REVIEW: The Molten Globule Concept: 45 Years Later
V. E. Bychkova*, G. V. Semisotnov, V. A. Balobanov, and A. V. Finkelstein
Institute of Protein Research, 142290 Pushchino, Moscow Region, Russia; E-mail: uralm62@rambler.ru* To whom correspondence should be addressed.
Received June 15, 2017; Revision received July 18, 2017
In this review, we describe traditional systems where the molten globule (MG) state has been detected and give a brief description of the solution of Levinthal’s paradox. We discuss new results obtained for MG-mediated folding of “nontraditional” proteins and a possible functional role of the MG. We also report new data on the MG, especially the dry molten globule.
KEY WORDS: protein self-organization folding, phase transitions in proteins, kinetics of protein folding, molten globule, formation and association of protein secondary structure, free-energy barrier, Levinthal’s paradoxDOI: 10.1134/S0006297918140043
Abbreviations: ANS, 8-anilino-1-naphthalenesulfonic acid; apoMb, apomyoglobin; ASA, accessible surface area; Bα-LA, bovine alpha-lactalbumin; β-LG, beta-lactoglobulin; DHFR, dihydrofolate reductase; FRET, fluorescence resonance energy transfer; Hα-LA, human alpha-lactalbumin; HSQC, heteronuclear single-quantum correlation (of NMR spectra); MG, molten globule; N, I, U are native, intermediate, unfolded state, respectively; TS, transition state; TTET, triplet–triplet energy transfer; WMG and DMG, “wet” and “dry” molten globule, respectively.
To the memory of Oleg B. Ptitsyn
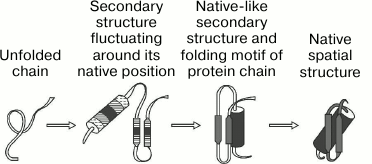
Forty-five years ago, Oleg B. Ptitsyn proposed a hypothesis of a stepwise mechanism of protein self-organization [1]. The available data on renaturation of globular proteins indicated that the primary structure of a protein carries information about its spatial organization [2, 3]. However, the protein chain is simply unable to sample all possible conformations in search for its unique spatial structure during a reasonable time (this is the so-called “Levinthal’s paradox” [4]). Therefore, it was only natural to assume that protein self-organization is a directed multi-step process (Fig. 1). With almost no experimental results to proceed from at the time (1973), Ptitsyn proposed that the key step in the process is the formation of what we now call the “molten globule” (MG) [1].
Fig. 1. The stepwise model of protein folding [1]. Secondary structure elements are shown: α-helices (cylinders) and β-sequences (arrows). Both predicted intermediates were later experimentally demonstrated and termed the “pre-molten” and “molten” globules.
In 1995 Ptitsyn published his extensive, detailed, and most informative review on the “Molten Globule and Protein Folding” [5], where this intermediate was shown to occur not only in the course of protein folding, but also during its denaturation, interaction with membranes, protein–protein interactions, etc. That review also covered the equilibrium molten globule, phase transitions in proteins, kinetics and mechanisms of protein folding, and the physiological role of the molten globule.
The years after publication of this review have brought better understanding of how a polypeptide chain finds the right way to its native, functional structure, which intermediates and transitional states it must overcome, and what is the energy of this process (see reviews [6-17] and references therein). Novel methods have been developed and applied to protein folding studies; they have revealed various details of the process of protein structure formation. In the same years, the attention of researchers was shifted towards protein “misfolding”, especially amyloid formation [6].
HISTORICAL EXCURSUS: THE MOLTEN GLOBULE IS A COMMON INTERMEDIATE
IN THE PATHWAY OF GLOBULAR PROTEIN SELF-ORGANIZATION
This section of the current review relates to the 50th anniversary of the Institute of Protein Research, Russian Academy of Sciences, and to the same anniversary of the Protein Physics Laboratory that Ptitsyn founded at the Institute. Here we focus on Ptitsyn’s concept (1973) that self-organization of a protein molecule necessarily results from a directed process encoded in its amino acid sequence and realized in time as a series of steps.
At that time, Ptitsyn’s team was developing a theory of formation of the secondary structure of globular proteins [18-25]. They showed that the stabilization of helical and elongated β-structural regions in the protein globule is determined by interactions between distant chain regions rather than local ones. However, local interactions alone are good enough for finding the secondary structure localization because they are related to long-range interactions that stabilize helical and β-structural regions exactly at the sites of local interactions within the unfolded chain. (Later, it was shown that statistically reliable matching of different interactions is necessary for the stability of a protein structure [26].) The further developed theories on other secondary structure elements, such as bends and loops, showed that their localization also originates from local interactions and are then stabilized by long-range interactions during formation of the globular structure [1, 18-25]. In other words, the type and localization of protein secondary structures are largely determined by the local amino acid sequence, although this structure is stabilized mostly by long-range rather than local interactions between chain regions.
Ptitsyn’s hypothesis implied that the fluctuating secondary structure seeds originate mostly from local interactions (primarily H-bonding) within the unfolded protein chain. Then they coalesce to form a compact globular structure (Fig. 1), which is not yet the unique native structure, but only an intermediate resulting mostly from nonspecific interactions between amino acid residues and the surrounding medium (water and the hydrophobic core of the incipient globule) [1, 21].
According to Ptitsyn’s hypothesis, further formation of the unique native structure consists of adjusting this intermediate, where the main role should be played by specific forces of interaction between adjacent amino acid residues. This hypothesis is based on the theory of helical structure of globular proteins, which indicated that the stabilization of helical regions in compact globules is determined mainly by long-range interactions rather than by local interactions, whose strength and specificity is not large [19, 20, 23, 25].
Secondary structure. The formation of secondary structure is of great importance because it not only decreases the chain volume and helps its transition from the aqueous medium to the globule, but it also contributes to correct topology of the chain (its spatial architecture), which is already an element of the tertiary structure. Experiments in support of this concept were reported later ([27], see also [47, 75, 78-80]) showing that H-bonding leads to protein compaction when a urea-unfolded chain is transferred into a “poor” solvent (water in this case) [27, 28]. At early stages of protein folding, seeds of secondary structure that is afterwards observed in the native protein usually appear. An interesting exception is exemplified by beta-lactoglobulin (β-LG) [29-32]. It has an early kinetic intermediate with a nonnative α-helix whose presence there before the globule has been formed is not a great surprise, because the β-LG site containing this helix is known to encode precisely an α-helical structure; further transition of this helix to β-structure is dictated by tertiary interactions. This example shows that the mentioned coordination of different interactions in a protein has a statistical rather than absolute character [26].
The MG, an equilibrium intermediate state. Ptitsyn’s hypothesis proposed in 1973 was developed in his subsequent works [21, 23, 33-35]. Concurrently, experimental studies of protein denaturation revealed an equilibrium intermediate [36] termed the “molten globule” (MG) [37] that showed properties of the folding intermediate predicted in 1973 (see [10, 34, 36], [38] and references therein, [39]). Studies of protein folding kinetics also confirmed the existence of kinetic intermediates of the MG type (see [5] and references therein, [34, 35, 38, 40, 41]).
The Protein Physics Lab was broadly involved in these studies ([41-44], also see references in review [45]). Specifically, a method using MG staining with ANS dye was proposed to test the accessibility of the hydrophobic MG core for solvents [43, 44, 46]. This method proved to be most useful in identifying the equilibrium MG [46].
As emphasized by Ptitsyn’s hypothesis, the formation of a native-like topology is a must for the existence of the MG. This requirement was later demonstrated for bovine alpha-lactalbumin (Bα-LA) [47, 75, 78], a cold shock protein [48], and other proteins [10, 49].
Proteins folding with intermediates (multi-state proteins) and without intermediates (two-state proteins). Studies of protein folding kinetics revealed that proteins mostly fold via their intermediate states formed in the folding pathway [41, 49, 50]. Proteins having more than two states (native, one, or several intermediates, and fully unfolded) are called “multi-state proteins”. In their folding pathways, there are not one, but at least two free-energy barriers (Fig. 2).
Fig. 2. Free-energy profiles for proteins having in their folding pathway two (a), three (b), four (c), and five (d) states (free-energy minima), i.e. one, two, three, or four kinetic stages, respectively. TS or TS*, the main rate-limiting folding intermediate (main free-energy maximum) is always positioned before the tightly packed, native, or “almost native” state, and the transition between them is often associated with proline isomerization in the tightly packed globule [41].
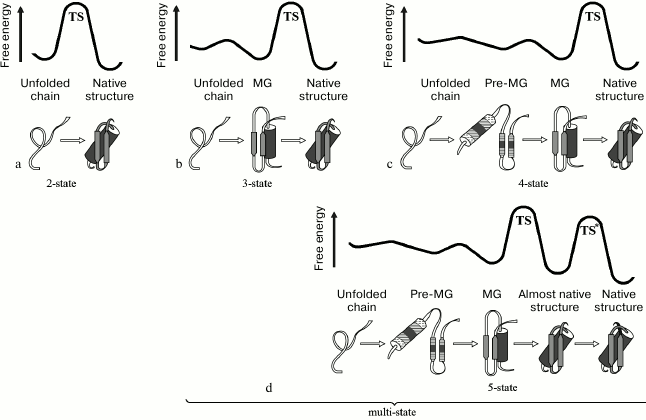
However, some proteins, which are usually small in size, fold via one barrier, i.e. they have only two states – native and unfolded (two-state proteins) (Fig. 2a) [49-51]. Galzitskaya and colleagues [52] and then Kamagata and colleagues [49, 50] have analyzed the data on folding kinetics of both protein types. Their conclusion was that the folding rate constants of multi-state proteins (significantly different in size) correlate well with the protein chain length. On the other hand, differences in the folding rates of small two-state proteins correlate with the backbone native topology (more precisely, with the average distance between interacting amino acid residues) [53], but not with the backbone length, although the latter becomes important in large two-state proteins (few in number) [54]. Kamagata and colleagues concluded [49] that the multi-state protein folding (Fig. 2, b and c) through accumulation of the productive kinetic intermediate (i.e. MG) represents a common model of the protein folding mechanism, while the intermediate-free folding of two-state proteins is a simplified version (Fig. 3) of this mechanism.
