REVIEW: Studies of the Process of Amyloid Formation by Aβ Peptide
O. V. Galzitskaya*, E. I. Galushko, and O. M. Selivanova
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: ogalzit@vega.protres.ru* To whom correspondence should be addressed.
Received July 5, 2017; Revision received August 2, 2017
Studies of the process of amyloid formation by Aβ peptide have been topical due to the critical role of this peptide in the pathogenesis of Alzheimer’s disease. Many articles devoted to this process are available in the literature; however, none of them gives a detailed description of the mechanism of the process of generation of amyloids. Moreover, there are no reliable data on the influence of modified forms of Aβ peptide on its amyloid formation. To appreciate the role of Aβ aggregation in the pathogenesis of Alzheimer’s disease and to develop a strategy for its treatment, it is necessary to have a well-defined description of the molecular mechanism underlying the formation of amyloids as well as the contribution of each intermediate to this process. We are convinced that a combined analysis of theoretical and experimental methods is a way for understanding molecular mechanisms of numerous diseases. Based on our experimental data and molecular modeling, we have constructed a general model of the process of amyloid formation by Aβ peptide. Using the data described in our previous publications, we propose a model of amyloid formation by this peptide that differs from the generally accepted model. Our model can be applied to other proteins and peptides as well. According to this model, the main building unit for the formation of amyloid fibrils is a ring-like oligomer. Upon interaction with each other, ring-like oligomers form long fibrils of different morphology. This mechanism of generation of amyloid fibrils may be common for other proteins and peptides.
KEY WORDS: Aβ peptide, oligomer, amyloid, X-ray, electron microscopy, polymorphismDOI: 10.1134/S0006297918140079
Abbreviations: a.a., amino acid residue; AD, Alzheimer’s disease; AFM, atomic force microscopy; AMP, antimicrobial peptide; APP, amyloid precursor protein; APPsα, soluble extracellular fragments of APP; cryo-EM, cryoelectron microscopy; EM, electron microscopy; HN, humanin; NUCB1, calcium-binding protein nucleobindin-1; ThT, thioflavin T.
Misfolding of proteins results in the development of many systemic and
neurodegenerative diseases called proteinopathies [1, 2]. In such pathologies,
proteins have an irregular structure (conformation different from the
native structure), become functionally inactive, toxic, and prone to
aggregation and deposition in various organs and tissues. The
hypothesis is now widely accepted, confirmed by the results of many
studies, according to which the primary cytotoxic agents in the
development of proteinopathies are oligomers of proteins prone to
aggregation [3-7].
The proteinopathy group includes Parkinson’s disease, Creutzfeldt–Jakob disease, type-2 diabetes mellitus, and many others. However, the most famous example is senile dementia of Alzheimer’s type, or Alzheimer’s disease (AD).
AD is one of the most common neurodegenerative diseases and the source of 50-70% of cases of acquired dementia [8]. The disease usually appears after 65 years of age, but rare early hereditary forms of AD occur when point mutations are found in the gene encoding the precursor protein of Aβ peptide (APP, amyloid precursor protein) [9]. In addition, it is noted that the development of AD can be triggered by various neuroinflammations, impaired regulation of the immune system [10], trauma, and other factors. The etiopathological mechanisms of the development of AD have not been adequately studied. The key features of the disease are accumulation of aggregates of Aβ peptides in the form of amyloid plaques in brain tissues as well as neurofibrillary tangles consisting of hyperphosphorylated tau protein [11]. Over the past two decades, a working hypothesis has been developed, known as the “amyloid cascade”, suggesting that progressive accumulation/oligomerization/aggregation of Aβ peptides in brain regions that are responsible for learning and memory is the main cause of neurodegeneration and death of brain cells in AD.
The pathogenesis of AD is an active area of research. This is due, in part, to the fact that life expectancy increases, and consequently the frequency of occurrence of this disease increases. According to the forecast for 2050, the number of patients suffering from AD will exceed 100 million people [12]. In addition, AD requires large financial investments for diagnosis and development of drugs that prevent the progression of the disease.
There are several characteristics indicating that the investigated fibrils are amyloids. First, the ability to interact with hydrophobic dyes for staining tissue sections: Congo red (birefringence) and thioflavin T and S (fluorescence spectroscopy). Second, there is a cross-β structure. X-ray diffraction analysis reveals two characteristic reflections: meridional 4.5-4.8 Å and equatorial 8-12 Å [13-18]. Third, upon direct visualization of preparations with microscopic methods (electron microscopy, EM; atomic force microscopy, AFM; cryoelectron microscopy, cryo-EM), fibrils with a diameter of about 10 nm and a length of up to 10-15 µm are observed [19]. Fourth, upon formation of fibrils, the so-called lag-period is detected, during which fibril nuclei are formed (nucleation stage, which is concentration dependent) and a period of rapid growth [20-24].
The study of the process of amyloid formation/fibrillation of the Aβ peptide, as well as the structure of its aggregates, can contribute to the understanding of pathophysiological mechanisms of development of AD and facilitate the search for an effective drug against aggregation of isoforms of the Aβ peptide.
AMYLOID PRECURSOR PROTEIN: PATHWAYS OF PROCESSING,
FUNCTIONS
The precursor of the main constituent of amyloid plaques found in AD, Aβ peptide, is the transmembrane protein APP (amyloid precursor protein). The APP gene is located on chromosome 21 and contains at least 19 exons, which can lead to the appearance of APP isoforms of various lengths with molecular mass from 100 to 140 kDa [25, 26].
Proteins of the APP family (695-770 a.a.) consist of a hydrophilic N-terminal extracellular domain, a hydrophobic transmembrane domain, and a C-terminal cytoplasmic domain [27] (Fig. 1). Comparison of human and other mammalian DNAs shows a high degree of conservatism of the APP gene: 100% identity has been established between APP-695 from the human and monkey brains. In the neurons of the central nervous system, the isoform of APP-695 predominates, and the forms APP-770 and APP-751 are present in trace amounts [28, 29]. APP is synthesized and glycosylated in the endoplasmic reticulum, then transferred to the Golgi complex for maturation before transport to the cell surface [30].
Fig. 1. Pathways of APP processing [11]. The protein of the amyloid precursor is preferably cleaved along the non-amyloidogenic pathway by α-secretase to form a large ectodomain, APPsα, and a C-terminal tail, from which peptide p3 is then cleaved by γ-secretase. APP can be cleaved along the amyloidogenic pathway by β-secretase to form the ectodomain of APPsβ and a longer C-terminal fragment. This fragment is cleaved by γ-secretase, resulting in the generation of Aβ peptides (672-713 corresponds to Aβ(1-42) and 672-711 corresponds to Aβ(1-40)). rAβ corresponds to regions of future molecules of Aβ in the structure of the APP molecule.
