REVIEW: Thirty Years of Studies of Qβ Replicase: What Have We Learned and What Is Yet to Be Learned?
A. B. Chetverin
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: alexch@vega.protres.ru
Received July 28, 2017; Revision received August 2, 2017
Qβ replicase (RNA-directed RNA polymerase of bacteriophage Qβ) has an unsurpassed capacity to amplify polynucleotides in vitro. In 1986, the Group of Viral RNA Biochemistry was organized at the Institute of Protein Research in order to exploit this property for the synthesis of messenger RNAs to be used in cell-free translation systems. Although the task has not been implemented in full, this work has led to a number of unexpected important results including uncovering the nature of the “template-free” RNA synthesis by Qβ replicase, discovering the ability of RNA molecules for spontaneous recombination, revealing the unusual mechanism Qβ replicase uses to discriminate between its proper and improper templates, and discovering a new function of the largest ribosomal protein S1, that is also one of the replicase subunits. Finally, our work resulted in the invention of the molecular colonies technique that has become the basis for the next generation sequencing methods and provided a new insight into the origin of life. However, Qβ replicase has not yet revealed all its secrets, and its studies promise further interesting findings.
KEY WORDS: RNA-directed RNA polymerase, bacteriophage Qβ, RNA replication, spontaneous RNA synthesis, RNA recombination, molecular colonies, ribosomal protein S1DOI: 10.1134/S0006297918140031
In 1965, Spiegelman and Haruna published a sensational paper showing that Qβ replicase (RNA-dependent RNA polymerase of bacteriophage Qβ) is capable, in their words, to “autocatalytically” synthesize RNA in vitro [1]. This mode of synthesis is also known as exponential, because in each round of replication, the number of templates doubles and increases 2n, where n is the number of completed rounds. This is because a complementary copy synthesized on the original template also serves as a template in the next round. Given the time of duplication of the Qβ RNA (4217-nt bacteriophage Qβ genomic RNA) of about 2 min at 37°C [2], Qβ replicase can synthesize a billion of copies (2.5 ng of RNA) in an hour. To synthesize 1 kg of Qβ RNA, the replicase would need a little more than 2 h if, of course, it continues to remain in a molar excess over RNA during the entire period of time. This record of the in vitro genetic material amplification rate is still unsurpassed.
Qβ replicase created another sensation at the end of 1983. The Science journal published an editorial in which it announced the birth of a fundamentally new technology, called recombinant RNA technology, and predicted that Qβ replicase would become “a molecular workhorse in a major industry” [3]. The paper commented on the work just published in the Journal of Molecular Biology [4] that was carried out under the leadership of F. R. Kramer, a former student of Spiegelman. The key point of that work was as follows.
Qβ replicase has natural templates, which are shorter than the genomic Qβ RNA, but often are more efficiently replicated [5]. These are bacteriophage Qβ satellite RNAs [6]. The authors had chosen one of these RNAs designated MDV-1 [7], embedded the A10 (decaadenylate) oligonucleotide into its sequence [8], and demonstrated that the resulting recombinant RNA is amplified by Qβ replicase as efficiently as the original MDV-1 sequence [4]. It was this finding that inspired the enthusiasm of the Science editor [3], because it was presumed that now any sequences could be made amplifiable by Qβ replicase using MDV-1 or another Qβ phage satellite RNA molecule as a vector.
In 1985, the Director of the Institute of Protein Research of the USSR Academy of Sciences Alexander Sergeevich Spirin visited the United States and met with F. R. Kramer who told him about that work. Being inspired by the Kramer’s story, A. S. Spirin came back to the Soviet Union and invited me to tackle this problem. At that time, I had finished my work on Na,K-ATPase and defended the PhD thesis, so I accepted his invitation.
This gave rise to a project, probably the first scientific project of this kind in our country, which I defended before the Scientific Council of the Institute of Protein Research in June of 1986. It was titled “Development of a cell-free RNA replication system” and its aim was “to develop a universal technique for enabling any RNA to be amplified in the Qβ replicase system” in order “to produce any RNA in virtually unlimited amounts”. The principal customer and potential consumer of these RNAs was Alexander Sergeevich Spirin, because at that time, his laboratory was developing continuous flow cell-free translation systems and needed large amounts of various mRNAs. To execute the project, the Director issued Order No. 72-k of June 19, 1986, establishing a scientific group named “Transient Interlaboratory Group of Viral RNA Biochemistry”. Over time, the group became permanent, and on November 23, 1998, it received the status of a laboratory. I must say that the project has not been fully implemented so far. Therefore, this review, that was presented at the Anniversary Conference of the Institute of Protein Research on June 7, 2017, may be considered an interim report on the 1986 project.
TEMPLATE-FREE RNA SYNTHESIS BY Qβ REPLICASE
The first problem we encountered was RNA synthesis by Qβ replicase in the absence of an added template. Even if we could make any RNA amplifiable by Qβ replicase, then, according to the 1975 paper by Manfred Eigen’s colleagues from the Max Planck Institute, Göttingen, Qβ replicase might ignore the added template and instead amplify its own RNAs, which it generates de novo [9].
The key point of the paper was as follows. The authors purified Qβ replicase to a state when the addition of even several molecules of an efficient template stimulated RNA synthesis in a standard 200-µl incubation mixture. Therefore, the replicase preparation that was added to the mixture contained no more than a few molecules of replicating RNAs. When the reaction volume (and therefore, the amount of added replicase) was reduced 10,000 times, the authors still observed spontaneous RNA synthesis in each of the microscopic 0.02-µl aliquots; hence, they concluded that Qβ replicase does not need any template. The authors concluded that, when incubated for several hours (and occasionally even within 1 h), the enzyme generated efficient templates that may overgrow the added RNA. Obviously, if we unable to find a different explanation of these results, any further work on the project would have lacked any sense.
The result of our first experiment on the template-free RNA synthesis was totally unexpected. Figure 1a (curve 1) displays the time course of the labeled nucleotide incorporation in the absence of the added RNA in the reaction mixture. We did not observe any RNA synthesis up to 4 h of incubation, although according to the Eigen’s group [9], RNA should have appeared at least by the end of the second hour. At the same time, not prominent but significant RNA synthesis was seen in a tube to which we added total RNA extracted from infected cells (Fig. 1a, curve 2). Just in case, we left the tubes overnight on the bench and the next morning were surprised: to find a large amount of incorporated label in the test tube with no added RNA (the 20-h point on curve 1). Analysis of the reaction mixture by gel electrophoresis showed a variety of the synthesized RNAs. On the same day, we carried out another experiment (Fig. 1a, curve 3). This time, and in all subsequent experiments on the template-free RNA synthesis, there was no long lag period – RNA synthesis started quickly in the absence of the added template (as in experiments of the Eigen’s group), and approximately the same sets of RNAs were synthesized.