Fig. 3. Scheme demonstrating why the same protein can show either three-state or two-state behavior depending on conditions, although its local free-energy minimum always corresponds to the folding intermediate. The main folding-rate-limiting maximum of free energy (TS) precedes the N-state. a) The free-energy profile for transition from the unfolded (U) state to the native (N) state through an intermediate (I) for the case when N is much more stable than U. Here, in comparison with U, I is stable in the wild-type (WT) protein and unstable in the mutant (mut); this difference is shown in the gray I area as a solid or dotted line, respectively. This figure illustrates the situation when only I is affected by the mutation. The U-to-N transition is a two-stage process (U → I → N) in case of the WT protein (behaving like a three-state protein) because the TS-preceding I is more stable than U. However, for a mutant behaving like a two-state protein, the folding seems to be one-stage (U → N) because in comparison with U, the I-state is unstable, and therefore cannot be observed in an experiment. b) The U-to-N transition for the case when N is only slightly more stable than U. With increased free energy, N and U stabilities become still closer to each other, thereby making the I-state unstable, which results in one-stage character of the U → N transition in both the WT and mut proteins. In other words, here the WT protein (and the mut too) behaves like a two-state protein. c) The N-to-U transition for the case when U is more stable than N. Then, this N → U transition is a one-stage process (like that of the “all-or-none” type [55] in two-state proteins) because the overall free-energy maximum (TS) is positioned between N and I, thus allowing the slow N → I transition to mask the rapid I → U transition.
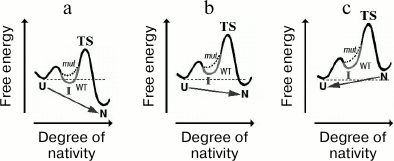
One review [11] reports that intermediates are present even in the folding pathway of proteins that initially were believed to be two-state proteins. Figure 3 demonstrates why the same protein can show both two-state and multi-state behavior depending on conditions.
It is stated in [11] that the nonlinear character of the dependence of the folding rate logarithm on the final denaturant concentration (Fig. 4), which is sometimes observed in folding kinetics of two-state proteins, often results from the presence of intermediates that are stable (in comparison with U) only at low concentrations of the denaturant; with increasing denaturant concentration their stability decreases, and they cannot be observed. This study also reports that the model of successive protein folding predicts stabilization of intermediates under specific conditions, supposedly due to changes in solvent composition. For example, the addition of sodium sulfate (which increases the stability of protein structures) leads to the appearance of an additional “burst” phase (reflecting the presence of intermediates) in mutants of ubiquitin [56] and tendamistat [57], while a change in pH causes the three-state folding of hisaxtophilin [58]. For protein G and the immune modulator Im, a pH change together with the addition of sodium sulfate also results in the appearance of folding intermediates [59, 60]. As mentioned above, the presence of intermediates is manifested as nonlinearity of the dependence of the folding rate constants on the final denaturant concentration (chevron plots, see Fig. 4).
Fig. 4. Typical view of a “chevron plot”, i.e. denaturant concentration dependence of the rate of approximation to equilibrium (kapp) by the native and denatured conformations of a protein molecule.
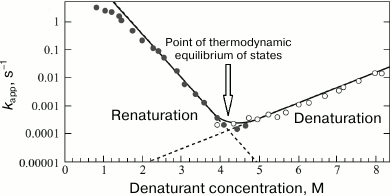
This behavior of the kinetic curves was observed to result from change in temperature and from a mutation made in the “WW domain” [61, 62]. The authors of those studies emphasize that the difference between the two-state and multi-state protein folding is related to the difference in stability of partially folded intermediates. It is noted that the above-mentioned curvature of the left side of the chevron plot (Fig. 4) is caused exclusively by the intermediates (due to changes in the rate-limiting step), whose stability increases with decreasing denaturant concentration. In addition, the authors suggested that the presence of intermediates is of importance for the folding process, because the conformational space becomes narrower, thus facilitating the search for the native contacts [63].
Folding intermediates arising in different conditions belong to one and the same region of the diagram of protein conformations. It often comes into question whether intermediates observed under different conditions (pH, denaturant concentration, temperature) are distinct, or just variants of the same intermediate. To answer this question, a study of denaturation of sperm whale apomyoglobin was made, which yielded a 3D diagram of its conformations in three coordinates, namely, pH, temperature, and denaturant (urea) concentration. This led to the conclusion that all apomyoglobin intermediates belong to one and the same region of the diagram, with no transitions between them [64].
MULTIPLICITY OF INTERMEDIATE STATES IN GLOBULAR PROTEIN FOLDING:
NEW RESULTS ON MOLTEN GLOBULES, INCLUDING “DRY” MOLTEN
GLOBULES
The problem of intermediate diversity is closely connected with that of protein folding. As early as 1989, Finkelstein and Shakhnovich proposed a theory of thermodynamic states of a protein chain [65-67]. According to this theory, in the folding chain the stable (or metastable) MG must occur between the N- and U-states. In addition, the theory distinguished “wet” MG (WMG) and “dry” MG (DMG); unlike the latter, the WMG implies that the solvent penetrates pores of the melted (loosened) protein (Fig. 5). It was proposed [65-67] that in most cases a WMG is more stable than a DMG. And it is the WMG that is commonly observed in proteins [68].
Fig. 5. Compact stable states of a protein globule (tightly packed N and melted (loosened) MG), unstable TS′ (needed for globule melting) between them, and the unfolded chain (U). MG can be either “wet” (with solvent molecules present in pores of the loosened protein) or “dry” (without solvent molecules). The MG pores are sufficiently large to ensure free fluctuations of the side groups. The transition state required for native-state melting is shown only schematically (as a homogeneous structure whose pores are smaller than those of the MG; being large enough to disturb intraglobular interactions, these pores are yet too small for free fluctuations of the side groups, which underlies the instability of TS′ [65, 66]). TS′ is heterogeneous in itself: it comprises a native-like part and a denatured part (see further [92]). a) TS′ is the main transition state in the protein denaturation pathway (the case considered in [65-67]). b) TS′′ is the main transition state during the denaturation of the protein with water penetrating the globule (which was not considered in [65-67] but was reported in several other studies [69, 70]).
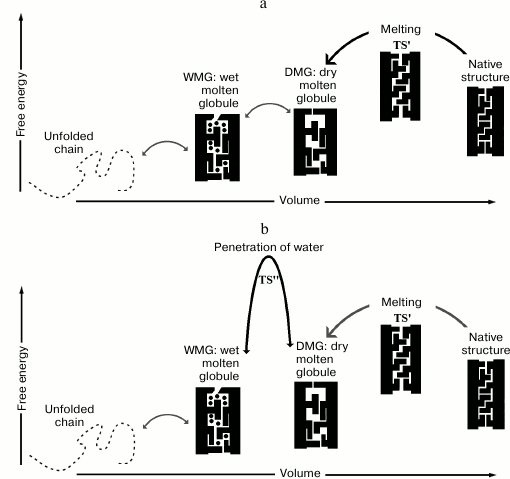
The DMG was detected experimentally using newly developed, sensitive techniques. An example of a DMG is the intermediate observed during the unfolding of ribonuclease A (RNase) [69]. The title of this study describes well its result: “Direct NMR evidence for an intermediate preceding the rate-limiting step in the unfolding…”. Importantly, the authors declare that this intermediate is exactly the DMG predicted earlier [66]! Thus, contrary to the assertion [65-67] that the main TS is located between N and MG (see Fig. 5a), it appears to be sometimes positioned between the DMG and the WMG (see Fig. 5b).
More direct evidence for the existence of a DMG in a protein (monellin) during unfolding has been reported [70]. Both equilibrium and kinetic GdmCl-induced denaturation of monellin was observed using FRET, CD, and ANS-binding techniques. It was shown that in the initial stage of the unfolding process the distance between the C-terminus of the single helix and Trp of monellin located near its N-terminus changed dramatically. However, Trp fluorescence and ANS-binding show that water was still away from the protein nucleus. This is direct evidence for the presence of a DMG at the initial stage of monellin unfolding. At this stage, the volume of the molecule increases, provoking rupture of the native contacts, but without water penetration into the hydrophobic protein nucleus. The authors concluded that denaturation of the native protein structure is a cooperative process, while the subsequent swelling of the globule occurs gradually (as predicted in [67]).