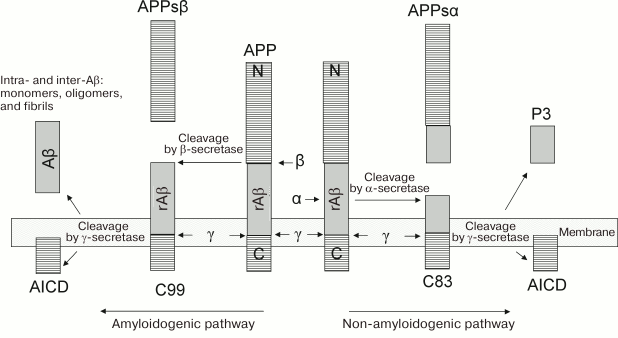
Mutations in some regions of the APP gene are the cause of the hereditary predisposition to AD. Several mutations of the Aβ peptide (Flemish (A21G), Italian (E22K), Arctic (E22G), Dutch (E22Q), and Aiova (D23N) [31]) accelerate its oligomerization (E22Q and E22G [32]) and fibrillogenesis (E22Q and D23N [31]). The exact mechanism by which these mutations contribute to the pathogenesis of AD is unknown.
APP is involved in adhesion, migration, and cell proliferation and provides cholesterol and copper homeostasis [33]. In the brain, APP is involved in the development of neurons, the formation and repair of synapses, and the provision of synaptic plasticity. However, detailed mechanisms of these effects are not well understood [34].
More and more data point to the neuroprotective role of this membrane protein in metabolic stress situations. By itself or its soluble extracellular fragments (APPsα), it can contribute to the survival of neurons. Indeed, various models of acute hypoxia–ischemia of animals, craniocerebral trauma, and excitotoxicity demonstrate the protective effects of APP or APPsα. The general mechanisms include APP-mediated regulation of calcium homeostasis through NMDA receptors, calcium potential-activated channels, or an internal calcium depot (store). In addition, APP affects the expression of genes associated with survival or apoptosis [35].
Amyloidogenic and non-amyloidogenic pathways of APP processing. Posttranslational modifications of APP include glycosylation, phosphorylation, sialylation, and limited proteolysis. In the non-amyloidogenic pathway, the protein is cleaved by proteases of the secretase family (α-secretase and β-secretase), which remove almost the entire extracellular domain with the release of C-terminal fragments attached to the membrane, and it can be associated with apoptosis (Fig. 1). In the amyloidogenic pathway, after digestion of APP by β-secretase, its further cleavage by γ-secretase in the region of the membrane-binding domain results in the appearance of peptide fragments of Aβ peptide of different lengths (39-43 a.a.), the two main forms of the peptides being variants of Aβ(1-40) and Aβ(1-42) (40 and 42 a.a., respectively). Although the Aβ(1-42) peptide variant is only 10% of the total amount of Aβ secreted from cells, it is the main protein component of amyloid plaques. In vitro, this type of Aβ peptide forms aggregates much faster than Aβ(1-40) peptide [21].
In both amyloid and non-amyloid processing of APP, the effect of γ-secretase leads to the formation of an intracellular terminal fragment (AICD) (Fig. 1), which is a transcription factor and probably regulates the level of expression of both APP by a feedback principle and the amyloid-degrading enzyme neprilysin [36, 37].
Gamma-secretase is a large multisubunit complex consisting of four individual proteins, including presenilin-1 or -2, whose genes have been identified as the main genetic risk factors for developing AD [38]. Mutations of the presenilin-1 and presenilin-2 genes are accompanied by an increase in the level of Aβ(1-42) compared to Aβ(1-40), although the total amount of Aβ peptide formed remains constant [39]. Recently, 138 mutations registered for human presenilin-1 have been analyzed [40]. About 90% of these mutations result in a reduced level of Aβ(1-42) and Aβ(1-40). It is noteworthy that 10% of these mutations lead to a decrease in the ratio of Aβ(1-42)/Aβ(1-40). There is no statistically significant correlation between the ratio of Aβ(1-42)/Aβ(1-40), which is associated with a variant of γ-secretase containing a specific mutation of the presenilin-1 gene, and the average age of the patients at the time when this mutation was detected.
Amyloidogenic processing of APP is associated with its presence on lipid rafts. When APP molecules occupy the lipid raft of the membrane region, they become more readily available for β-secretase cleavage, whereas APP molecules outside the raft are differentially cleaved by α-secretase. The activity of γ-secretase is also associated with lipid rafts. Since cholesterol is involved in the organization of a lipid raft, its high level, as well as the genotype of apolipoprotein E4, can be considered as risk factors for developing AD [41].
Recently, an alternative pathway for APP processing was discovered using intramembrane rhomboid protease of mammals (RHBDL4). The rhomboid protease cleaves APP several times in the ectodomain region, resulting in the formation of several N- and C-terminal fragments that do not degrade with classical secretases. Thus, APP does not participate in the amyloidogenic pathway of processing, which leads to a decrease in the level of Aβ peptide [42]. In addition, there is η-secretase, which forms amyloid-η fragments that inhibit neuronal functions [43].
STRUCTURAL FEATURES AND FIBRILLOGENESIS OF ISOFORMS OF Aβ
PEPTIDE
The main isoforms of the Aβ peptide are represented by the 40-a.a. peptide Aβ(1-40) and the 42-a.a. peptide Aβ(1-42). The amino acid sequence of Aβ(1-42) is: (NH2)-DAEFRHDSGYEFHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA-(COOH).
The function of the isoforms of Aβ peptide in the human body remains unexplored. In the brain of patients suffering from AD, this peptide can form the so-called amyloid plaques formed from fibrillar deposits. Both peptides can form amyloids, and at the initial stages of fibrillation aggregate (associate) with the formation of oligomeric structures [44]. Various modifications of the Aβ peptide including oxidation, phosphorylation, nitration, racemization, isomerization, pyroglutamylation, and glycosylation result in the appearance of peptides with various physiological and pathological properties that can affect the course of the disease [45-47].