Fig. 1. First experiments on the “template-free” RNA synthesis by Qβ replicase carried out at the Institute of Protein Research. a) Time course of RNA synthesis in test tubes containing Qβ replicase and labeled NTPs in the first experiment in the absence (1) or presence (2) of the total RNA isolated from infected E. coli cells and in the second experiment in the absence of the added template (3). Closed tubes were incubated at 37°C; at certain time points, the tubes were opened to withdraw aliquots of the reaction mixture that were analyzed for the label incorporation. b) Electrophoretic analysis of the RNAs synthesized in tube 1. The gel was stained with ethidium bromide. Arrow marks the RNA that had been sequenced. c) Alignment of nucleotide sequences of the sequenced RNA with the sequences of E. coli tRNA1Asp and the Qβ phage genomic RNA coat protein cistron [10].
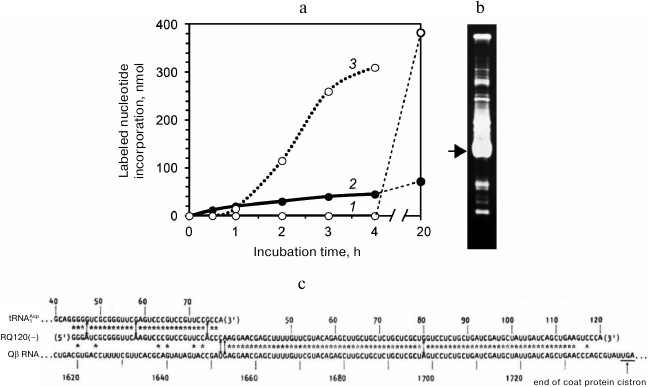
Then we sequenced the major RNA band indicated by the arrow in Fig. 1b. The results were published in Nature [10] as the first paper of the group. In that paper we introduced an acronym RQ (for Replicable by Qβ replicase) for the designation of a satellite RNA followed by a number indicating the length of its sequence in nucleotides. In accordance with the suggested nomenclature, the sequenced RNA was named RQ120. This RNA, which had been synthesized in a nominally template-free reaction, turned out to be a perfect recombinant of two known RNAs. One part of its sequence matched a ~80-nt fragment of the Qβ RNA coat protein cistron, while the other part matched a 33-nt fragment of the E. coli aspartyl tRNA (Fig. 1c).
Two fundamental conclusions followed immediately from this fact: (i) RQ120 RNA could not be synthesized without a template, because such a long homology could not have occurred accidentally; (ii) in bacterial systems, RNA recombination does take place, since one of the recombination substrates (Qβ RNA) exists in nature in the form of RNA only. The first conclusion contradicted the concept of de novo RNA synthesis [9], while the second one opposed the statement that in bacterial systems, there is no recombination between RNA molecules, as follows from unsuccessful attempts to detect recombination between RNA-containing phages [11].
In the project report for year 1987, we came to the conclusion that RNA synthesis observed in our experiments in the absence of an added template was actually template-directed. We suggested that “the main cause of the uncontrolled synthesis is contamination of the reaction mixture with replicating RNAs present in the air”.
However, how could this be proven? Long ago, a similar problem had been solved by Louis Pasteur who investigated why a meat broth became spoiled [12]. To get the answer, he boiled the broth in a flask made in a shape of a swan: its neck was drawn and bended downwards so that vertically falling particles could not enter the broth, but settled within the bend. In such a flask, the broth did not spoil indefinitely long. However, if the flask had been tilted so as to rinse the bend with the cooled broth, the broth became spoiled soon. Hence, Pasteur came to conclusion that “no life could arise in a boiled meat broth unless solid particles heavier than air were allowed to enter”.
We could not follow the Pasteur’s procedure, since boiling the reaction medium would kill Qβ replicase. The problem seemed unsolvable. Then we carried out the following experiment. It was a desperate move, as there was very little hope of success. We mixed molten low-temperature gelling agarose with Qβ replicase and nucleoside triphosphates (RNA synthesis substrates) and poured the mixture into a Petri dish. After 1 h, the gel was moistened with ethidium bromide, a dye that increases its fluorescence upon intercalation between the stacked bases in a polymer RNA. We saw what we were afraid to hope for: the gel contained fluorescent spots (Fig. 2a). It became immediately clear for us that these were RNA colonies formed by amplification of the replicating molecules. Since they amplified in a gel rather than in a liquid, the daughter molecules did not spread throughout the reaction volume, but concentrated around the original templates. Thus, we had acquired a tool enabling us to detect single molecules of replicating RNAs.
Fig. 2. Detection of RNA molecules by the molecular colony technique. a) The first experiment on the growth of RNA colonies in an agarose gel containing Qβ replicase and NTPs carried out in January, 1988 [45]. b) Detection of replicating RNA molecules in the air [19]. RNA colonies grown in dishes that were left for 1 h closed (left) or open (right) before pouring the second agarose layer (see text).
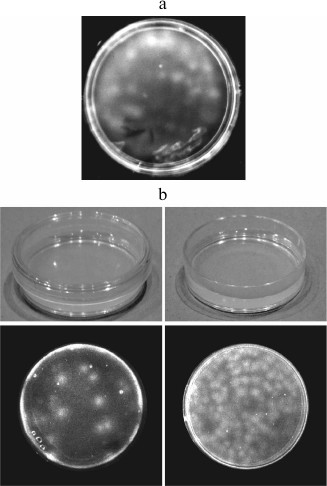
Having slightly changed the design, we carried out another experiment. In each of two Petri dishes, two layers of agarose gel were cast: first, the layer containing nucleotides and, above it, the layer containing Qβ replicase. The placement of the enzyme and its substrates in different layers made it possible to delay RNA amplification until the gel was formed and, hence, to avoid amplification in the liquid phase. Before pouring the second layer, the dishes were left on a bench for 1 h, one being closed and the other being open. One hour after pouring the second layer, we stained the gels in both dishes with ethidium bromide and saw exactly what was expected: a much greater number of RNA colonies in the open dish than in the closed one (Fig. 2b). Therefore, we concluded that the cause of the so-called “template-free” synthesis is the entry of RNA molecules from the air into the reaction mixture [6].
APPLICATIONS OF MOLECULAR COLONIES
The January 1988 experiment on the growth of RNA colonies (Fig. 2a) has been the first example of the genetic material amplification in a form of molecular clones [13]; never before clonal nucleic acid amplification had been carried out in vitro.
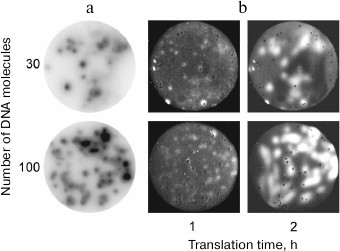
Later, we succeeded in growing DNA colonies by performing PCR in a polyacrylamide gel [14, 15], as well as in the DNA colony transcription and translation in situ [16] (Fig. 3). This demonstrated that, in a certain sense, molecular colonies are analogs of living cells since they can be a site for the genetic material replication and expression, including synthesis of active proteins.