Works by Baldwin and Rose (see reviews [13, 15] and references therein) contribute to further development of this issue. They emphasize that the discovery of the DMG destroys the concept that the two-state protein folding is a one-stage process. They note that so far, the DMG has been found in the unfolding pathway of only a few proteins, namely, RNase A [69] (see above), dihydrofolate reductase (DHFR) (for the study of which five tryptophan residues were replaced by [19F]tryptophan, and NMR in stopped-flow mixing was used) [71], monellin (see above) [70], and villin (HP35) [72]. For the study of HP35, the TTET method (triplet–triplet energy transfer) was used. A reversible equilibrium between tightly packed and loosely packed states was found to depend on temperature and GdmCl concentration. The observed conformational fluctuations in this protein destroyed the van der Waals contacts between its Trp residues and the introduced label.
This issue was further developed in a detailed study of human serum albumin (HSA) [73]. During the pH-induced unfolding of this protein, its equilibrium intermediate was identified as a DMG by a variety of techniques, including FRET, dynamic fluorescence quenching, and near- and far-UV CD. The data indicated that two of the three HSA domains exhibit DMG properties, specifically, a larger volume in comparison with the native protein and disturbed tight packing of the side groups without solvation of the hydrophobic core (it remained dry). In these domains of HSA, side group disbonding and core solvation occurred at different stages of the pH-induced unfolding, contrary to the common unfolding mode [73].
DMG, WMG, and a solvated transition state between them were observed during the urea-induced unfolding of barstar [74]. Using various fluorescence and CD techniques, it was found that although the native-like intermediate showed weakened tertiary interactions, the core structure remained intact, thus preventing water penetration. This suggests the following pathway of barstar folding: a WMG forms rapidly from the U-state; then the folding process slows, thereby implying that the rate-limiting step might be positioned between the WMG and the DMG. However, in this situation it is difficult to study the DMG because its formation occurs after the rate-limiting step [74].
NEW DATA REPORTED FOR “TRADITIONAL” PROTEINS
New data about proteins traditionally used for MG studies. Over the years after publication of Ptitsyn’s comprehensive review on the molten globule (1995), many new studies have been reported in the literature. When focus on proteins traditionally used in MG research, these studies consider details of MG structure and peculiarities in the behavior of proteins either with point mutations or under drastically changing conditions.
α-Lactalbumin (α-LA). a) Kuwajima’s team published a review covering the kinetic and hydrodynamic aspects of the formation of the MG of α-lactalbumin [10, 75]. The available data indicate that the α-LA MG is heterogeneous, that is its alpha domain is structured, while its beta domain is significantly unfolded. However, its backbone fold is already native-like. The formation of a MG from a fully unfolded chain occurs in milliseconds [10, 75]. The structural stability of the kinetic intermediate is the same as that of the equilibrium MG, and this kinetic intermediate is mandatory for the folding pathway of this protein.
b) α-LA is a Ca2+-binding protein. Wu and colleagues showed [76] that Ca2+ binds to the β-domain of α-LA, which leads to the cooperative formation of tertiary interactions promoting a more rapid MG-to-N transition.
c) NMR studies of α-LA folding kinetics showed that as much as 85% of its helical structure forms at an early folding stage, and the compactness of this structure is almost native [47]. According to H/D exchange, mass-spectrometry, and NMR, this structure originates mostly in the α-helical domain. The rest of the protein (β-domain) folds more slowly, and addition of Ca2+ stabilizes the already formed structure and accelerates the MG-to-N transition. For the MG state, this study clearly demonstrated the native-like character of the main chain topology, especially in the rapidly folding α-domain, while subsequent deceleration of the process is attributed to structural arrangement of the β-domain and the transition state [10, 75].
d) Quezada et al. [77] used the 15N-H 2D HSQC NMR technique to study the effect of mutations, especially Pro substitutions, on cooperativity of folding/unfolding of the human lactalbumin (Hα-LA) MG. The appearance of well-defined resonances suggested noncooperative unfolding induced by denaturants [77]. At low denaturant concentrations, the resonances corresponded to the Hα-LA MG segments in the least stable regions of the structure. In experiment, Hα-LA with one disulfide bond (Cys28–Cys111) was used. As shown, mutations in the A-helix strongly affected stability of the Hα-LA MG. It was demonstrated that the regions containing the Cys28–Cys111-bonded peptides 1-38 and 95-120 are not only highly helical, but they also show typical features of a MG, including binding to the hydrophobic probe ANS. The authors concluded that the MG is more likely stabilized by a relatively large number of nonspecific hydrophobic interactions rather than a small number of highly specific interactions [77].
e) For bovine α-LA, a MG can be induced by heat, various alcohols, and even by added oleic acid. Since the latter induces apoptosis of cancer cells, the here MG can be considered functional. Fontana’s team comparatively analyzed Bα-LA by circular dichroism and limited proteolysis. The Bα-LA intermediates observed at 45°C and neutral pH or with 15% trifluoroacetic acid or 7.5 equivalents of oleic acid appeared to be structurally similar to the Bα-LA MG at pH 2.0. Limited proteolysis using proteinase K revealed that peptide disbonding occurred mostly in the β-domain, while the proteolytic fragments were linked by four S–S bonds. The main conclusion was that the Bα-LA MG retained the native-like tertiary fold manifested in the well-structured α-domain [78].
f) Development of NMR techniques, and specifically photo-CIDNP (chemically-induced dynamic nuclear polarization using pulse labeling), allowed the study of side-group conformations in partially folded states and in kinetic intermediates during the real folding time [79]. This technique was used to characterize various MGs of bovine and human α-LA at the level of individual amino acid residues. Photo-CIDNP can distinguish interactions between aromatic residues such as Trp, Tyr, and to a lower degree His. It was found that the main backbone fold is stabilized by a rather small number of solvent-inaccessible groups, and that contacts between the hydrophobic residues involved in this process are not necessarily native.
g) The Raman optical activity (ROA) technique developed by Barron’s team was used for characterization and comparison of various MGs. The vibrational optical activity can be measured due to small differences in the scattering intensity of right- and left-polarized light, thus distinguishing in ROA spectra bands typical for loops and turns, as well as for secondary structure elements and side chains [80]. This gives information about the backbone fold and protein structure dynamics. ROA spectra of calcium-binding equine lysozyme were compared with those of bovine α-LA; hydration of their α-helices was also revealed. Together, these results indicate that the equine lysozyme MG has a more structured core than that of the α-LA MG, including three interacting protected helices against two in α-LA. In addition, it was reported that the structure of the Ca2+-binding loop remained unchanged even in the absence of Ca2+ [80].
β-Lactoglobulin (β-LG). a) The folding of β-LG is an interesting case: although it is mostly a β-structured protein [29, 30, 32], a nonnative α-helical sequence was observed during the “burst” phase where an intermediate is typically formed. Stopped-flow CD and optical absorption demonstrated that the main properties of this intermediate closely resemble those of other proteins. Submillisecond observations of the β-LG folding by CD and SAXS (small-angle X-ray scattering) indicated that the conformation of its early intermediate was almost as compact as that of the native form [32]. A peculiar feature of β-LG is the presence of a residual β-sequence in its unfolded chain, while its intermediate conformation has a nonnative α-helix whose localization was encoded by local interactions.
b) It was suggested by Sakurai and colleagues [30] that α-helix is required to accelerate the folding of β-LG and to prevent its aggregation. In the context of Lim’s hypothesis on the formation of highly helical intermediate globules [81], it was suggested that a nonnative α-helix would promote long-range contacts with the native helix, thereby stabilizing the intermediate conformation and facilitating its transformation into the correctly folded β-structure of the native protein [32].
c) On the other hand, it was reported that short N-terminal fragments of apomyoglobin, a completely helical protein, coalesce to form a β-structure or possibly even amyloids [82]. This is explained by the fact that these short helical fragments cannot be effectively packed into a hydrophobic core at the intramolecular level, and therefore self-association (or interactions with chaperones) is required to screen hydrophobic groups.
Apomyoglobin (apoMb). a) Folding/unfolding of apomyoglobin, a completely helical protein, attracts great attention. The most intensive studies of its folding kinetics have been reported by Wright’s team (see review [17] and references therein). It has been shown that destabilization of the H-helix and substitution of Gly for its amino acids cause an altered folding pathway of apoMb [83]. New techniques provided detailed information about early events in apoMb folding. During the dead time (<300 µs), rapidly formed helices and structural collapse were registered by a continuous-flow technique combined with CD and SAXS [84]. H/D exchange and 2D NMR techniques used to monitor rapid apoMb folding (during 400 µs) revealed formation of a compact state that included the major parts of the A, G, and H-helices already inaccessible for H/D exchange, while the B, E, and C-helices underwent H/D exchange later and became stabilized within milliseconds, thereby confirming the hierarchical character of the folding of apoMb [85].
b) Measurements of NMR line-broadening for spin-labeled samples revealed time-varying hydrophobic interactions between different regions of the polypeptide chain that arise in the initial stage of protein folding [86]. Considering the average surface embedded into the structure during protein folding, numerous native and nonnative hydrophobic clusters of different stability were identified. Using directed spin-labeling, an increase in paramagnetic relaxation of the unfolded protein was observed. The localization of spin-labeled residues was determined using 1H-15N HSQC NMR for apoMb at acidic pH. In the same report [86], the role of the embedded surface in initiation of hydrophobic collapse is discussed.