In the process of fibrillation, two stages are distinguished: the lag-period, in which the preparation for growth of fibrils takes place (the formation of nuclei and oligomers of different sizes), and the growth of fibrils. Particular attention is paid to the study of oligomeric formations as the initial stage of fibril formation. This point is important for understanding the mechanism of fibrillation. Oligomers are small multimers that do not yet have the ability to lengthen at the same rate as fibrils. Oligomers of different sizes, starting with dimers, are the most toxic intermediate components in the pathway of fibril formation in comparison with mature fibrils [6].
Structural features of oligomers and fibrils are actively studied. The rate of formation of both oligomers and fibrils and their morphology are affected by the ionic composition, pH, temperature, activation of the process due to drug mixing, etc. For example, for oligomers of Aβ(1-42) at low temperature (4°C) and ionic strength, pentameric and hexameric disc-shaped oligomers of 10-15 nm in diameter are formed, with almost no β-structure, of which mature fibrils consisting of β-sheets are formed upon further incubation [48]. In contrast, soluble globular oligomers formed in the presence of aliphatic detergents contain mixed parallel and antiparallel β-sheet structures other than fibrils [49].
It is known that the formation of fibrils is preceded by the appearance of metastable oligomers of different sizes [50, 51], and at the same time it is not clear which of the oligomers formed can serve as the nucleus for the subsequent growth of the fibril. It is possible that different morphologies of fibrils are a consequence of similar but not identical pathways of forming mature fibrils. Differences can be laid at the beginning of the pathway of formation of fibrils, including the nucleation stage. In the recently published work devoted to the process of amyloid formation of Aβ peptide [52], it was shown that the nucleus could be an oligomer whose size varies from 2 to 3 monomers. The authors also demonstrated a strong dependence of the form of the kinetic curve on the experimental conditions (in particular, pH). At the same time, the energy barrier for the transition of the monomer/oligomer was determined, it was 5 kcal/mol, while for the monomer/fibril transition the height of the barrier was 12.1 kcal/mol.
Both isoforms of Aβ peptide can form fibrils, but they differ in morphology. The Aβ(1-40) peptide basically forms fibrils with parallel folding in the form of ribbons of different diameters and twists with different periods, as well as bundles of different diameters, and Aβ(1-42) usually forms fibrils with a rough surface of different diameter and often demonstrate branching under the same conditions [24, 53, 54]. It was shown that in the initial period of fibril formation, intermediate aggregates of peptides are formed, which often have a rounded morphology and a diameter similar to that of single fibrils. EM images show that such oligomeric complexes have a ring-like morphology. With prolonged incubation time of peptide preparations, oligomeric complexes disappear, and fibrils persist. Their length increases, and polymorphism increases. Large clusters of fibrils of different diameters appear [54-56].
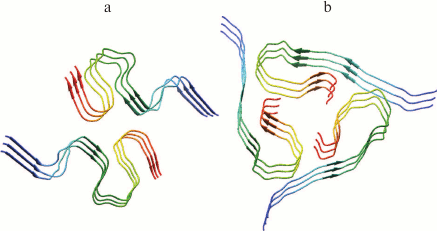
Amyloid fibrils are characterized by polymorphism, when fibrils acquire a different morphology (ribbons, bundles, films, clusters of fibrils). Polymorphism of amyloid fibrils is an obstacle for their crystallization, so other biophysical methods are used to study their structure. Most information on their structural organization is obtained mainly by solid-state NMR, EM, AFM, cryo-EM, X-ray diffraction analysis, and also using theoretical research methods, for example, molecular dynamics and molecular modeling. In particular, models of Aβ(1-40) and Aβ(1-42) peptide fibrillar structures have been obtained using the solid-state NMR method. It has been shown that the structure of Aβ(1-42) (Fig. 2a) [57] has significant differences from the structure of Aβ(1-40) (Fig. 2b) [58], because according to the authors the extension of the C-terminus by the 2-a.a. residue leads to acceleration of the fibrillation process [59].
Fig. 2. Three-dimensional structural models of amyloid fibrils developed based on solid-state NMR spectroscopy data. a) The 3D structure of Aβ(1-42) consists of two peptide molecules per fibril layer and forms a double horseshoe structure of cross-β sheet with submerged fibril hydrophobic side-chain groups (pdb, 2NAO [57]). b) The structure of Aβ(1-40) fibril obtained by incubating an extract posthumously taken from the brain of a patient suffering from AD (pdb, 2M4J [58]).
The question of the size of the nuclei of fibrils of various proteins and peptides remains open. With the nucleation mechanism, fibril generation begins with thermodynamically unprofitable steps, which results in the formation of a “critical nucleus” consisting of a definite number of monomers. We proposed a kinetic model of the process of amyloid formation that makes it possible to calculate the size of the nucleus according to kinetic data [22-24, 55]. In addition to the primary nucleation stage, which is believed to be characteristic of the amyloid formation process, the models considered include various growth regimes of the aggregate. The growth regimes following the nucleation stage can be divided into two types: the “linear” growth regime of protofibrils, where the possible number of growth points (the place where monomers can attach) is proportional to the number of nuclei, and the “exponential” growth regime, where the possible number of growth points during aggregation, the number of nuclei, may be much higher. Analysis of the kinetic curves of amyloid formation showed that in experiments the regime of exponential growth is realized more often. The implementation of the exponential growth in the experiment can be different, but in general, everything can be reduced to three scenarios: fragmentation, growth from the surface, and bifurcation. In the case of fragmentation, the number of growth points increases due to the appearance of additional fibril ends. In the case of bifurcation, deformations on the surface of fibrils can serve as new growth points or on the surface of a growing aggregate the nuclei of secondary nucleation can be formed, also serving as new growth points. The case of growth from the surface is unusual: upon such growth, the entire surface is the point of growth, and the shape of the aggregate will not be fibrillar [22, 23]. Such growth in aggregates was noted for the human prion [60].