Fig. 3. Gene cloning and expression in molecular colonies [16]. a) Amplification of the green fluorescent protein (GFP) gene as DNA colonies by PCR in a polyacrylamide gel. Colonies were detected by hybridization with a radioactive probe. b) Expression of the GFP gene in molecular colonies by using the coupled transcription-translation system. Colonies were detected by the GFP fluorescence.
The ability of molecular colonies to compartmentalize biochemical reactions makes them likely candidates for the role of cell precursors in the process of life origin (molecular colonies might have grown in porous minerals, such as clay, rather than in a gel) [17, 18] and a convenient experimental model for assembling living cells de novo [19].
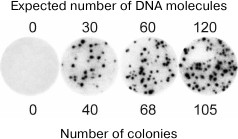
Molecular colonies have been used by us to develop molecular diagnostics methods with the absolute sensitivity. Figure 4 shows the result of an experiment in which a known amount of viral DNA was added to the human blood, and then the total DNA was isolated and used to grow the virus-specific molecular colonies [20]. It can be seen that within the statistical error, the number of colonies matched the number of viral DNA molecules present in the analyzed sample. Hence, the method was able to detect even a single molecule of the target DNA. It is important that this result was achieved against the background of a trillion-fold excess of non-viral DNAs present in the blood sample without any preliminary enrichment of the target DNA. In comparison, even a 1000-fold lesser amount of non-target DNA suppresses amplification of small quantities of target DNA in liquid PCR due to the synthesis of non-specific products [21].
Fig. 4. The use of molecular colonies for detection of hepatitis B virus (HBV) DNA in human blood (from [20]; © 2009 BioTechniques. Used by permission). Total RNA and DNA were isolated from 100-µl blood samples, to which 0, 50, 100, or 200 molecules of HBV DNA had been added. A part (60%) of the resulting preparations were used for PCR in a polyacrylamide gel. Colonies were detected by hybridization with a radioactive probe.
Finally, DNA amplification in a form of molecular clones is used by the next generation genome and transcriptome sequencing. In particular, the popular technology of total sequencing developed by Illumina [22] is based on the amplification of DNA fragments in the form of molecular colonies, which in this case are termed “clusters”.
REPLICATION OF RECOMBINANT RQ-mRNAs
After we had sorted out the mystery of the template-free synthesis, we could approach the main objective of the project: make any RNA amplifiable by Qβ replicase using the strategy proposed by the Kramer’s group [4]. By utilizing genetic engineering methods, we constructed a recombinant RNA using RQ135 (another replicating RNA that was isolated and sequenced by us [23]) as a vector. Similarly to MDV-1 RNA (RQ223), RQ135 was also efficiently amplified by Qβ replicase. mRNA for dihydrofolate reductase (DHFR) was inserted into the loop of one of the internal RQ135 hairpins. The resulting RNA was designated RQ-DHFR. As a control, we used MDV-CAT RNA from the Kramer’s group; the RNA comprised chloramphenicol acetyltransferase (CAT) mRNA inserted into the MDV-1 RNA sequence and, according to the authors, was amplified exponentially by Qβ replicase [24].
To our disappointment, none of the RNAs tested proved to be capable of exponential amplification. Qβ replicase only made a single copy of each of these templates [25]. For both RNAs, the newly synthesized complementary strand formed a duplex with the template and the process ceased because double stranded RNA could not serve as a template for Qβ replicase. However, we were able to enhance the RNA synthesis by supplementing the reaction mixture with a complete translation system that allows the protein-encoding (+) strand to be read by the ribosomes. In this case, most of the synthesized RNA molecules were single-stranded (Fig. 5a); furthermore, the majority of them were (+) strands, i.e., those being translated (Fig. 5b).
Fig. 5. Mutual enhancement of replication and translation of the recombinant RQ-DHFR RNA [25]. a) Electrophoretic analysis of the products of RNA synthesis by Qβ replicase in the presence of [32P]UTP using DHFR-mRNA (1) or RQ-DHFR RNA (2, 3) as a template in the absence (1, 2) or in the presence (3) of complete E. coli cell-free translation system. Symbols “ss” and “ds” indicate positions of the single-stranded and double-stranded forms of the RQ-DHFR RNA, respectively. b) Time course of the synthesis of RQ-DHFR RNA (+) strands (1) and (−) strands (2) by Qβ replicase in the presence of E. coli cell-free translation system. c) Time course of DHFR synthesis on the RQ-DHFR RNA in the E. coli cell-free translation system in the absence (2) or presence of 1 µg (1, 3) or 2 µg (4) of Qβ replicase and in the absence (2-4) or presence (1) of 0.5 mM puromycin.
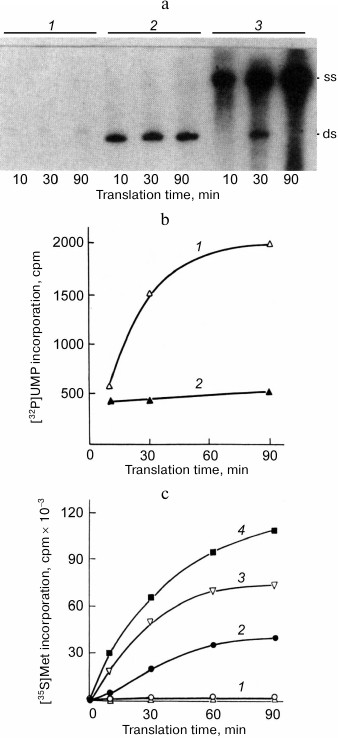
These results were explained as follows. Translating ribosomes have two mutually opposite effects on the RQ-DHFR RNA replication. On the one hand, they compete with the replicase for the (+) strands, along which they move in the 5′ → 3′ direction, while replicase goes in the opposite direction (3′ → 5′), and this suppresses synthesis of the (−) strands. On the other hand, they prevent annealing of the (+) strands with the (−) strands, allowing the (−) strands to be produced in a single-stranded form and hence to serve as templates for the synthesis of new (+) strands, thereby promoting the replication. As soon as a newly synthesized (+) strand appears, it most likely enters translation and quits replication. At the same time, the (−) strand is incapable of translation and serves as a template for the synthesis of (+) strands. This leads to the observed asymmetry in the synthesis of the strands with the opposite polarity, which is similar to the asymmetry of the (+) and (−) Qβ RΝΑ strand synthesis in E. coli cells infected with RNA-containing bacteriophages [26].
Therefore, coupling replication with translation enables, in principle, replication of a recombinant RQ-mRNA in vitro (though not exponential), which in turn increases the synthesis of a protein encoded by the (+) strand (Fig. 5c). mRNA replication and translation in a cell-free system mutually enhance each other.
THE MECHANISM OF TEMPLATE RECOGNITION BY Qβ REPLICASE
Our results showed that the strategy proposed by the Kramer’s group did not work. A recombinant RQ-mRNA cannot be amplified by Qβ replicase in the exponential mode or even cannot be copied more than once in the absence of a coupled translation system. To achieve the project’s goals, it was necessary to understand why Qβ replicase accepts certain, but not other, RNAs as templates.