c) In NMR experiments 15N, 1HN, and 13CO relaxation dispersion profiles were used to study spontaneous processes of folding and unfolding of apoMb. At pH above 5.0, the dispersion is determined by fluctuations of the F-helix region, which cannot be seen on NMR spectra. The measured relaxation values of the residues in contact with the F-helix in its native state were the basis for identifying a transition state that resulted from the local unfolding of the F-helix and its undocking from the core. This transition state closely resembled the equilibrium MG of apoMb observed at high temperature and low pH [87]. Analysis of these data led to the conclusion that in this state changes occur in the protein backbone and in other parts of the chain, thus indicating significant rearrangements in the native and MG states or their folds. The authors of this report believe that NMR relaxation dispersion can be used under moderately destabilizing conditions when the folding and unfolding kinetics can be measured in the experiment. It is also applicable to characterization of the early unfolding intermediate, which is of importance for understanding the processes of protein aggregation and amyloid formation [88].
d) The MG of apoMb is “wet”; water dynamics in the nonpolar MG core have been considered (see [88] and references therein). Hydration dynamics were studied using dynamic nuclear polarization and EPR. The former method reveals the local dynamics of the solvent within 10 Å of a spin label, whereas the latter tests the polarity near the label and the mobility of the spin-labeled protein. It was found that in the MG state, the nonpolar apoMb core is hydrated with water molecules whose mobility was only 4-6 times lower than that in the free solvent, while in the unfolded state at pH 2, the hydration water diffuses rapidly. The conclusion was that in apoMb, the water dynamics are site-specific and folding degree-dependent; the residues located on the native protein surface may appear to be less hydrated in the MG state than in the N-state. The presence of diffusing (albeit slowly) hydration water can facilitate conformational rearrangements, which consolidate the loosely packed MG side chains into a compact, water-free native hydrophobic core ([88] and references therein).
e) In 1999, Ptitsyn compared the sequences of 728 globins and found that they had only six conserved residues not bound to heme, located pairwise on the A-helix (Ala10 and Trp14), G-helix (Ile111 and Leu115), and H-helix (Met131 and Leu135) [89]. It was suggested that these nonfunctional conserved residues were required for fast and correct folding of apoMb into its stable 3D structure. Appropriate mutants were synthesized in Ptitsyn’s lab, along with six others which, though carrying Ala substitutions for nonconserved residues, were theoretically important for protein folding, and specifically for the folding rate of apoMb [90, 91]. These residues belonged to the B-helix (I28, F33), C-helix (L40), D-helix (M55), and E-helix (L61, L76). The pH-induced unfolding of all these mutants was studied, including determination of thermodynamic characteristics of their U → I and I → N transitions, as well as Fersht’s parameter φ (reflecting comparative contribution of the replaced side groups in I-state stabilization [92]). It was concluded that for conserved residues, the interaction force shown by side groups in the MG state does not exceed 50% of that in the native state, while for nonconserved residues it is close to 0% [93, 94]. This supports Ptitsyn’s hypothesis that conserved residues are important for the apoMb folding intermediate, whose structure serves as the basis for formation of other helices and then the native fold of this protein.
For the same mutant proteins, kinetics of their folding over a wide range of urea concentrations was studied using Trp fluorescence. Solely kinetic results showed that the apoMb folding involves one intermediate that is similar to the equilibrium MG. The study of 12 mutant proteins clarified the role of the selected amino acid residues in protein stability and the rate of formation of the native fold. It was found that the introduced mutations strongly affected stability of the N-state of the mutant, while the stability of the I-state was influenced only slightly. As shown, amino acid residues of the A,G,H-helical complex contribute to stability of the intermediate and the folding nucleus in the U → I → N transition. On the other hand, in the I → N transition, the folding nucleus arises after the A,G,H-complex has been formed and includes mostly residues belonging to the B-, C-, and E-helices [95]. The φ-values (Fersht’s parameter) were obtained for the barrier existing in both the I → N and U → I → N transition of apoMb.
Together, these results support Ptitsyn’s hypothesis about a significant role of conserved residues in the U → I transition [89] and show that these residues are of importance for the intermediate formation at an early stage of the folding of apoMb. In the folding process, the side group interaction force increases step-by-step and reaches its maximum in the native state of apoMb.
FUNCTIONAL ROLE AND OCCURRENCE OF THE MOLTEN GLOBULE STATE IN
LIVING SYSTEMS: NONTRADITIONAL SYSTEMS WITH THIS STATE OBSERVED
It seems important and interesting that the area of identification of the molten globule expands with time. Also, it appears that in some cases a MG with its fluctuating tertiary structure has functional significance, thereby supporting the hypothesis proposed in 1988 [96, 97] and further developed in numerous studies on “intrinsically disordered” proteins (see, e.g. [98-101]), where properties of a MG are often demonstrated under physiological conditions. The functional specificity of intrinsically disordered proteins is manifested mostly in the mechanism of their interaction with ligands.
a) When in the MG state, some proteins can undergo co-folding and co-binding to a substrate, coupled with effective catalysis. Hilvert’s group [102] reported synthesis of a monomeric form of chorismate mutase (mCM) by introducing an additional loop into the molecule that otherwise exists as a dimer. The resulting structure was characterized by fast H/D exchange, mass-spectroscopy, CD, NMR spectroscopy, and ANS binding. The monomeric enzyme appeared to have not a rigid, but rather a MG-like structure and to exhibit high activity. Upon substrate binding, its MG structure became tightly packed in many respects. This “enzymatic molten globule” was studied by 3D NMR [103]. Kinetics and thermodynamics of ligand binding to mCM showed that the binding/release process of this ligand was much more rapid than that typical for the native dimeric form of CM [104]. These results suggest that MGs may be promising for generating new catalysts.
b) The case of ligand binding to a partially unfolded protein was considered using dihydrofolate reductase (DHFR) from Escherichia coli [105]. Binding of NADP+ (a functional ligand) to DHFR showed that the partially unfolded enzyme could bind the substrate in a manner similar to that of the native enzyme. Also, it was found that DHFR could be partially unfolded without ligand release, although its binding capacity was reduced. Analysis of the crystal structure of the DHFR–NADP+ complex and free energy required for the partial unfolding of DHFR at various NADP+ concentrations suggested that the adenine-binding domain of the partially unfolded protein retained its structure and the ability to bind adenine.
c) Ubiquitin (Ub) is another interesting case. It participates in many cellular events, from cell degradation to DNA reparation and chromatin rearrangement [106], which requires a certain plasticity of its structure. It was shown that at 3-kbar pressure the native structure of ubiquitin undergoes a transition to an intermediate state with properties close to a MG. The study of Ub mutants by NOE, 15N spin relaxation, HSQC, and 15N-1H NMR revealed that the Q41N mutant of ubiquitin had similar structural changes. What is still more interesting, similar changes in ubiquitin structure were observed upon its binding to E2-ubiquitinating enzyme [106]. The functional role of the MG state is important not only for the E2-ubiquitinating enzyme, but also for other modifying proteins involved in E1–E2–E3 cascade reactions [106].
d) Apart from mCM, periplasmic binding proteins should be mentioned here [107]. They are carriers of specific ligands Leu, Ile, maltose, and ribose that they bind both in the native state and when MG state (at pH 3-4), though in the latter case their binding efficiency is reduced. These states are stabilized only by hydrophobic interactions, hydrogen bonding, and specific packing in the absence of prosthetic groups. This might be explained by the necessity to undergo translocation through a membrane. The intermediate of the maltose-binding protein interacts with the chaperone SecB. The ligand binding by these transport proteins involves rather distant residues. Therefore, it can be performed by proteins in the MG state if this state retains native-like topology [107].
e) Interesting data were reported on the folding of staphylococcal nuclease (SNR121) fragment 1-121 synthesized on the ribosome [108]. This fragment showed all properties of a MG, unlike fragments of larger or smaller size. The authors concluded that the folding of this protein proceeds step-by-step: from accumulation of the secondary structure through different structured states to the native structure, and that continuous adjustment of its conformation is necessary for its correct folding and active expression.
Appearance of the MG in different systems. a) In a study of measles virus, an intermediate in the folding of the tenth domain of phospholipid P was identified and its structure characterized [109]. Some of its mutants followed the two-state folding pattern, while others reached their native state through intermediates with a native-like α-helical structure.
b) An MG-like intermediate was identified by NMR in the DNA-binding domain of protein p53 upon its interaction with the chaperone Hsp90. Intensive H/D exchange was observed in this complex, while in solution protons of p53 itself exchanged only slightly, and Hsp90 exhibited H/D exchange only in the binding cavity [110].
c) To understand the formation of cataracts, urea-induced unfolding of the mutant cataract-associated V75D γD-crystallin was studied [111]. Combined NMR and SAXS analysis showed that at 4.2 M urea, this protein was in an intermediate state where its C-terminal domain was rather structured, while its N-terminal domain had no distinct structure. The authors suggest that these conformations can contribute to the formation of early cataracts also under physiological conditions.
d) A MG state was observed for [3Fe4S] [4Fe4S] ferredoxin upon heating at pH 2.5. These conditions led to loss of Zn2+ and formation of apoferredoxin, in which the MG state was observed upon cooling [112]. The authors suggested that the apo-ferredoxin MG might be physiologically functional because its flexibility can facilitate incorporation of the [3Fe4S] [4Fe4S] complex into the apoferredoxin structure.
e) Another example is one of the α-galactosidases, a widespread enzyme catalyzing the hydrolysis of the terminal α-galactosyl moieties from glycolipids and glycoproteins. The enzyme is used in therapy of Fabry’s disease, and it is effective in conversion of group B blood into group O blood. It is also used for transglycosylation and reversibility of hydrolytic reactions. For a legume α-galactosidase, it was shown [113] that at pH 2.0 it forms a MG-like state that retains the secondary structure and a screened Trp residue. Because loss of activity is associated with disturbed secondary structure, this enzyme remains active over a wide range of denaturant concentrations. Therefore, the study of its folding is aimed at synthesizing a recombinant enzymatic protein with improved stability that is useful for biotechnological application.
f) We now consider two cases of the folding of recognized two-state proteins in which later studies revealed an intermediate; these are acyl-CoA-binding protein and the SH3 domain of PI3 kinase.