Our theory of amyloid formation can be used to calculate the size of the nucleus (the most unstable state on the monomer → fibril pathway) from which the fibril growth begins in the presence of appropriate experimental data [22]. To estimate the size of fibril nuclei and the possible scenario for which aggregates are formed, it is necessary to perform a number of kinetic experiments, where the only variable parameter is the monomer concentration. For each curve obtained during the experiment, the characteristic times Tlag (lag time), T2 (the transition time of all monomers to the aggregate), and Lrel (the relative lag-period), which is defined as the Tlag/T2 ratio, are calculated. To determine when it is possible to talk about the realization of the linear growth regime in the experiment, and in which the exponential one, a new quantity, the relative lag-period Lrel, was introduced. It was found that Lrel can also be used to calculate the sizes of nuclei of primary and secondary nucleation. It was shown [22] that the dependences of lnT2 and Lrel on ln[MΣ], the logarithm of the initial monomer concentration, are linear, and the values of the corresponding slope coefficients for each of the dependences can be used to calculate the fibril nuclei size (including non-amyloid type) and determining the mechanism by which the aggregate is formed. At the same time, the analysis has shown that not all the results of kinetic experiments can be unambiguously interpreted to determine the aggregation mechanism. In such cases, direct experimental methods such as EM and X-ray diffraction analysis should be used, with the help of which the fibrillation mechanism can be distinctively determined. At the same time, the nature of the behavior of the quantities Lrel, Tlag, and T2 allows us to narrow the range of possible mechanisms in such uncertain cases.
The structure of amyloids during their formation has been studied for both synthetic and recombinant Aβ(1-40) and Aβ(1-42) preparations [20, 21, 54, 61, 62]. We were the first to determine the sizes of nuclei of amyloid fibrils for Aβ(1-40) and Aβ(1-42) peptides [24] using reported kinetic data [20, 21]. It should be noted that the sizes of nuclei (both the primary and secondary nucleation) for Aβ(1-42) fibrils were larger than for Aβ(1-40) fibrils. It was shown that the size of the primary nucleus for Aβ(1-42) fibrils corresponds to three monomers, and the size of the secondary nucleus for this peptide is two monomers. Similarly, it was determined that the size of the primary nucleus for Aβ(1-40) is two monomers, and the size of the secondary nucleus is one monomer. Knowledge of the sizes of the nuclei for the fibrillation process necessary to halt the further growth of fibrils is required to search for new drugs to correct the misfolded forms of proteins [63].
Molecular mechanism of amyloid formation by Aβ peptide. Since the 1960s, intensive studies have been carried out using electron microscopy of various amyloid deposits localized in various organs (liver, spleen, heart, skin, and brain) [64-73]. Gradually, an understanding of the structural organization of amyloid fibrils has been formed from a combination of EM and X-ray diffraction analysis. According to the data of Chiti and Dobson [19], amyloid fibrils have an average diameter of about 10 nm, and the length can reach up to 15 µm. Fibrils can consist of 2-6 filaments and have different morphologies. They can associate laterally into ribbons, bundles, and twists with different periods. Amyloid structures in X-ray diffraction analysis show the presence of cross-β structure. Such structure is interpreted so that the amyloid fibrils are constructed from β-sheets passing along the entire axis of the fibril. The β-sheet itself is constructed of β-strands passing perpendicular to the axis of the fibril [17, 18, 74]. This interpretation of the structural organization of fibrils dominates in the literature. Until now, this interpretation of the molecular structure of amyloid fibrils for various proteins/peptides has been refined [57, 58]. However, there are other ideas about the structural organization of amyloid fibrils. In [75], based on X-ray diffraction analysis at small angles, it is assumed that the fibril can be constructed from tubular cylinders.
Oligomers are an invariable participant in the fibrillation process. In many works, where the EM method has been used to analyze amyloid formation, the presence of annular oligomeric particles with a diameter of about 10 nm can be seen on images (even if they are not mentioned in the articles) [76, 77]. However, if the authors pay attention to them, they rarely describe the method of fibril formation through the association of precisely oligomeric particles into extended polymers. Nevertheless, an example of the use of EM for studying the fibrillogenesis process for Aβ(1-42) peptide has been published [78]. It was shown that fibril formation occurs through association of oligomeric particles having the diameter of a mature fibril. A new mechanism for the formation of α-synuclein fibrils according to which the fibril is formed by association of the formed oligomeric granules, which are the building unit of the fibril (a double-concerted fibrillation model), has been proposed [79]. The assembly of fibrils from oligomers was demonstrated for lysozyme [80]. On EM and AFM images from the works of various authors, one can notice rounded oligomers of approximately the same diameter with thin (single mature) fibrils and the oligomeric particles often have a cavity inside, i.e. they are ring-like [77, 81]. Several schemes for assembling amyloid fibrils for amyloid proteins have also been proposed, in which the formation of fibrils occurs due to the interaction of oligomeric particles in different ways [82-85]. Our experience in analyzing various amyloidogenic proteins (insulin, Aβ peptides and their fragments, amyloidogenic fragments of Bgl2 protein) shows that at the beginning of fibril formation there are many oligomeric ring structures and a small number of short thin (single) fibrils with a diameter comparable to that of oligomers [24, 54, 56, 86-88]. With increasing incubation time, the number of oligomeric particles decreases, the fibrils grow to several microns in length, and fibrils of different morphologies emerge. With an increasing magnification of EM images, one can note that the fibrils are constructed from ring-like oligomers associated in different ways, which is the reason for polymorphism of fibrils under the same conditions (Fig. 3). Despite the different amino acid composition of the peptides, the average diameter of a ring-like oligomer is about 10 nm (similar to the diameter of mature fibrils). However, we believe that each peptide has its peculiarities when forming an oligomer. The outer and inner diameters of the ring, its height, and the number of monomers and way of its molecular packing in oligomers can vary [54, 56, 62, 87, 88]. To correctly interpret the experimental data obtained using EM methods, it becomes evident that the X-ray diffraction analysis method (the preparation method and the correct interpretation of the diffraction patterns obtained), solid-state NMR, and the involvement of bioinformatics research methods play an indispensable role for the final interpretation of the results.
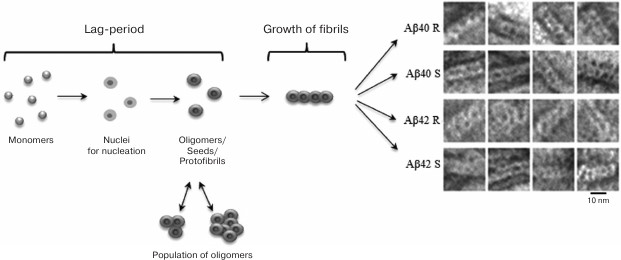
The theoretical and experimental results allowed us to offer a new model of the structural organization of amyloid fibrils and the mechanism of fibril formation. The model assumes that the formation of fibrils occurs according to the following simplified scheme: monomer → ring-like oligomer → mature fibril constructed of ring-like oligomers as the main building blocks (Fig. 4) [24, 54, 56, 86].