Comparison of the primary structures of Qβ RNA and a number of sequenced RQ RNAs did not reveal any common elements except for the terminal triplets GGG and CCC at the 5′ and 3′ ends, respectively. Nothing like a promoter for the template recognition was found. Nevertheless, the specificity of Qβ replicase is extremely high: only a few RNA species selected by the SELEX procedure from a population of 1012 random sequences of 50-70 nt long RNAs flanked by 5′-GGG and 3′-CCC were replicable to some degree [27].
Using Qβ RNA as a template, Weissmann et al. showed that no double-stranded intermediates are formed in the course of exponential amplification. All previously reported double-stranded structures appeared to be artifacts of the RNA isolation procedure and were generated following addition of any agent that compromised the integrity of replicase, such as phenol, detergent, or protease. Moreover, Qβ replicase proved to be unable to read RNA duplexes [2].
On the other hand, Palmenberg and Kaesberg demonstrated that despite high template specificity, Qβ replicase was able to make a complementary copy of almost any RNA, regardless of its terminal sequences, especially when Mg2+ was replaced with Mn2+. However, unlike the copy of an exponentially amplified RNA, the reaction product was resistant to the cleavage with pancreatic ribonuclease, i.e., was double-stranded [28].
Thus, there are two types of Qβ replicase templates that we named legitimate and illegitimate templates. The differences between these types of templates [29] are summarized in the table. Unlike illegitimate templates, legitimate templates always have CCC at the 3′-end, and initiation of RNA synthesis on these templates requires GTP. If at the initiation step GTP is replaced with ITP (inosine triphosphate, capable of pairing with the template cytosine), an illegitimate template will be copied, but a legitimate template will not. Most importantly, the full-size complementary strand can be synthesized on a legitimate template in the presence of aurintricarboxylic acid (ATA), if the latter was added after completion of initiation, i.e., at the elongation step. On the contrary, ATA completely suppressed the copying of illegitimate templates regardless of the reaction step at which it was added. Since ATA is a potent inhibitor of the RNA–protein interactions [30], the observed insensitivity to ATA means that, unlike illegitimate templates, legitimate templates and their products cannot leave the replicase active site during the elongation step. Thus, we concluded that elongation of a legitimate template takes place in a closed conformation of Qβ replicase, in which any exchange of large molecules (RNA, polymeric ATA) between the enzyme active site and its environment is precluded, whereas during copying of illegitimate templates, the transition into the closed conformation does not occur [29]. Finally, the product synthesized on a legitimate template is single-stranded, while the product synthesized on an illegitimate template is double-stranded.
Two types of the Qβ replicase templates [29]

Thus, it was found that unlike DNA-directed RNA polymerases, the template specificity of Qβ replicase is determined not by the recognition of a specific template structure (promoter) at the onset of the replication initiation, but by the enzyme ability to transit into the closed conformation after the initiation step. Qβ replicase recognizes and begins to copy both legitimate and illegitimate templates, but only legitimate templates can induce the replicase transition into the closed conformation (in the presence of GTP), in which RNA synthesis is resistant to the ATA. Probably, the transition into the closed conformation enables Qβ replicase to keep the replicative complex single-stranded [29].
Then we learned that legitimate templates of Qβ replicase have the property of functional circularity [31]. Point mutations at the 5′-end of a legitimate template reduce the rate and the yield of initiation at the 3′-end and also enhance the dependence of initiation on the Mg2+ concentration. Moreover, these mutations decrease stability of the post-initiation replicative complex in the presence of ATA. Therefore, Qβ replicase simultaneously interacts with the 3′- and 5′-ends of a legitimate template during and after replication initiation.
We have also established another fact. All legitimate templates can potentially fold into a structure, in which the 3′- and 5′-ends are paired, and form the terminal helix that could lock the molecule in a circular structure [32]. In some instances (e.g., in the case of RQ135 RNA [23]), the presence of such helix has been verified by probing the structure with ribonucleases capable of cleaving either single-stranded or double-stranded RNA regions.
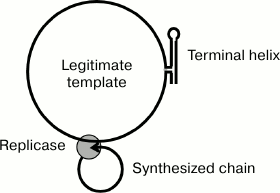
The circular configuration of the template can contribute to maintaining the replication intermediate single-stranded. Together with the hypothetical ability of the replicase to simultaneously interact with the growing 3′-end and the 5′-end of the nascent RNA strand during the entire elongation step [2], this would result in both the template and its complementary copy forming two circles (Fig. 6) unable to generate an extended double helix due to topological constraints.
Fig. 6. Model explaining the single-stranded structure of the replicative complex due to the ability of the terminal helix and the replicase to lock the template and the synthesized strand, respectively, in a circular form.
STRUCTURE OF THE Qβ REPLICASE CORE
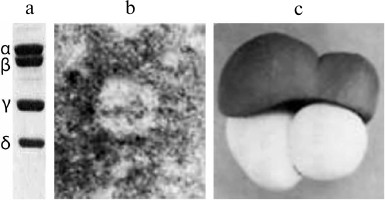
To understand how Qβ replicase recognizes legitimate templates and discriminates between different RNA molecules, it is important to know the structure of the replicase itself and of its complexes with legitimate templates. For a long time, there had been no progress in this direction. In 1988, the same year when we published the Nature paper on recombinant RQ120 RNA, we published together with V. D. Vasiliev an article on the electron microscopy of Qβ replicase [33]. Figure 7 presents a model of the replicase molecule proposed by V. D. Vasiliev based on the obtained images. The most surprising result here is that all subunits of Qβ replicase look like globules, despite the fact that the enzyme includes, in addition to the Qβ phage genome-encoded β-subunit and translation elongation factors EF-Tu and EF-Ts, the ribosomal protein S1 (Fig. 7a), whose molecule has always been considered highly elongated.
Fig. 7. The molecule of Qβ replicase (from [33] with kind permission from John Wiley and Sons; ©1988 Federation of European Biochemical Societies). a) SDS-electrophoresis of Qβ replicase subunits: α, ribosomal protein S1; β, catalytic subunit encoded by the Qβ phage genome; γ and δ, translation elongation factors EF-Tu and EF-Ts, respectively. b) Electron micrograph of the Qβ replicase molecule (negatively stained sample). c) Model of Qβ replicase molecule based on the electron microscopy data.
However, all attempts to crystallize the Qβ replicase holoenzyme had been unsuccessful. Finally, in 2010 the Qβ replicase core was crystallized (the core is Qβ replicase lacking the protein S1 but still capable of RNA synthesis). For crystallization, the enzyme molecule was additionally stabilized by either replacing E. coli EF-Ts with its homolog from Thermus thermophilus [34] or fusing EF-Tu and EF-Ts into a single polypeptide [35]. In the second case, the crystals diffracted to a higher resolution and were used for solving the Qβ replicase structure.