Acyl-CoA-binding protein. A modified continuous-flow technique (dead time 70 µs) in Trp fluorescence experiments showed that during the folding of this protein a dramatic increase in energy migration could be registered by FRET after 100 µs. This suggests the presence of a previously unknown partially folded intermediate with about 1/3 of the solvent-accessible surface embedded in the structure [114].
SH3 domain of PI3 kinase. The folding/unfolding of this protein previously used in TS studies as a two-state protein was then studied by a combination of H/D exchange and electrospray mass spectrometry [115]. In the pathway of its guanidine hydrochloride-induced unfolding, there first appeared an intermediate with five exchangeable amide protons, and then the remaining 14 protons underwent exchange. During its folding, the reverse order was observed.
g) Discussion of T4 lysozyme concludes this section devoted to interesting cases of folding pathway intermediates. T4 lysozyme has two domains, N- and C-terminal. For the latter, an additional “hidden” intermediate not revealed by common kinetic techniques was detected. Equilibrium urea-induced unfolding of T4 lysozyme showed that its commonly observed intermediate consisted of an unstructured N-domain and a structured C-domain. On the other hand, kinetic H/D exchange studies revealed an intermediate where both domains were structurally disturbed [116]. To resolve this contradiction, kinetics of the folding and unfolding of this protein was explored to yield chevron plots for its mutant variants with turns on both of their branches. Thorough analysis of the H/D data showed two intermediates, one on each side of the major TS, which adequately explained these experimental results. The detected intermediate (termed “hidden” for going unobserved by kinetic techniques) was structured solely in the C-domain. However, in the case of T4 lysozyme, kinetic techniques were useful in detecting this intermediate, thus making a considerable contribution to understanding the role of intermediates observed on the both sides of the I-state barrier during folding of the protein [116].
SOLUTION OF LEVINTHAL’S PARADOX
Despite great success in studies of intermediates occurring in protein folding pathways, there remained an unanswered question associated with Levinthal’s paradox – how does a protein chain selects its native structure from a huge number of possible variants during a short “biologically reasonable” time? There have been many attempts to explain or reject this paradox (which, using Shakhnovich’s mot juste, “played the role of Fermat’s Big Theorem for Protein Science”), but the results gave only hints in the search for a solution, but not the solution as such. Since several recent books and reviews published by our research team are devoted to the analysis of these results [117-121] (see the latest of them in this issue of Biochemistry (Moscow)), we take the liberty not to dwell on them in detail but to describe in outline the solution. A theoretical estimate of the time sufficient for a chain to do so was obtained by approaching Levinthal’s paradox “from the other end”, that is, by identifying the optimal way of protein folding and then considering its unfolding [122-124]. The theory used only two parameters: the time required for amino acid residue transition from one conformation to another (~10 ns) and the average energy required for its transition from the unfolded to the globular state (~5 kJ/mol). This was used to estimate the height of the free-energy barrier in the protein folding pathway, thereby giving the time range theoretically sufficient for the folding of single-domain globular proteins of any size and any stability of native structure. It appeared that all proteins whose folding rates were measured both under physiological conditions and near the point of equilibrium between their native and denatured state (these are over a hundred already) fall into a “golden triangle” with the following coordinates: protein size – logarithm of its folding rate [124]. This region is permitted theoretically (and confirmed experimentally) for both two-state and multi-state proteins. The size of such a self-organizing protein domain (again experimentally confirmed) is limited to ~500 amino acid residues. Longer sequences should be divided into domains. Also, the theory shows that the folding of proteins containing less than ~80-100 residues is solely under thermodynamic control, while that of larger proteins is more likely controlled by kinetics, because its task is not only to achieve a stable structure, but also in a “biologically reasonable” time.
So, the sequence length dependence of folding rates (from ~exp(0.5L2/3) to ~exp(1.5L2/3) ns) has been determined, and Levinthal’s paradox has been solved in principle. However, this solution is in terms of energy and time, and not in terms of sampling of protein chain structures to identify the most stable structure, which is not in terms of Levinthal’s paradox as such.
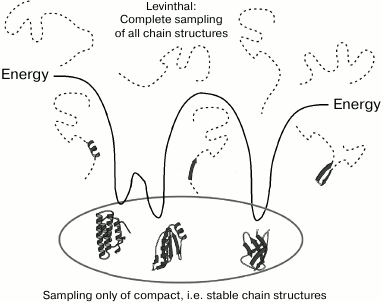
The solution in terms of the necessary sampling was obtained only recently [119-121, 125] (although seemingly, this could have been done by Ptitsyn and Finkelstein as early as 40 or even 45 years ago, when working on studies described in [22] and [1]). For this purpose, the necessary sampling was estimated not at the level of all chain conformations in general (Levinthal’s approach), but at the level of the complete sampling of deep energy minima; in other words, only compact (globular, see Fig. 6) ensembles of secondary structures were considered (likewise [1]). The sampling estimate appeared to be approximately (to the order of magnitude) equal to LN, where L is the number of amino acid residues, and N is the number of secondary structure elements in the sequence. As a result, for a chain containing L ~ 80-100 residues and typically N ~4-6, the sampling is restricted to ~1010 variants (in contrast to ~10100 Levinthal variants) and requires only minutes or hours at the most, and not the entire life time of the Universe.
Thus, the final solution of Levinthal’s paradox has been obtained.
Fig. 6. Solution of Levinthal’s paradox “in Levinthal’s terms” was obtained at the level of the complete sampling of all deep energy minima, i.e. not all the chain structures in general, but only all compact ensembles of secondary structures.
In conclusion, we can say that Ptitsyn’s hypothesis has received its experimental verification. Protein folding follows the pattern encoded in its primary structure; importantly, according to recent data [126, 127], the in vivo folding of at least small proteins closely resembles that observed in vitro. During folding, elements of secondary structure are formed in the initially unfolded protein chain, resulting first from local interactions and then from long-range interactions. The formation of the secondary structure dramatically reduces the conformational space, thereby provoking the appearance of a compact chain structure with fluctuating tertiary structure. In turn, this ensures the selection of the correct topology that is also encoded by the amino acid sequence and realized through linking all the secondary structure elements into a single chain. In the course of protein folding, the fluctuating tertiary structure of an intermediate (molten globule) is preserved for some time, thus facilitating the correct packing of the secondary structure elements and their mutual adjustment. The duration of this process depends on the chain length and sometimes on the necessity of interactions with other folding participants. After the adjustment of the tertiary structure stabilized by S–S bonding (at times) and addition of cofactors or other stabilizing systems, the protein acquires its functional tertiary structure in the right place and at the right time.
Acknowledgments
We thank E. V. Serebrova for help in manuscript preparation. We apologize to the colleagues whose works have not been referred to because of volume restrictions of the current review.
This study was supported by the Russian Science Foundation (project No. 14-24-00157).
REFERENCES
1.Ptitsyn, O. B. (1973) Step-wise mechanism of
self-organization of protein molecules, Dokl. Akad. Nauk USSR,
210, 1213-1215.
2.Anfinsen, C. B., Haber, E., Sela, M., and White, F.
H., Jr. (1961) The kinetics of formation of native ribonuclease during
oxidation of the reduced polypeptide chain, Proc. Natl. Acad. Sci. USA,
47, 1309-1314.
3.Gutte, B., and Merrifield, R. B. (1969) The total
synthesis of an enzyme with ribonuclease A activity, J.
Am. Chem. Soc., 91, 501-502.
4.Levinthal, C. (1969) How to fold graciously, in
Mössbauer Spectroscopy in Biological Systems: Proc. Meeting held
at Allerton House, Monticello, Illinois (Debrunner, P., Tsibris, J. C.
M., and Munck, E., eds.) University of Illinois Press,
Urbana-Champaign, pp. 22-24.
5.Ptitsyn, O. B. (1995) Molten globule and protein
folding, Adv. Protein Chem., 47, 83-229.
6.Dobson, C. M. (1995) Finding the right fold,
Struct. Biol., 2, 513-517.
7.Creighton, T. E. (1997) Protein folding: does
diffusion determine the folding rate? Curr. Biol., 7,
R380-R383.
8.Dobson, C. M., and Karplus, M. (1999) The
fundamentals of protein folding: bringing together theory and
experiment, Curr. Opin. Struct. Biol., 9, 92-101.
9.Baldwin, R. L., and Rose, G. D. (1999) Is protein
folding hierarchic? II. Folding intermediates and transition states,
Trends Biochem. Sci., 24, 77-83.
