxypeptidases, J. Biol. Chem.,
276, 16177-16184.