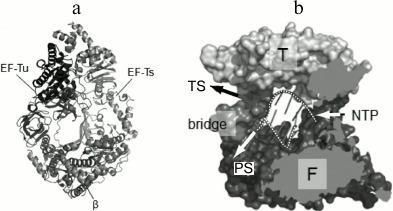
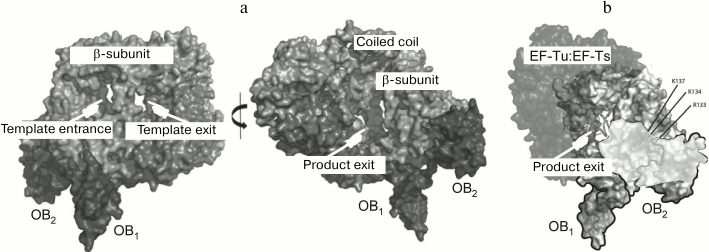
The general architecture of the Qβ replicase core proved to be similar to that of other viral RNA-directed RNA polymerases (Fig. 8a) except for one important feature. At the exit from the active site, there is a septum termed “bridge”, that divides the exit into two openings – for the template and for the nascent strand (Fig. 8b) – and thereby separates the double helix into constituent strands by acting like the slider of a zipper [35].
Fig. 8. Crystal structure of the Qβ replicase core [35] (from PhD thesis of N. N. Vasilyev, Institute of Protein Research [46]). a) Molecule of the monomer enzyme consisting of the β-subunit and elongation factors EF-Tu and EF-Ts. b) Model of the Qβ replicase active site formed by the β-subunit. Arrows indicate the directions of movement of the template strand (TS), the product strand (PS), and nucleotides (NTP); F and T, Fingers and Thumb domains of the β-subunit, respectively; dashed white lines, sugar-phosphate backbones of the template and product strands.
THE ROLE OF RIBOSOMAL PROTEIN S1 IN RNA REPLICATION
Having determined the structure of the core, we wanted to make sure that the core is, as Weissmann wrote [36], a fully active enzyme capable of the exponential RNA synthesis that requires protein S1 only for the recognition of the Qβ RNA (+) strand during initiation of its replication. To our surprise, this turned out not to be true [37].
We found that replication of RQ135 RNA, which is not related to the phage genome and has a quite dissimilar primary structure, was enhanced 10 times in the presence of protein S1, if the replication was monitored at the exponential phase, i.e., when Qβ replicase was present in a molar excess over RNA (Fig. 9a). At the linear phase (when the RNA was in excess), the effect of protein S1 was almost undetectable. Analysis of the replication products by a non-denaturing gel electrophoresis revealed that proportion of the single strands (ss) was significantly higher in the presence of protein S1 than in its absence (Fig. 9b).
Fig. 9. Ribosomal protein S1 acts as a termination factor in RNA synthesis by Qβ replicase [37]. a) Amplification of RQ135 RNA (1.66 nM) by the Qβ replicase core (360 nM) in the absence (1) and presence (2) of 360 nM protein S1. The amount of the full-sized product is expressed as percent of the maximum value synthesized in the presence of protein S1. b) Single-stranded (ss) and double-stranded (ds) forms in the products of RQ135 RNA amplification by the Qβ replicase core in the absence (1) and presence (2) of protein S1. 32P-labeled products were extracted with phenol and separated by non-denaturing gel electrophoresis. c) Distribution of the 32P-labeled product between ss- and ds-forms during Qβ RNA replication at 30°C in the absence (1) and presence (2) of protein S1. Prior to phenol extraction, the samples were either treated with ribonuclease T1 (+) or left intact (−). d) Domain structure of protein S1 and its N-terminal fragment OB1-2. The OB domains are numbered in white; amino acid residues are numbered in black. e) Distribution of the 32P-labeled product between ss- and ds-forms during Qβ RNA replication in the absence (1) and presence (2) of the S1 fragment OB1-2. Prior to phenol extraction, the samples were either treated with ribonuclease T1 (+) or left intact (−).
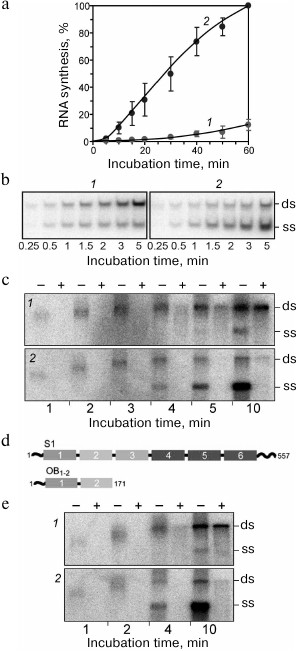
Then we decided to investigate the replication cycle in detail using the Qβ RNA (+) strand as a template. This RNA is more than 4000 nt in length, and its replication takes a few minutes, thereby allowing observation of individual stages of the cycle. Here we used an approach that needs to be explained to make the results of the experiment clear. A portion of the reaction mixture taken at a specified time was directly extracted with phenol and analyzed by non-denaturing electrophoresis (samples marked “−” in Fig. 9c), whereas the other portion was first treated with ribonuclease T1, which cleaves single-stranded RNA, but not double-stranded RNA, and only then extracted with phenol (samples marked “+”). This allowed us to determine the true state of the replicative complex at a given time.
Figure 9c shows that in the untreated samples, the electrophoretic mobility of the labeled RNA reduced over time. This is because the nascent (−) strand annealed to the template during phenol extraction, and its mobility approached that of the double-stranded (ds) Qβ RNA as the elongation proceeded. By the end of the fourth minute, the electrophoretic mobility of the product became equal to the mobility of the double-stranded Qβ RNA, indicating that elongation was completed. During these four minutes, the mobility of the reaction product decreased at the same rate in both the samples containing or lacking protein S1. Therefore, protein S1 had no effect on the elongation rate of the (−) strand. Moreover, regardless of the protein S1 presence, the replicative complex was sensitive to ribonuclease T1 (“+” samples), hence, remained single-stranded this entire time. However, in the presence of protein S1, the product left the replicative complex as soon as the elongation ended and, therefore, was single-stranded (ss) after phenol extraction (“−” samples), whereas in the absence of protein S1, the amount of free product strand was negligible, and the product accumulated in the form of double strands (ds) resistant to ribonuclease T1 (“+” samples).
From this observation, we concluded that protein S1 acts as the RNA synthesis termination factor. It promotes the release of the single-stranded product from the replicative complex at the termination step [37].
We also found that the termination function of S1 does not require entire protein and could be ensured by the N-terminal fragment consisting of two of the six OB domains (Fig. 9d). It is commonly accepted that the acronym “OB” stands for “Oligonucleotide/oligosaccharide Binding”. However, as A. V. Finkelstein told me recently, the OB fold was named by A. G. Murzin after his teacher Oleg Borisovich Ptitsyn; i.e., it is “Oleg Borisovich” domain. We found that the OB1-2 fragment, though being three times smaller than S1, provides the same function as the full-size protein [37], i.e., stimulates the termination of RNA synthesis (Fig. 9e).