10.Arai, M., and Kuwajima, K. (2000) Role of the
molten globule state in protein folding, Adv. Prot. Chem.,
53, 209-282.
11.Sanchez, I. E., and Kiefhaber, T. (2003) Evidence
for sequential barriers and obligatory intermediates in apparent
two-state protein folding, J. Mol. Biol., 325,
367-376.
12.Sinha, K. K., and Udgaonkar, J. B. (2009) Early
events in protein folding, Curr. Sci., 96, 1053-1070.
13.Baldwin, R. L., Frieden, C., and Rose, G. D.
(2010) Dry molten globule intermediates and the mechanism of protein
unfolding, Proteins, 78, 2725-2737.
14.Jha, S. K., and Udgaonkar, J. B. (2010) Direct
evidence for a dry molten globule intermediate during the unfolding of
a small protein, Proc. Natl. Acad. Sci. USA, 106,
12289-12294.
15.Baldwin, R. L., and Rose, G. D. (2013) Molten
globules, entropy-driven conformational change and protein folding,
Curr. Opin. Struct. Biol., 23, 4-10.
16.Camilloni, C., Bonetti, D., Morrone, A., Giri,
R., Dobson, C. M., Brunori, M., Gianni, S., and Vendruscolo, M. (2016)
Towards a structural biology of the hydrophobic effect in protein
folding, Sci. Rep., 6, 1-9.
17.Nishimura, C. (2017) Folding of apomyoglobin:
analysis of transient intermediate structure during refolding using
quick hydrogen deuterium exchange and NMR, Proc. Jpn. Acad. Ser.
B, 93, 10-27.
18.Ptitsyn, O. B., Lim, V. I., and Finkelstein, A.
V. (1972) Secondary structure of globular proteins and principle of
concordance of local and long-range interactions, in Analysis and
Simulation of Biochemical Systems (Hess, B., and Hemker, H. C.,
eds.), Proc. 8th FEBS Meeting, North Holland Publishing Company,
Amsterdam, Vol. 25, pp. 421-431.
20.Finkelstein, A. V., and Ptitsyn, O. B. (1978) The
theory of self-organization of the protein secondary structure:
dependence of the native globule structure on the unfolded chain
secondary structure, Dokl. Akad. Nauk USSR, 242,
1226-1228.
21.Ptitsyn, O. B., and Finkelstein, A. V. (1979)
Mechanism of protein folding, Int. J. Quant. Chem., 16,
407-418.
22.Ptitsyn, O. B., and Finkelstein, A. V. (1980)
Similarities of protein topologies: evolutionary divergence, functional
convergence or principles of folding? Quart. Rev. Biophys.,
79, 137-138.
23.Ptitsyn, O. B., and Finkelstein, A. V. (1981) The
directed mechanism of protein self-organization: a generalized model,
Kristallografiya, 26, 1066-1073.
24.Ptitsyn, O. B., and Finkelstein, A. V. (1983)
Theory of protein secondary structure and algorithm of its prediction,
Biopolymers, 22, 15-25.
25.Ptitsyn, O. B. (1992) Secondary structure
formation and stability, Curr. Opin. Struct. Biol., 2,
13-20.
26.Finkelstein, A. V., Badretdinov, A. Ya., and
Gutin, A. M. (1995) Why do protein architectures have a Boltzmann-like
statistics? Proteins, 23, 142-150.
27.Holthauzen, L. M., Rosgen, J., and Bolen, D. W.
(2010) Hydrogen bonding progressively strengthens upon transfer of the
protein urea-denatured state to water and protecting osmolytes,
Biochemistry, 49, 1310-1318.
28.Auton, M., Rosgen, J., Sinev, M., Holthauzen, L.
M., and Bolen, D. W. (2011) Osmolyte effects on protein stability and
solubility: a balancing act between backbone and side-chains,
Biophys. Chem., 159, 90-99.
29.Kuwajima, K., Yamaya, H., and Sugai, S. (1996)
The burst-phase intermediate in the refolding of beta-lactoglobulin
studied by stopped-flow circular dichroism and absorption spectroscopy,
J. Mol. Biol., 264, 806-822.
30.Sakurai, K., Fujioka, S., Konuma, T., Yagi, M.,
and Goto, Y. (2011) A circumventing role for the non-native
intermediate in the folding of beta-lactoglobulin, Biochemistry,
50, 6498-6507.
31.Matsumura, Y., Shinjo, M., Kim, S. J., Okishio,
N., Gruebele, M., and Kihara, H. (2013) Transient helical structure
during PI3K and Fyn SH3 domain folding, J. Phys. Chem. B,
117, 4836-4843.
32.Konuma, T., Sakurai, K., Yagi, M., Goto, Y.,
Fujisawa, T., and Takahashi, S. (2015) Highly collapsed conformation of
the initial folding intermediates of beta-lactoglobulin with non-native
α-helix, J. Mol. Biol., 427, 3158-3165.
33.Ptitsyn, O. B. (1981) Protein folding: general
physical model, FEBS Lett., 131, 197-202.
34.Ptitsyn, O. B. (1987) Protein folding: hypotheses
and experiments, J. Protein Chem., 6, 273-293.
35.Ptitsyn, O. B. (1991) How does protein synthesis
give rise to the 3D-structure? FEBS Lett., 285,
176-181.
36.Dolgikh, D. A., Gilmanshin, R. I., Brazhnikov, E.
V., Bychkova, V. E., Semisotnov, G. V., Veniaminov, S. Yu., and
Ptitsyn, O. B. (1981) alpha-Lactalbumin: compact state with fluctuating
tertiary structure? FEBS Lett., 136, 311-315.
37.Ohgushi, M., and Wada, A. (1983)
“Molten-globule state”: a compact form of globular proteins
with mobile side chains, FEBS Lett., 164, 21-24.
38.Ptitsyn, O. B. (1992) The molten globule state,
in Protein Folding (Creighton, T. E., ed.) W. H. Freeman and
Company, N.Y., pp. 243-300.
39.Dolgikh, D. A., Abaturov, L. V., Bolotina, I. A.,
Brazhnikov, E. V., Bychkova, V. E., Bushuev, V. N., Gilmanshin, R. I.,
Lebedev, Yu. O., Semisotnov, G. V., Tiktopulo, E. I., and Ptitsyn, O.
B. (1985) Compact state of a protein molecule with pronounced
small-scale mobility: bovine alpha-lactalbumin, Eur. Biophys.
J., 13, 109-121.
40.Ptitsyn, O. B. (1994) Kinetic and equilibrium
intermediates in protein folding, Protein Eng., 7,
593-596.
41.Ptitsyn, O. B., and Semisotnov, G. V. (1991) The
mechanism of protein folding, in Conformations and Forces in Protein
Folding (Nall, B. T., and Dill, K. A., eds.) American Association
for the Advancement of Science, Washington, D. C., pp. 155-168.
42.Gilmanshin, R. I., and Ptitsyn, O. B. (1987) An
early intermediate of refolding alpha-lactalbumin forms within 20 ms,
FEBS Lett., 223, 327-329.
43.Semisotnov, G. V., Rodionova, N. A., Kutyshenko,
V. P., Ebert, B., Blanck, J., and Ptitsyn, O. B. (1987) Sequential
mechanism of refolding of carbonic anhydrase B, FEBS Lett.,
224, 9-13.
44.Ptitsyn, O. B., Pain, R. H., Semisotnov, G. V.,
Zerovnik, E., and Razgulyaev, O. I. (1990) Evidence for a molten
globule state as a general intermediate in protein folding, FEBS
Lett., 262, 20-24.
45.Ptitsyn, O. B. (1997) Structures of folding
intermediates, Curr. Opin. Struct. Biol., 5, 74-78.
46.Semisotnov, G. V., Rodionova, N. A., Razgulyaev,
O. I., Uversky, V. N., Gripas, A. F., and Gilmanshin, R. I. (1991)
Study of the “molten globule” intermediate state in protein
folding by a hydrophobic fluorescent probe, Biopolymers,
31, 119-128.
47.Forge, V., Wijesinha, R. T., Balbach, J., Brew,
K., Robinson, C. V., Redfield, C., and Dobson, C. M. (1999) Rapid
collapse and slow structural reorganization during the refolding of
bovine alpha-lactalbumin, J. Mol. Biol., 288,
673-688.
48.Magg, C., Kubelka, J., Holtermann, G., Haas, E.,
and Schmid, F. X. (2006) Specificity of the initial collapse in the
folding of the cold shock protein, J. Mol. Biol., 360,
1067-1080.
49.Kamagata, K., Arai, M., and Kuwajima, K. (2004)
Unification of the folding mechanisms of non-two-state and two-state
proteins, J. Mol. Biol., 339, 951-965.
50.Kamagata, K., and Kuwajima, K. (2006)
Surprisingly high correlation between early and late stages in
non-two-state protein folding, J. Mol. Biol., 357,
1647-1654.
51.Jackson, S. E. (1998) How do small single-domain
proteins fold? Fold. Des., 3, R81-R91.
52.Galzitskaya, O. V., Garbuzynskiy, S. O., Ivankov,
D. N., and Finkelstein, A. V. (2003) Chain length is the main
determinant of the folding rate for proteins with three-state folding
kinetics, Proteins, 51, 162-166.