Fig. 3. EM images of polymorphic forms of fibrils of recombinant Aβ(1-40) peptide (0.2 mg/ml, 27 h incubation at 25°C, 50 mM Tris-HCl, pH 7.5). a) Single fibrils; b) fibrils in the form of ribbons; c) fibrils in the form of bundles.
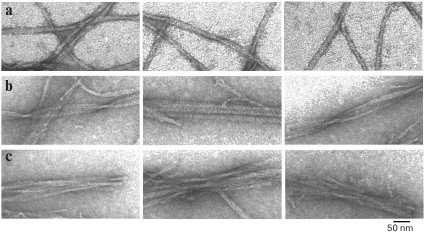
Fig. 4. Schematic representation of the fibrillation process. The building block for assembling fibrils is a ring-like oligomer. Ring-like oligomers interact with each other side-by-side, slightly overlapping each other. The fibril formation process is the same for Aβ(1-40) and Aβ(1-42) peptides. R and S are recombinant and synthetic samples, respectively [54, 89].
TOXICITY OF FIBRILS AND OLIGOMERS FORMED BY Aβ
PEPTIDE
It is believed that insoluble fibrillar aggregates of Aβ peptide found in senile plaques trigger neurodegenerative processes in AD. However, there is no clear correlation between the number and size of amyloid plaques and the severity of memory impairment or cellular dysfunction [90]. The accumulated data indicate that prefibrillar soluble Aβ peptide oligomers induce synaptic dysfunction [91, 92]. In the literature, data are found indicating that the size of oligomers of the Aβ peptide is distributed over a wide range of molecular weights (from 10 to 100 kDa), and structural polymorphism of oligomers of Aβ peptides is mentioned [93]. Studies have shown that Aβ peptide can accumulate both inside and outside cells [94, 95]. However, despite numerous published data confirming the critical role of Aβ oligomers in synaptic dysfunction and cell death, exact mechanisms of the toxic effect of amyloid oligomers are still unclear [96].
Aβ peptides can have a negative effect on neurons and other types of brain cells. It is known that Aβ peptide in the process of self-aggregation promotes the development of reactive oxygen species. Aggregation of Aβ peptides on the neuronal membrane causes lipid peroxidation and the formation of a toxic aldehyde called 4-hydroxynonenal, which in turn weakens the function of ATP-dependent ion channels, glucose transporters, and glutamate. As a result, the Aβ peptide promotes depolarization of the synaptic membrane, excessive influx of calcium, and disruption of mitochondrial structure [97].
As a highly toxic peptide, oligomeric Aβ peptide directly stimulates neuronal apoptosis by interacting with cell surface receptors. Moreover, long-term accumulation of toxic species of Aβ peptide in the parenchyma also leads to oxidative damage to DNA and proteins, to physical damage to cellular organelles, and to disruption of intracellular calcium homeostasis. Each of these factors can provoke cell death [98].
The inadequacy of autophagy and other ways of controlling the quality of proteins contributes to the dysfunction of neurons and glial cells. Activation and proliferation of glial cells promote inflammation. Several mechanisms affect GABAergic signaling and contribute to the loss of inhibitory tone [96].
Previously, it was suggested that soluble Aβ peptide oligomers are the main cause of synaptic dysfunction and memory loss in AD. To clarify this issue, the neurotoxicity of various Aβ isoforms on cultures of PC12 cells was analyzed [99]. The results showed that Aβ(1-42) peptide can form oligomers much faster than Aβ(1-40) peptide, and Aβ(1-43) and Aβ(1-42) peptides exhibit the highest level of neurotoxicity. In general, these data demonstrate the high pathogenicity of Aβ(1-42) peptide among the three Aβ isoforms and support the idea that Aβ(1-42) oligomers are involved in pathological processes leading to neurodegeneration in AD.
In addition, based on EM and AFM data it is assumed that oligomeric particles having annular morphology can form pore-like structures (annular pores) on the cell surface (membrane pores). This can lead to perforation of the cell membrane and disruption of cellular metabolism, which results in cell death [81, 100].
Possible physiological role of Aβ peptide. Aβ peptide is most often characterized as a product of APP processing that plays no physiological role. However, it has been shown that the Aβ peptide is a specific ligand for several different receptors and molecules that are activated in response to exogenous stress factors, and it can induce inflammatory processes. It has been found that Aβ peptides have a striking similarity to peptide LL-37 of the cathelicidin family. This peptide is prone to formation of cytotoxic soluble oligomers and insoluble fibrils, as well as to staining with Congo red, which is considered to be a characteristic of amyloid fibrils. The LL-37 peptide is an antimicrobial peptide (AMP), a component of the innate human immune system. Because of the similarity of Aβ and LL-37, the potential antimicrobial activity of Aβ(1-42) and Aβ(1-40) was investigated. It turned out that these peptides suppress the growth of 8 of 12 investigated and clinically significant microorganisms, such as C. albicans, E. coli, S. aureus, etc. The minimum inhibitory concentration of Aβ peptides is comparable, and in some cases, is less than that for LL-37. Homogenates of tissues of the frontal lobes of the brain from patients who died of AD also suppressed the growth of microorganisms. The antimicrobial activity of tissue samples can be reduced by inactivating the peptides with anti-Aβ antibodies, which also corresponds to the assumption of Aβ-mediated antimicrobial activity [101]. It can be assumed that Aβ peptides are AMPs and play an important role in nonspecific immune response in brain tissues. Alzheimer’s disease can be a consequence of a disturbance in the regulation of APP processing, the production of Aβ peptide, and an increase in the Aβ(1-40)/Aβ(1-42) ratio.
INTERACTION OF Aβ PEPTIDE WITH OTHER MOLECULES
Influence of molecular chaperones on amyloid formation of Aβ peptide. Chaperones are unique remodeling proteins that participate in a variety of intracellular processes and are involved in the adjustment of protein structure, prevention of their aggregation, destruction of protein aggregates, and the unfolding of native target proteins for translocation through membranes. In addition, chaperones participate both in the disassembly of active oligomeric structures to the state of inactive unfolded monomers for their subsequent proteolytic degradation, and in the formation of specific complexes and protein ensembles. Chaperones are part of a large family of heat shock proteins (hsp), the synthesis of which in cells increases significantly in response to heat shock or other types of cell stress. However, in the absence of stressful effects, most of the proteins of this family are synthesized quite intensively.
Different molecular chaperones interact with proteins prone to aggregation, and they partially control or prevent accumulation of aggregates in cells. Molecular chaperones of bacterial origin can also interact with fibrillogenic proteins.