STRUCTURE OF THE Qβ REPLICASE CORE COMPLEX WITH THE PROTEIN
S1 N-TERMINAL FRAGMENT
By using the compact OB1-2 fragment instead of protein S1, we have solved the crystal structure of the fully active Qβ replicase [38] which, in addition to initiation and elongation, is also capable of releasing the synthesized RNA in a single-stranded form (Fig. 10a). While analyzing the electrostatic potential distribution, we found that the surface of the second OB domain contains a positively charged area immediately adjacent to a similarly charged area in the β-subunit. These two areas form a tract that leads from the opening through which the synthesized RNA strand presumably leaves the active site (Fig. 10b). It is possible that this positively charged tract somehow assists the release of the product from the enzyme and, hence, its termination.
Fig. 10. Crystal structure of the Qβ replicase core in a complex with the S1 fragment OB1-2 [38]. a) Two views rotated approx. 180° relative to each other. b) Positively charged tract (shown in a lighter tone) located on the adjacent surfaces of the β subunit and the S1 domain OB2 next to the product outlet.
RNA RECOMBINATION
As mentioned above, our first article reported the discovery of the recombinant RQ120 RNA in the products of the “template-free” RNA synthesis. This finding indicated that in the bacterial systems, recombination can occur at the RNA level [10]. However, it remained unclear whether this RNA had originated due to the action of some cellular enzymes or of Qβ replicase itself, or RNA molecules had recombined due to their own catalytic activity.
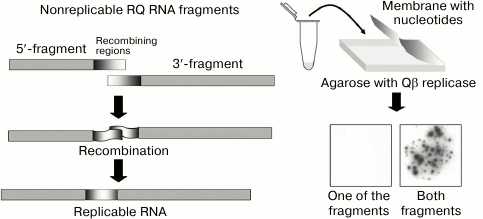
To explore the issue, we have developed a cell-free RNA recombination system using Qβ replicase and its templates. To selectively visualize recombinant molecules without seeing the nonrecombinant ones, we used the molecular colony technique [39]. We chose the natural replicating RNA, RQ135, and cut it roughly in two halves. None of the resulting fragments could be amplified by Qβ replicase and form an RNA colony. However, if the two fragments recombined into a replicating molecule, a colony would grow around it. By counting the colonies, we could determine the number of replicable recombinant molecules. Indeed, when the fragment mixture was plated on the Qβ replicase-containing agarose, numerous RNA colonies showed up, whereas none of the fragments gave rise to the colonies by itself (Fig. 11).
Fig. 11. Detection of recombination events using the molecular colony technique [19]. See the text for explanation.
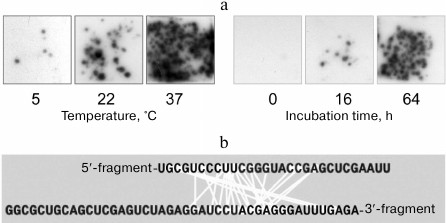
Using this system for detecting recombinant molecules, we discovered that RNA molecules can recombine by themselves, i.e., spontaneously change their primary structure [40]. If a mixture of fragments was incubated under different conditions for various time intervals and, before being plated on the agarose, was oxidized with periodate to suppress further recombination (see below), the number of RNA colonies increased with the time of the fragment mixture incubation prior to the contact with Qβ replicase, as well as with the increase in the incubation temperature (Fig. 12a). Sequencing of the recombinants had shown that recombination occurred between RNA internal sites more or less randomly (Fig. 12b).
Fig. 12. Spontaneous recombination between RNA molecules. a) Dependence of the recombinant yield on the temperature and duration of incubation [19]. b) Nucleotides brought close to each other (connected by light lines) within the recombinant molecules (from [47] with kind permission from John Wiley and Sons; ©2004 Federation of European Biochemical Societies).
We also found that Qβ replicase promotes RNA recombination, but by a different mechanism. First, the recombination rate in the presence of Qβ replicase was several orders of magnitude higher than the rate of spontaneous recombination. Second, the recombinants formed in a way that the 5′ fragment became entirely incorporated and attached to the 3′ fragment at various positions (Fig. 13a). It is interesting that Qβ replicase completely ignored the homology between the foreign extensions intentionally introduced into the fragments to facilitate replicase switching between the templates [39]. This has been the first example of RNA recombination by a mechanism other than template switching previously considered to be the only possible option for the RNA recombination [41]. Later it was shown in collaboration with the laboratory of V. I. Agol that even in the poliovirus, a classic example of the template switch recombination [42], non-homologous recombination of the non-replicative type can occur as well [43].
Fig. 13. RNA recombination catalyzed by Qβ replicase. a) Nucleotides that neighbor each other (connected by light lines) in the recombinant molecules. Identical nucleotides in the foreign sequences of recombining RNA fragments are shown in gray (from [47] with kind permission from John Wiley and Sons; ©2004 Federation of European Biochemical Societies). b) To recombine, the 5′-fragment must have free 3′-hydroxyl group (from [48] with kind permission from John Wiley and Sons; ©1999 Federation of European Biochemical Societies).
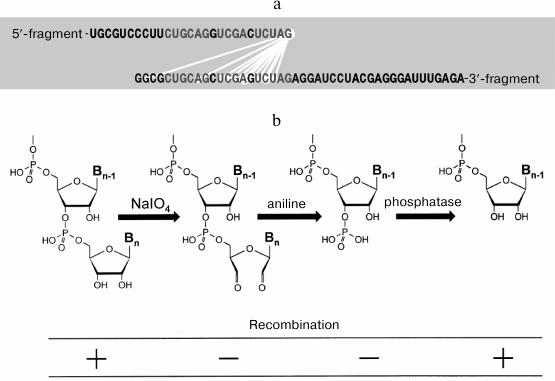
Moreover, it was found that removal of the 3′-hydroxyl by oxidation of the 5′-fragment with periodate makes this fragment incapable of recombination. If the oxidized RNA is treated with aniline so that the opened ribose ring is eliminated leaving the phosphate group at the next 3′-hydroxyl, the recombination still does not occur. However, if the phosphate group is removed with phosphatase, the ability of the 5′-fragment to recombine is restored (Fig. 13b). Therefore, we hypothesized that Qβ replicase catalyzes transesterification reaction, in which the 3′-hydroxyl of the 5′ fragment attacks the phosphate groups of the 3′-fragment [39]. However, later the picture proved to be not that simple. We found that recombination requires the 3′-fragment to be copied by Qβ replicase [44]. This fact does not fit into the proposed model; for now, we can only say that further studies are needed.
WHAT HAVE WE LEARNED?
1. The “template-free” synthesis of RNA by Qβ replicase is caused by RNA templates present in the air.
2. When single molecules of RNA or DNA are amplified in a gel (or another porous medium), they form colonies (molecular clones).
3. Genes can be expressed in molecular colonies to produce active proteins. In this sense, molecular colonies can be considered functional analogs of living cells.
4. Molecular colonies can serve as a tool for the absolute molecular diagnostics.
5. Artificial recombinant RQ-mRNAs are not amplified by Qβ replicase, but can be replicated by this enzyme in the presence of a translation system. This in turn leads to the increase in the encoded protein synthesis.