53.Plaxco, K. W., Simons, K. T., and Baker, D.
(1998) Contact order, transition state placement and the refolding
rates of single domain proteins, J. Mol. Biol., 277,
985-994.
54.Ivankov, D. N., Garbuzynskiy, S. O., Alm, E.,
Plaxco, K. W., Baker, D., and Finkelstein, A. V. (2003) Contact order
revisited: influence of protein size on the folding rate, Protein
Sci., 12, 2057-2062.
55.Privalov, P. L. (1979) Stability of proteins:
small globular proteins, Adv. Protein Chem., 33,
167-241.
56.Khorsanizadeh, S., Peters, I. D., and Roder, H.
(1996) Evidence for a three-state model of protein folding from kinetic
analysis of ubiquitin variants with altered core residues, Nat.
Struct. Biol., 3, 193-205.
57.Bachmann, A., and Kiefhaber, T. (2001) Apparent
two-state tendamistat folding is a sequential process along a defined
route, J. Mol. Biol., 306, 375-386.
58.Houliston, R. S., Liu, C., Singh, L. M., and
Meiering, E. M. (2002) pH and urea dependence of amide
hydrogen-deuterium exchange rates in the beta-trefoil protein
hisaxtophilin, Biochemistry, 41, 1182-1194.
59.Park, S. H., Shastry, M. C., and Roder, H. (1999)
Folding dynamics of the B1 domain of protein G explored by ultrarapid
mixing, Nat. Struct. Biol., 6, 943-947.
60.Gorski, S. A., Capaldi, A. P., Kleanthous, C.,
and Radford, S. E. (2001) Acidic conditions stabilize intermediates
populated during the folding of Im7 and Im9, J. Mol. Biol.,
312, 849-863.
61.Jager, M., Nguyen, H., Crane, J. C., Kelly, J.
W., and Gruebele, M. (2001) The folding mechanism of a beta-sheet: the
WW domain, J. Mol. Biol., 311, 373-393.
62.Jager, M., Deechongkit, S., Koepf, E. K., Nguyen,
H., Gao, J., Powers, E. T., Gruebele, M., and Kelly, J. W. (2008)
Understanding the mechanism of beta-sheet folding from a chemical and
biological perspective, Biopolymers, 90, 751-758.
63.Wagner, C., and Kiefhaber, T. (1999)
Intermediates can accelerate protein folding, Proc. Natl. Acad. Sci.
USA, 96, 6716-6721.
64.Balobanov, V. A., Katina, N. S., Finkelstein, A.
V., and Bychkova, V. E. (2017) Intermediate states of apomyoglobin: are
they parts of the same area of conformations diagram? Biochemistry
(Moscow), 82, 625-631.
65.Shakhnovich, E. I., and Finkelstein, A. V. (1982)
On the theory of cooperative transitions in protein globules, Dokl.
Akad. Nauk USSR, 267, 1247-1250.
66.Shakhnovich, E. I., and Finkelstein, A. V. (1989)
Theory of cooperative transitions in protein molecules. I. Why
denaturation of globular proteins is a first-order phase transition,
Biopolymers, 28, 1667-1680.
67.Finkelstein, A. V., and Shakhnovich, E. (1989)
Theory of cooperative transitions in protein molecules. II. Phase
diagram for a protein molecule in solution, Biopolymers,
28, 1681-1694.
68.Kharakoz, D. P., and Bychkova, V. E. (1997)
Molten globule of human alpha-lactalbumin: hydration, density, and
compressibility of the interior, Biochemistry, 36,
1882-1890.
69.Kiefhaber, T., Labhardt, A. M., and Baldwin, R.
L. (1995) Direct NMR evidence for an intermediate preceding the
rate-limiting step in the unfolding of ribonuclease A, Nature,
375, 513-515.
70.Jha, S. K., and Udgaonkar, J. B. (2009) Direct
evidence for a dry molten globule intermediate during the unfolding of
a small protein, Proc. Natl. Acad. Sci. USA, 106,
12289-12294.
71.Hoeltzli, S. D., and Frieden, C. (1995)
Stopped-flow NMR spectroscopy: real-time unfolding studies of
6-19F-tryptophan-labeled Escherichia coli
dihydrofolate reductase, Proc. Natl. Acad. Sci. USA, 92,
9318-9322.
72.Reiner, A., Henklein, P., and Kiefhaber, T.
(2010) An unlocking/relocking barrier in conformational fluctuations of
villin headpiece subdomain, Proc. Natl. Acad. Sci. USA,
107, 4955-4960.
73.Acharya, N., Mishra, P., and Jha, S. K. (2016)
Evidence for dry molten globule-like domains in the pH-induced
equilibrium folding intermediate of a multidomain protein, J. Phys.
Chem. Lett., 7, 173-179.
74.Sarkar, S. S., Udgaonkar, J. B., and
Krishnamoorthy, G. (2013) Unfolding of a small protein proceeds via dry
and wet globules and a solvated transition state, Biophys. J.,
105, 2392-2402.
75.Kuwajima, K. (1996) The molten globule state of
alpha-lactalbumin, FASEB J., 10, 102-109.
76.Wu, L. C., Schulman, B. A., Peng, Z., and Kim, P.
S. (1996) Disulfide determinants of calcium-induced packing in
alpha-lactalbumin, Biochemistry, 35, 859-863.
77.Quezada, C. M., Schulman, B. A., Froggatt, J. J.,
Dobson, C. M., and Redfield, C. (2004) Local and global cooperativity
in human alpha-lactalbumin molten globule, J. Mol. Biol.,
338, 149-158.
78.Polverino de Laureto, P., Frare, E., Gottardo,
R., and Fontana, A. (2002) Molten globule of bovine alpha-lactalbumin
at neutral pH induced by heat, trifluoroethanol, and oleic acid: a
comparative analysis by circular dichroism spectroscopy and limited
proteolysis, Proteins, 49, 385-397.
79.Mok, K. H., Nagashima, T., Day, I. J., Hore, P.
J., and Dobson, C. M. (2005) Multiple subsets of side-chain packing in
partially folded states of alpha-lactalbumins, Proc. Natl. Acad.
Sci. USA, 102, 8899-8904.
80.Blanch, I. W., Morozova-Roche, L. A., Hecht, L.,
Noppe, W., and Barron, L. D. (2000) Raman optical activity
characterization of native and molten globule states of equine
lysozyme: comparison with hen lysozyme and bovine alpha-lactalbumin,
Biopolymers, 57, 235-248.
81.Lim, V. I. (1975) Structural transitions in the
protein chain during formation of native globules. A hypothesis of
“excess” helices, Dokl. Akad. Nauk USSR, 222,
1467-1469.
82.Chow, C. C., Chow, C., Raghunathan, V., Huppert,
T. J., Kimball, E. B., and Cavagnero, S. (2003) Chain length dependence
of apomyoglobin folding: structural evolution from misfolded sheets to
native helices, Biochemistry, 42, 7090-7099.
83.Cavagnero, S., Nishimura, C., Schwarzinger, S.,
Dyson, H. J., and Wright, P. E. (2001) Conformational and dynamic
characterization of the molten globule state of an apomyoglobin mutant
with an altered folding pathway, Biochemistry, 40,
14459-14467.
84.Uzawa, T., Akiyama, S., Kimura, T., Takahashi,
S., Ishimori, K., Morishima, I., and Fujisawa, T. (2004) Collapse and
search dynamics of apomyoglobin folding revealed by submillisecond
observations of alpha-helical content and compactness, Proc. Natl.
Acad. Sci. USA, 101, 1171-1176.
85.Uzawa, T., Nishimura, C., Akiyama, S., Ishimori,
K., Takahashi, S., Dyson, H. J., and Wright, P. E. (2008) Hierarchical
folding mechanism of apomyoglobin revealed by ultra-fast H/D exchange
coupled with 2D NMR, Proc. Natl. Acad. Sci. USA, 105,
13859-13864.
86.Felitsky, D. J., Lietzow, M. A., Dyson, H. J.,
and Wright, P. E. (2008) Modeling transient collapsed states of an
unfolded protein to provide insights into early folding events,
Proc. Natl. Acad. Sci. USA, 105, 6278-6283.
87.Meinhold, D. W., and Wright, P. E. (2011)
Measurements of protein unfolding/refolding kinetics and structural
characterization of hidden intermediates by NMR relaxation dispersion,
Proc. Natl. Acad. Sci. USA, 108, 9078-9083.
88.Armstrong, B. D., Choi, J., Lopez, C., Wesener,
D. A., Hubbell, W., Cavagnero, S., and Han, S. (2011) Site-specific
hydration dynamics in the nonpolar core of a molten globule by dynamic
nuclear polarization of water, J. Am. Chem. Soc., 133,
5987-5995.
89.Ptitsyn, O. B., and Ting, K.-L. H. (1999)
Non-functional conserved residues in globins and their possible role as
a folding nucleus, J. Mol. Biol., 291, 671-677.
90.Galzitskaya, O. V., and Finkelstein, A. V. (1999)
A theoretical search for folding/unfolding nuclei in three-dimensional
protein structures, Proc. Natl. Acad. Sci. USA, 96,
11299-11304.
91.Garbuzynskiy, S. A., Finkelstein, A. V., and
Galzitskaya, O. V. (2005) On the prediction of folding nuclei in
globular proteins, Mol. Biol. (Moscow), 39,
1032-1041.