GroEL from E. coli is the best-studied member of the family of chaperones, which ensure the correct folding of partially unfolded proteins. GroEL operates in conjunction with the cochaperone GroES. GroEL consists of two ring structures, each being constructed of seven monomeric subunits. Each subunit of the heptameric “ring” GroEL (57 kDa) consists of three domains: the apical (Ap) domain containing the common binding center of the non-native proteins and the cochaperone GroES; the hinge intermediate domain (In), and the C-terminal equatorial domain (Eq) with an ATPase center (Fig. 5). Equatorial domains provide the main intersubunit contacts both inside the heptameric ring and between the rings of the chaperone. The interaction of the equatorial domains of the two GroEL rings leads to the formation of a mirror-symmetric toroid with two isolated hydrophobic cavities (trans-state of the rings), whose entrance openings are formed by apical domains (cavity diameter about 45 Å) [102]. The 10-kDa subunits of the cochaperone GroES also form a cyclic heptameric dome complex that can cover one of the GroEL toroid ends. The interaction of GroEL chaperone with GroES leads to significant conformational changes in the chaperone, an increase in cavity size up to 60 Å, and its hydrophilization (cis-state of the ring) (Fig. 5) [103]. There are two viewpoints about the operation of the GroEL complex: (i) the peptide/protein cannot aggregate, and it acquires a native conformation upon entering the cavity [104]; (ii) the peptide/protein does not enter the cavity, but it interacts with the outer part of the complex [105].
Fig. 5. Arrangement and linear dimensions of the heptameric GroEL ring: a) open conformation (without GroES), PDB 4AAQ [102]; b) closed conformation (associated with GroES), PDB 3WVL [103]. The figures indicate the equatorial domain (1-136, 410-525 a.a., light gray), the apical domain (191-370 a.a., dark gray), and the intermediate domain (137-190, 371-409 a.a., black). Oligomeric complexes of GroEL are presented in the section.
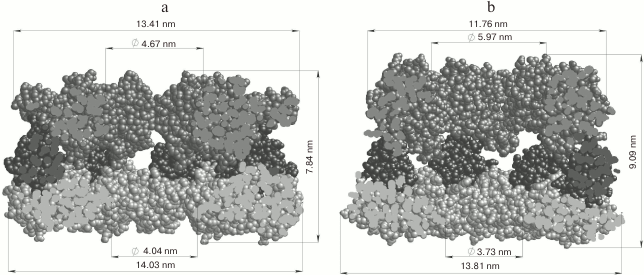
NMR studies of the interaction of the GroEL substrate with the Aβ peptide as a model ligand showed that GroEL can suppress the formation of amyloid Aβ(1-40) by interacting with its two hydrophobic segments, Leu17-Ala21 and Ala30-Val36, which contain the key amino acid residues for fibrillogenesis. Also, the binding site with Aβ(1-40) was identified on the GroEL mini-chaperone molecule (apical domain from 193 to 375 a.a.) by the NMR method. It includes two α-helices, H and I, located in the apical domain [106].
It has been found that the isolated apical domain of the E. coli GroEL complex subunit can also suppress irreversible fibrillation of numerous amyloid-forming polypeptides, including Aβ(1-40) and α-synuclein. It was shown that the affinity of GroEL with respect to fibrillogenic polypeptides grew in accordance with the increase in surface area for van der Waals interactions of the side-chain substituent [107]. The most effective fibrillation suppressor compared to the GroEL wild phenotype was the mutant protein with the Gly192Trp replacement, that is, with the largest substituent. This is explained by the fact that the presence of side chains of a larger amino acid leads to a slope of the apical domain, as a result of which the hydrophobic regions are opened for interaction with the unfolded polypeptide [107]. These data indirectly indicate that interaction with ligands can occur at the surface of the complex and calls into question the need for the peptide to enter the internal cavity of the GroEL complex.
Crystallins are proteins that form the structure of the eye lens. αA-crystallin (αA) and αB-crystallin (αB) are small molecular chaperones that are necessary to maintain the quasi-crystalline structure and transparency of the lens [108]. In the eye lens, they exist in the form of an αAB-complex at αA/αB ratio 3 : 1. An increased level of α-crystallins is found in oligodendrocytes and astrocytes of the frontal and temporal lobes of the brain from AD patients.
The level of αB-crystallin increases in response to different stress stimuli, and it can be identified with β-amyloid fibrils in extracellular plaques that are characteristic of AD. The ability of αB-crystallin to interact with amyloid fibrils in vitro has been studied. It was found that αB-crystallin binds to wild-type Aβ(1-42) fibrils with micromolar affinity, and it also binds to fibrils formed from mutant Aβ(1-42) with the E22G replacement. Immuno-electronic microscopy confirms that the binding takes place over the entire length. Studies of the effect of αB-crystallin on seeds of growing Aβ peptide fibrils in solution showed that the binding of αB-crystallin to seed fibrils strongly inhibits their elongation [109].
Studies were also conducted on cell cultures of PC12. An attempt was made to determine whether αB-crystallin inhibits the formation of Aβ(1-40) fibrils, and whether this affects the toxicity of the Aβ peptide. To this end, fibrillogenesis of Aβ(1-40) peptide was induced and PC12 cells were treated with this preparation to assess their viability. As expected, mature fibrils and smaller oligomers were more toxic to culture cells than non-induced Aβ(1-40) peptide. At the same time, αB-crystallin completely inhibited the formation of Aβ(1-40) fibrils at a molar ratio of 1 : 100 (αB-crystallin/Aβ(1-40) peptide), which was substantiated by attenuation of thioflavin T (ThT) fluorescence and the absence of fibrils in images obtained by EM. When Aβ(1-40) samples incubated with αB-crystallin were added to PC12 cells, a significant increase in cell survival was observed [110].
Recently, a new class of chaperone-like amyloid-binding proteins was discovered that interferes with the aggregation of proteins and peptides. It was found that one of these proteins, the calcium-binding protein nucleobindin-1 (NUCB1), is a new chaperone-like amyloid-binding protein. NUCB1 has been shown to inhibit the aggregation of amylin associated with type-2 diabetes, the alpha synuclein associated with Parkinson’s disease, the Val30Met transthyretin mutant associated with hereditary amyloid polyneuropathy, and Aβ(1-42), by stabilizing their intermediate protofibrils. It was suggested that NUCB1 binds to the cross-β structure of protofibril aggregates and stabilizes soluble macromolecular complexes through their “capping”. Interestingly, NUCB1 prevented the toxicity of Aβ(1-42) protofibrils in cell cultures. The authors suggested that NUCB1-stabilized amyloid protofibrils can be used as immunogens for production of conformationally specific antibodies and can be used as new tools for creating anti-protofibril diagnosis and therapy [111].