6. The template specificity of Qβ replicase is determined by the enzyme ability to acquire the closed conformation as a result of initiation, rather than by the promoter recognition.
7. The closed conformation of Qβ replicase is induced during the initiation on legitimate templates that have the property of functional and structural (presumably noncovalent) circularity.
8. Solving the crystal structure of the Qβ replicase core (the β subunit complex with EF-Tu and EF-Ts) has led to the discovery of the “bridge” capable of separating the template strand and its nascent complementary copy.
9. The replicase core is capable of transition into the closed conformation in the absence of protein S1 and maintaining the single-stranded form of the replicative complex during elongation.
10. Protein S1 is a termination factor for RNA replication; it catalyzes the exit of the single-stranded product from the active site. This function is performed by the S1 fragment OB1-2 composed of the first two OB domains.
11. The crystal structure of the Qβ replicase core complex with the OB1-2 fragment was determined. The OB2 domain was found to be involved in the formation of positively charged tract on the way of the product strand from the enzyme active site.
12. Recombination between RNA molecules can occur in bacterial systems.
13. RNA molecules are able to spontaneously change their primary structure as a result of self-catalyzed recombination events.
14. In addition to amplification of RNA molecules, Qβ replicase catalyzes their recombination at a rate that is much higher than the spontaneous recombination rate. It uses a previously unknown mechanism.
WHAT IS TO BE LEARNED?
The project was started in 1986 and has not been finished yet. There remain important unanswered questions, in particular:
1. What are the structures of the Qβ replicase closed conformation and the replicative complex? This needs to be figured out to understand the ability of Qβ replicase to discriminate between its proper (legitimate) and improper (illegitimate) templates.
2. What is the mechanism of termination of the product strand and its catalysis by protein S1? At present, we only know that such catalysis exists, but its mechanism remains a mystery.
3. What is the role of proteins EF-Tu and EF-Ts in RNA replication? A completely obscure question, yet no one has come close to answering it.
4. What is the mechanism of RNA recombination catalysis by Qβ replicase?
5. What is the role of protein S1 in protein synthesis on the ribosome besides mRNA binding? Whether it performs in the ribosome a function similar to that performed in the Qβ replicase at the termination step?
Acknowledgments
I thank, for their contribution to the project, the members of the group/laboratory of Viral RNA Biochemistry: Elena Vladimirovna Chetverina, Leonid Alexandrovich Voronin, Alexander Valerievich Munishkin, Viktor Ivanovich Ugarov, Natalia Henrikhovna Berestovskaya, Alexander Alexandrovich Volkov, Alexander Petrovich Alimov, Alexander Anatolievich Demidenko, Timur Rustemovich Samatov, Marina Vitalievna Falaleeva, Damir Sergeyevich Kopein, Alexandra Vladimirovna Kravchenko, Elena Alexandrovna Uzlova, Nikita Nikolaevich Vasilyev, Alexander Andreevich Gordeev, Zarina Shaimuratovna Kutlubaeva; our collaborators from the Institute of Protein Research: Alexander Sergeevich Spirin, Victor Dmitrievich Vasiliev, Igor Yurievich Morozov, Petr Nikolaevich Symonenko; our foreign collaborators: Marat Miratovich Yusupov, Lasse Jenner, Rune T. Kidmose, Gregers Rom Andersen, Charlotte R. Knudsen, Heidi Gytz, Durita Mohr, Paulina Seweryn, Yuichi Yoshimura, Fleur Dolman, Bosene Chelchessa, Frans A. A. Mulder, Ditlev E. Brodersen.
I also thank the Institute of Protein Research of the USSR Academy of Sciences, Russian Foundation for Basic Research, Molecular and Cell Biology Program, Russian Science Foundation, International Science Foundation, Howard Hughes Medical Institute, INTAS (International association for the promotion of co-operation with scientists from the New Independent States of the former Soviet Union) for their financial support of the project.
The present work was supported by the Russian Foundation for Basic Research (project No. 17-04-00597) and by the Program of the Presidium of the Russian Academy of Sciences for Basic Research in Molecular and Cell Biology and Postgenomic Technologies.
REFERENCES
1.Haruna, I., and Spiegelman, S. (1965) Autocatalytic
a synthesis of viral RNA in vitro, Science, 150,
884-886.
2.Weissmann, C., Feix, G., and Slor, H. (1968) In
vitro synthesis of RNA phage: the nature of the intermediates,
Cold Spring Harbor Symp. Quant. Biol., 33, 83-100.
3.Lewin, R. (1983) The birth of recombinant RNA
technology, Science, 222, 1313-1315.
4.Miele, E. A., Mills, D. R., and Kramer, F. R.
(1983) Autocatalytic replication of a recombinant RNA, J. Mol.
Biol., 171, 281-295.
5.Mills, D. R., Peterson, R. L., and Spiegelman, S.
(1967) An extracellular Darwinian experiment with a self-duplicating
nucleic acid molecule, Proc. Natl. Acad. Sci. USA, 58,
217-224.
6.Chetverin, A. B., Chetverina, H. V., and Munishkin,
A. V. (1991) On the nature of spontaneous RNA synthesis by Qβ
replicase, J. Mol. Biol., 222, 3-9.
7.Kacian, D. L., Mills, D. R., Kramer, F. R., and
Spiegelman, S. (1972) A replicating RNA molecule suitable for a
detailed analysis of extracellular evolution and replication, Proc.
Natl. Acad. Sci. USA, 69, 3038-3042.
8.Mills, D. R., Kramer, F. R., and Spiegelman, S.
(1973) Complete nucleotide sequence of a replicating RNA molecule,
Science, 180, 916-927.
9.Sumper, M., and Luce, R. (1975) Evidence for de
novo production of self-replicating and environmentally adapted RNA
structures by bacteriophage Qβ replicase, Proc. Natl. Acad.
Sci. USA, 72, 162-166.
10.Munishkin, A. V., Voronin, L. A., and Chetverin,
A. B. (1988) An in vivo recombinant RNA capable of autocatalytic
synthesis by Qβ replicase, Nature, 333, 473-475.
11.Horiuchi, K. (1975) Genetic studies of RNA
phages, in RNA Phages (Zinder, N. D., ed.) Cold Spring Harbor
Laboratory Press, N.Y., pp. 29-50.
12.Pasteur, L. (1860) Experiences relatives aux
generations dites spontanees, C. R. Acad. Sci., 50,
303-307.
13.Chetverina, H. V., and Chetverin, A. B. (1993)
Cloning of RNA molecules in vitro, Nucleic Acids Res.,
21, 2349-2353.
14.Chetverin, A. B., and Chetverina, H. V. (1995)
Method for Amplification of Nucleic Acids, Method for Expressing
Them, and Medium for Their Implementation, Russian Federation
Patent 2048522.
15.Chetverin, A. B., and Chetverina, H. V. (1997)
Method for Amplification of Nucleic Acids in Solid Media, U.S.
Patent 5,616,478.