92.Fersht, A. (1999) Structure and Mechanism in
Protein Science: A Guide to Enzyme Catalysis and Protein Folding,
Chaps. 2, 15, 18, 19, W. H. Freeman & Co, N.Y.
93.Baryshnikova, E. N., Melnik, B. S., Finkelstein,
A. V., Semisotnov, G. V., and Bychkova, V. E. (2005) Three-state
protein folding: experimental determination of free-energy profile,
Protein Sci., 14, 2658-2667.
94.Samatova, E. N., Katina, N. S., Balobanov, V. A.,
Melnik, B. S., Bychkova, V. E., and Finkelstein, A. V. (2009) How
strong are side chain interactions in the folding intermediate?
Protein Sci., 18, 2152-2159.
95.Samatova, E. N., Melnik, B. S., Balobanov, V. A.,
Katina, N. S., Dolgikh, D. A., Semisotnov, G. V., Finkelstein, A. V.,
and Bychkova, V. E. (2010) Folding intermediate and folding nucleus for
Ι → Ν and U → Ι → N transitions in
apomyoglobin: contributions by conserved and non-conserved residues,
Biophys. J., 98, 1694-1702.
96.Bychkova, V. E., Pain, R. H., and Ptitsyn, O. B.
(1988) The “molten globule” state is involved in the
translocation of proteins across membranes? FEBS Lett.,
238, 231-234.
97.Bychkova, V. E., and Ptitsyn, O. B. (1993) The
molten globule in vitro and in vivo, Chemtracts
Biochem. Mol. Biol., 4, 133-163.
98.Wright, P. E., and Dyson, H. J. (1999)
Intrinsically unstructured proteins: re-assessing the protein
structure-function paradigm, J. Mol. Biol., 293,
321-331.
99.Uversky, V. N., Gillespie, J. R., and Fink, A. L.
(2000) Why are “natively unfolded” proteins unstructured
under physiologic conditions? Proteins, 41, 415-442.
100.Dunker, A. K., Lawson, J. D., Brown, C. J.,
Williams, R. M., Romero, P., Oh, J. S., Oldfield, C. J., Campen, A. M.,
Ratliff, C. M., Hipps, K. W., Ausio, J., Nissen, M. S., Reeves, R.,
Kang, C., Kissinger, C. R., Bailey, R. W., Griswold, M. D., Chiu, W.,
Garner, E. C., and Obradovic, Z. (2001) Intrinsically disordered
protein, J. Mol. Graph. Model., 19,
26-59.
101.Dunker, A. K., Oldfield, C. J., Meng, J.,
Romero, P., Yang, J. Y., Chen, J. W., Vacic, V., Obradovic, Z., and
Uversky, V. N. (2008) The unfoldomics decade: an update on
intrinsically disordered proteins, BMC Genomics, 9,
Suppl. 2, S1.
102.Vamvaca, K., Voegeli, B., Kast, P., Pervushin,
K., and Hilvert, D. (2004) An enzymatic molten globule: efficient
coupling of folding and catalysis, Proc. Natl. Acad. Sci. USA,
101, 12860-12864.
103.Pervushin, K., Vamvaca, K., Voegeli, B., and
Hilvert, D. (2007) Structure and dynamics of a molten globular enzyme,
Nat. Struct. Mol. Biol., 14, 1202-1206.
104.Vamvaca, K., Jelesarov, I., and Hilvert, D.
(2008) Kinetics and thermodynamics of ligand binding to a molten
globular enzyme and its native counterpart, J. Mol. Biol.,
382, 971-977.
105.Kasper, J. R., and Park, C. (2015) Ligand
binding to a high-energy partially unfolded protein, Protein
Sci., 24, 129-137.
106.Kitazawa, S., Kameda, T., Yagi-Utsumi, M.,
Sugase, K., Baxter, N. J., Kato, K., Williamson, M. P., and Kitahara,
R. (2013) Solution structure of the Q41N variant of ubiquitin as a
model for alternatively folded N2 state of ubiquitin,
Biochemistry, 52, 1874-1885.
107.Prajapati, R. S., Indu, S., and Varadarajan, R.
(2007) Identification and thermodynamic characterization of molten
globule states of periplasmic binding proteins, Biochemistry,
46, 10339-10352.
108.Zhou, B., Tian, K., and Jing, G. (2000) An
in vitro peptide folding model suggests the presence of the
molten globule state during nascent peptide folding, Protein
Eng., 13, 35-39.
109.Bonetti, D., Camilloni, C., Visconti, L.,
Longhi, S., Brunori, M., Vendruscolo, M., and Gianni, S. (2016)
Identification and structural characterization of an intermediate in
the folding of the measles virus X domain, J. Biol. Chem.,
291, 10886-10892.
110.Park, S. J., Borin, B. N., Martinez-Yamout, M.
A., and Dyson, H. J. (2011) The client protein p53 forms a molten
globule-like state in the presence of Hsp90, Nat. Struct. Mol.
Biol., 18, 537-541.
111.Whitley, M. J., Xi, Z., Bartko, J. C., Jensen,
M. R., Blackledge, M., and Gronenborn, A. M. (2017) A combined NMR and
SAXS analysis of the partially folded cataract-associated V75D
gamma-D-crystallin, Biophys. J., 112, 1135-1146.
112.Leal, S. S., and Gomes, C. M. (2007) Studies of
the molten globule state of ferredoxin: structural characterization and
implications on protein folding and iron-sulfur center assembly,
Proteins, 68, 606-616.
113.Singh, N., Kumar, R., Jagannadham, M. V., and
Kayastha, A. M. (2013) Evidence for a molten globule state in cicer
α-galactosidase induced by pH, temperature, and guanidine
hydrochloride, Appl. Biochem. Biotechnol., 169,
2315-2325.
114.Teilum, K., Maki, K., Kragelund, B. B.,
Poulsen, F. M., and Roder, H. (2002) Early kinetic intermediate in the
folding of acyl-CoA binding protein detected by fluorescence labeling
and ultrarapid mixing, Proc. Natl. Acad. Sci. USA, 99,
9807-9812.
115.Wani, A. H., and Udgaonkar, J. B. (2009) Native
state dynamics drive the unfolding of the SH3 domain of PI3 kinase at
high denaturation concentration, Proc. Natl. Acad. Sci. USA,
106, 20711-20716.
116.Cellitti, J., Bernstein, R., and Marqusee, S.
(2007) Exploring subdomain cooperativity in T4 lysozyme II: uncovering
the C-terminal subdomain as a hidden intermediate in the kinetic
folding pathway, Protein Sci., 16, 852-862.
117.Finkelstein, A. V., and Ptitsyn, O. B. (2012)
Protein Physics [in Russian], 4th Edn., Chaps. 18-21, University
Publishing House, Moscow.
118.Finkelstein, A. V. (2014) Physics of Protein
Molecules [in Russian], Chap. 9, Izhevsk Institute of Computer
Science, Moscow-Izhevsk.
119.Finkelstein, A. V., and Ptitsyn, O. B. (2016)
Protein Physics. A Course of Lectures, 2nd Edn., Chaps. 7, 10,
13, 18, 19-21, Academic Press, an Imprint of Elsevier Science,
Amsterdam-Boston-Heidelberg-London-New York-Oxford-Paris-San Diego-San
Francisco-Singapore-Sydney-Tokyo.
120.Finkelstein, A. V., Badretdin, A. J.,
Galzitskaya, O. V., Ivankov, D. N., Bogatyreva, N. S., and
Garbuzynskiy, S. O. (2017) There and back again: two views on the
protein folding puzzle, Phys. Life Rev., 21, 56-71.
121.Finkelstein, A. V. (2018) 50+ years of protein
folding, Biochemistry (Moscow), 83, Suppl.1, S3-S18.
122.Finkelstein, A. V., and Badretdinov, A. Ya.
(1997) The physical basis of fast protein folding into a stable spatial
structure: a solution of Levinthal’s paradox, Mol. Biol.
(Moscow), 31, 391-398.
123.Finkelstein, A. V., and Badretdinov, A. Ya.
(1997) Rate of protein folding near the point of thermodynamic
equilibrium between the coil and the most stable chain fold,
Fold. Des., 2, 115-121.
124.Garbuzinskiy, S. O., Ivankov, D. N.,
Bogatyreva, N. S., and Finkelstein, A. V. (2013) Golden triangle for
folding rates of globular proteins, Proc. Natl. Acad. Sci. USA,
110, 147-150.
125.Finkelstein, A. V., and Garbuzinskiy, S. O.
(2015) Reduction of the search space for the folding of proteins at the
level of formation and assembly of secondary structures: a new view on
the solution of Levinthal’s paradox, ChemPhysChem,
16, 3375-3378.
126.Eichmann, C., Preissler, S., Riek, R., and
Deuerling, E. (2010) Cotranslational structure acquisition of nascent
polypeptides monitored by NMR spectroscopy, Proc. Natl.
Acad. Sci. USA, 107, 9111-9116.
127.Han, Y., David, A., Liu, B., Magadan, J. G.,
Bennink, J. R., Yewdell, J. W., and Qian, S.-B. (2012) Monitoring
cotranslational protein folding in mammalian cells at codon resolution,
Proc. Natl. Acad. Sci. USA,
109, 12467-12472.