Previously, the structure of the complex between Aβ(1-40) and the neuropeptide leucine-enkephalin was characterized. It was shown that leucine-enkephalin might provide a basis for the discovery of peptide-binding biotherapeutic treatments for Alzheimer’s disease [112]. Amino acid residues were substituted to analyze the complex in vitro. For Aβ peptide, the important residues are Asp1, Glu3, Phe4, Arg5, His6, Tyr10, Glu11, His13, His14, Gln15, K16, Glu22, Lys28, and Val40. Mass spectrometric measurements of ion mobility and modeling of molecular dynamics showed that the hydrophobic C-terminal of leucine-enkephalin (Phe-Leu) is crucial for the formation of peptide complexes. The authors suggested that Phe-Leu preferentially binds to the oligomer of Aβ peptide, and it binds to Aβ most specifically on residues Tyr10 and Gln15 in the region between the N-terminus and the hydrophobic nucleus. In addition, it was shown that Phe-Leu can prevent the formation of fibrils [112].
A 24-a.a. peptide humanin (HN) should be mentioned; it was proposed as a peptide-based inhibitor interacting directly with the Aβ oligomers and interfering with the formation of toxic Aβ peptide. Studies in vivo showed that both HN and its mutant HNG (the peptide with the Ser14Gly replacement) reduced the relative prevalence (the number) of prefibrillar oligomers and thereby reduced the level of toxicity in a similar manner. These results provide information on the mechanisms underlying the anti-oligomer effects of HN and HNG [113].
Action of amino acid derivatives (dopamine, L-DOPA, noradrenalin, adrenalin) on fibrillogenesis of Aβ peptide. Catecholamines (dopamine, L-DOPA, adrenaline) inhibit the formation of Aβ fibrils. ThT fluorescence, EM, and AFM data indicated that micromolar concentrations of dopamine, L-DOPA, norepinephrine, and epinephrine inhibit in vitro the formation of amyloid fibrils of Aβ peptide and α-synuclein depending on the dose, as well as disaggregating already formed fibrils. Light-scattering data was used to confirm the dopamine-induced dissolution of fibrils. The presence of dopamine caused a significant decrease in light scattering. Similarly, a decrease in ThT fluorescence was observed. Direct confirmation of fibril disaggregation was obtained using AFM and EM, the latter showing no fibril on images in the presence of catecholamines [114].
POSSIBLE MISTAKES IN INTERPRETATION OF EXPERIMENTAL DATA FOR
VARIOUS AMYLOIDOGENIC PROTEINS
Interpretation of experimental data requires great care, and many conclusions depend on how the data are analyzed.
Using X-ray diffraction analysis, the cross-β structure of amyloid formations (“pleated-sheet”) was first revealed in the study of amyloidosis-damaged liver and spleen tissues in 1968 [115]. Since then, X-ray diffraction analysis along with direct visualization of amyloids by EM, interaction with ThT, and birefringence with Congo red has become the main method in the study of amyloid structures. Using X-ray diffraction, it was shown that the presence of a cross-β structure is a general characteristic of amyloids [17, 18, 74]. The cross-β structure suggests that the fibrils are structures in which the polypeptide chain is organized so that β-sheets are formed running parallel to the long axis of the fibril at a distance of about 10 Å from each other. Such β-sheets are formed from β-regions located at a distance of about 4.7 Å and running perpendicular to the long axis of the fibril. With the beginning of the decoding of the amino acid sequences of the first amyloidogenic peptides and proteins, the determination of individual amyloidogenic proteins, their intensive study, and the search for mechanisms of fibril formation began. This led to the appearance of several models of stacking of β-sheets in the fibril as well as to the emergence of many models of fibril formation (Fig. 2) [57, 116-122]. However, for each research object (insulin, Aβ peptide, its various fragments, etc.), different models of fibril formation can be found. It has been suggested that fibrils can be formed from a different number of filaments (from 2 to 6), can associate with each other laterally with the formation of bands of different widths, can twist and form bundles of different diameters and periods, or associate with the formation of bundles. The tendency for polymorphism of amyloid fibrils is common to all studied amyloids. In this case, polymorphism is observed not only when the fibril formation conditions (pH, ionic conditions, temperature) change, but under the same conditions. For all amyloid fibrils, it has been noted that the average diameter of the thinnest polymers (single fibrils) is about 10 nm, and the length can reach several microns [19]. The question arises how the common cross-β structure and different ways of tertiary structure formation are preserved for different amino acids, with the same parameters of fibrils being preserved on average. In addition, considering that the simplified scheme of fibril formation destabilized monomer → oligomer → mature fibril, the main unclear point in all schemes is the transition from oligomers, often having an annular structure, to fibrils.
After the model of extended protofibrils twisted along the axis of the fibril was established in the field of amyloid protein research, most of the models followed exactly this interpretation of the structural organization of fibrils. However, the accumulated experimental data pointed to other possible models.
Studying the process of polymerization of insulin back in the 1940s, Waugh discovered that the process can be reversible, since during the disassembly of insulin fibrils, insulin molecules regain biological activity and the possibility of being crystallized [123]. Considering the reversibility of the process, Waugh suggested that fibrils are constructed only from globular, slightly deformed insulin molecules [124].
However, based on measurements of infrared dichroism, Elliott et al. (1951) concluded that insulin fibrils consist of polypeptide chains in a β-structural conformation and are located across the axis of the fibrils [125].
Nevertheless, Koltun et al. (1954), based on X-ray diffraction analysis, confirmed the presence in insulin fibrils of almost unchanged structural monomers of the protein, i.e. insulin in the globular conformation [126]. This analysis was also supported by Reitel (1963) in his review of the association of proteins [127], and then by several other authors [128].
Further, in 1972 Burke and Rougvie considered studying insulin fibril formation, since Koltun et al. (1954) obtained an unexpected meridional reflection at 48 Å on small-angle diffraction of insulin [129]. However, later, a similar meridional reflection (53 Å) was obtained by Kirschner (1993) for Aβ (1-40) peptide [14]. Kirschner et al. explained this reflection as follows: either this reflection displays repetition of the object along the fibril axis, or a repetition when the helix is twisted (periodicity). It should be noted that in subsequent works this reflection was not given due attention, although this may indicate the repetition of some oligomeric particles along the fibril axis.