16.Samatov, T. R., Chetverina, H. V., and Chetverin,
A. B. (2005) Expressible molecular colonies, Nucleic Acids Res.,
33, e145.
17.Szostak, J. W. (1999) Constrains on the sizes of
the earliest cells, in Size Limits of Very Small Microorganisms.
Proceedings of a Workshop, National Academy Press, Washington,
D.C., pp. 120-125.
18.Spirin, A. S. (2002) Omnipotent RNA, FEBS
Lett., 530, 4-8.
19.Chetverin, A. B. (2010) Can a cell be assembled
from its constituents? Paleontol. J., 44, 715-727.
20.Chetverina, H. V., Samatov, T. R., Ugarov, V. I.,
and Chetverin, A. B. (2002) Molecular colony diagnostics: detection and
quantitation of viral nucleic acids by in-gel PCR,
BioTechniques, 33, 150-156.
21.Chetverin, A. B. (2010) Molecular colonies,
Herald Russ. Acad. Sci., 80, 430-437.
22.Bentley, D. R., et al. (194 authors) (2008)
Accurate whole human genome sequencing using reversible terminator
chemistry, Nature, 456, 53-59.
23.Munishkin, A. V., Voronin, L. A., Ugarov, V. I.,
Bondareva, L. A., Chetverina, H. V., and Chetverin, A. B. (1991)
Efficient templates for Qβ replicase are formed by recombination
from heterologous sequences, J. Mol. Biol., 221,
463-472.
24.Wu, Y., Zhang, D. Y., and Kramer, F. R. (1992)
Amplifiable messenger RNA, Proc. Natl. Acad. Sci. USA,
89, 11769-11773.
25.Morozov, I. Yu., Ugarov, V. I., Chetverin, A. B.,
and Spirin, A. S. (1993) Synergism in replication and translation of
messenger RNA in a cell-free system, Proc. Natl. Acad. Sci. USA,
90, 9325-9329.
26.Billeter, M. A., Libonati, M., Vinuela, E., and
Weissmann, C. (1966) Replication of viral ribonucleic acid. X. Turnover
of virus-specific double-stranded ribonucleic acid during replication
of phage MS2 in Escherichia coli, J. Biol. Chem.,
241, 4750-4757.
27.Brown, D., and Gold, L. (1995) Selection and
characterization of RNAs replicated by Qβ replicase,
Biochemistry, 34, 14775-14782.
28.Palmenberg, A., and Kaesberg, P. (1974) Synthesis
of complementary strands of heterologous RNAs with Qβ replicase,
Proc. Natl. Acad. Sci. USA, 71, 1371-1375.
29.Ugarov, V. I., Demidenko, A. A., and Chetverin,
A. B. (2003) Qβ replicase discriminates between legitimate and
illegitimate templates by having different mechanisms of initiation,
J. Biol. Chem., 278, 44139-44146.
30.Gonzalez, R. G., Haxo, R. S., and Schleich, T.
(1980) Mechanism of action of polymeric aurintricarboxylic acid, a
potent inhibitor of protein–nucleic acid interactions,
Biochemistry, 19, 4299-4303.
31.Ugarov, V. I., and Chetverin, A. B. (2008)
Functional circularity of legitimate Qβ replicase templates, J.
Mol. Biol., 379, 414-427.
32.Chetverin, A. B. (2011) Paradoxes of replication
of RNA of a bacterial virus, Mol. Biol. (Moscow), 45,
127-137.
33.Berestowskaya, N. H., Vasiliev, V. D., Volkov, A.
A., and Chetverin, A. B. (1988) Electron microscopy study of Qβ
replicase, FEBS Lett., 228, 263-267.
34.Vasiliev, N. N., Jenner, L., Yusupov, M. M., and
Chetverin, A. B. (2010) Isolation and crystallization of a chimeric
Qβ replicase containing Thermus thermophilus EF-Ts,
Biochemistry (Moscow), 75, 989-994.
35.Kidmose, R. T., Vasiliev, N. N., Chetverin, A.
B., Andersen, G. R., and Knudsen, C. R. (2010) Structure of the Qβ
replicase, an RNA-dependent RNA polymerase consisting of viral and host
proteins, Proc. Natl. Acad. Sci. USA, 107,
10884-10889.
36.Kamen, R., Kondo, M., Romer, W., and Weissmann,
C. (1972) Reconstitution of Qβ replicase lacking subunit α
with protein-synthesis-interference factor i, Eur. J. Biochem.,
31, 44-51.
37.Vasilyev, N. N., Kutlubaeva, Z. S., Ugarov, V.
I., Chetverina, H. V., and Chetverin, A. B. (2013) Ribosomal protein S1
functions as a termination factor in RNA synthesis by Qβ phage
replicase, Nat. Commun., 4, 1781.
38.Gytz, H., Mohr, D., Seweryn, P., Yoshimura, Y.,
Kutlubaeva, Z., Dolman, F., Chelchessa, B., Chetverin, A. B., Mulder,
F. A., Brodersen, D. E., and Knudsen, C. R. (2015) Structural basis for
RNA–genome recognition during bacteriophage Qβ replication,
Nucleic Acids Res., 43, 10893-10906.
39.Chetverin, A. B., Chetverina, H. V., Demidenko,
A. A., and Ugarov, V. I. (1997) Nonhomologous RNA recombination in a
cell-free system: evidence for a transesterification mechanism guided
by secondary structure, Cell, 88, 503-513.
40.Chetverina, H. V., Demidenko, A. A., Ugarov, V.
I., and Chetverin, A. B. (1999) Spontaneous rearrangements in RNA
sequences, FEBS Lett., 450, 89-94.
41.Lai, M. M. C. (1992) RNA recombination in animal
and plant viruses, Microbiol. Rev., 56, 61-79.
42.Kirkegaard, K., and Baltimore, D. (1986) The
mechanism of RNA recombination in poliovirus, Cell, 47,
433-443.
43.Gmyl, A. P., Belousov, E. V., Maslova, S. V.,
Khitrina, E. V., Chetverin, A. B., and Agol, V. I. (1999)
Nonreplicative RNA recombination in poliovirus, J. Virol.,
73, 8958-8965.
44.Chetverin, A. B., Kopein, D. S., Chetverina, H.
V., Demidenko, A. A., and Ugarov, V. I. (2005) Viral RNA-directed RNA
polymerases use diverse mechanisms to promote recombination between RNA
molecules, J. Biol. Chem., 280, 8748-8755.
45.Chetverin, A. B., and Chetverina, H. V. (2007)
Scientific and practical applications of molecular colonies, Mol.
Biol. (Moscow), 41, 250-261.
46.Vasilyev, N. N. (2011) The Structure of the
Core of the Phage Qβ Replicase, Ph.D. Thesis, Moscow
University, Moscow, p. 135.
47.Chetverin, A. B. (2004) Replicable and
recombinogenic RNAs, FEBS Lett., 567, 35-41.
48.Chetverin, A. B. (1999) The puzzle of RNA
recombination, FEBS Lett., 460, 1-5.