It should be noted again that in many studies on insulin, it is suggested that the procedure of forming fibrils of a given protein/peptide at the molecular level can differ significantly from the structure of amyloid fibrils of several other fibrillar [130] and synthetic polypeptides [13]. For example, polypeptide chains can be stacked in planes parallel to the axis of the fibrils and form β-sheets that are perpendicular to the axis of the fibril. Then a reflection of 9.6 Å can be both meridional and equatorial. From this it follows that X-ray diffraction data can be ambiguous, and more precise interpretation is required. Recently, data have been obtained that suggest the protein transthyretin has a native conformation in amyloid fibrils [131].
It should be noted that in the 1980s, X-ray diffraction was widely used to study amyloid fibrils. X-ray patterns were obtained for several proteins and peptides, in which, in addition to the main reflections, many additional reflections were observed. Attempts were made to interpret all the observed reflections. However, in the construction of the model of amyloid fibrils, the interpretation was based on the generally accepted idea of the structure of fibrils built from long twisted filaments, and not from the point of view of other possible models.
The possibility of Aβ(1-40) serving as a seed for growth of Aβ(1-42) fibrils and vice versa [132] was discussed for a long time. Previously, many experimental works were published, but only in 2017 this point was called into question [133]. It was shown that both Aβ(1-40) and Aβ(1-42) can act as seeds. This suggests that the building block for constructing fibrils is structurally similar for these isoforms of the Aβ peptide, and does not differ, as shown in many works [121, 134, 135].
DATA OF EARLIER YEARS TO BE CONSIDERED
In this section, we analyze many historical data and contradictions that arose upon studying amyloid structures, and which are still not completely understood. As early as 1935, similar X-ray diffraction patterns were obtained in X-ray diffraction analysis of fibrillar proteins such as fibroin, β-keratin, β-myosin, fibrin, and fibrils from denatured globular protein egg albumin [13]. It was concluded that there is a unified principle at the molecular level for the organization of these proteins in fibrils. According to X-ray diffraction data, characteristic reflections of 9.8 and 4.65 Å were observed for fibrillar proteins and denatured globular proteins. However, it was noted that if for fibrillar proteins the reflection of 9.8 Å (“side-chain spacing”) is meridional, and 4.65 Å (“backbone spacing”) equatorial, then for egg albumin the situation is the reverse. Accordingly, two interpretations of the formation of fibrils at the molecular level have been given. In modern terms for denatured albumin in the fibril, the distance between the chains is 4.65 Å (β-strands) and the distance between the layers is 9.8 Å (between β-sheets). It is the latter interpretation that is currently generally accepted to explain the structure of amyloid proteins/peptides. Before the appearance of the term “cross-β structure”, there was the notion of “pleated sheet” and “rippled sheet”. The configuration of polypeptide chains in the fibrillar structure was described in these terms by Pauling and Corey in 1953 [136]. Only the term “pleated sheet” is now used for amyloid structures. The term cross-β structure first appeared in 1968 [115]. When studying amyloid structures from human liver and spleen tissues and from experimental animals using X-ray structural analysis of well-oriented samples, characteristic meridional reflections of 4.75 Å and equatorial reflections of 9.8 Å were obtained; they are perpendicular to each other (cross-location).
The invention of an electron microscope and its improvements made possible electron microscopic studies of amyloid formations in various tissues. Beginning in the late 1950s and early 1960s, tissue sections of various organs (spleen, liver, kidneys, skin) [64-67], and then amyloid extracts from these organs, were studied [68-73, 137]. Already in 1959, it was concluded that the morphology of amyloids from different organs is similar. The length of the fibrils is about 1.2-1.6 µm, and the diameter is 50-120 Å [64]. In 1963, Gueft and Ghidoni, based on EM of tissues, concluded that the fibrils consist of two filaments with the width of about 2.5 nm and the filament spacing of about 2.5 nm [65]. The total width of fibrils was 7.5 nm. Then one of the first works was published where in addition to EM studies of tissue sections, isolated amyloid studies were performed using negative contrasting [69]. It was concluded that the structures of amyloids in tissues and preparations of isolated amyloids are similar. It is clearly seen that the fibrils are constructed from structures that were interpreted as doughnut-like or annular. The diameter of these structures was about 10 nm. In addition, this diameter coincides with the diameter of the “native” fibrils in tissues. At higher magnification, it is seen that the ring structure consists of five subunits; such rings are combined in pairs (the height of a pair is 40 Å) and piled on top of each other in stacks, forming rod-like fibrils. Thus, it follows from these data that the main element of the fibril is the ring structure.
At the same time, similar research was conducted by Shirahama and Cohen [71]. They used a method for isolation of fibrils from amyloid formations of the liver and spleen that was different from that used by Benditt and Eriksen [69]. The morphology of both tissue sections and isolated fibrils was studied. According to their data, fibrils in damaged tissues consist of two or more filaments. The filament has a diameter of about 75-80 Å and length up to 1.5 µm. On sections of tissues, oligomeric structures of the same diameter are visible, which were interpreted as longitudinal sections of filaments. It is notable that on the section the filament looks like an annular structure consisting of five globular particles (pentagon) with the diameter of about 25-35 Å, and the central cavity of the ring also has the diameter of about 25-35 Å. Fibrils of similar morphology and parameters are also observed in studies of negatively stained preparations of isolated fibrils. This is one of the first attempts to describe the morphology (structure) of fibrils at the level of filaments and protofibrils. The filament (the thinnest fibrillar formation) is formed from five protofibrils associated with each other laterally along the long axis of the filament, so that a pentagonal structure with an empty middle is visible on the section. Thus, the structure of amyloid fibrils is very different from that described in [69]. However, the authors doubted the interpretation of the construction of fibrils from ring doughnut-like structures [69], since they did not observe such structures in their images. It was suggested that the use of different methods of isolation of fibrils from amyloidogenic tissues may result in different structures. Nevertheless, upon careful examination of images of negatively contrasted preparations of isolated amyloids, one can see separately lying ring structures with the diameter comparable to that of the thinnest fibrils (about 10 nm).
It should be noted that after the papers by Shirahama [71] and Glenner [137] were published, in which two methods of isolation of amyloid fibrils from damaged tissues of the liver, spleen and kidneys were used, according to the method of isolation both ring-like oligomers and those constructed from them have rod-like structures, as in [69], or elongated amyloid fibrils described by Shirahama. After this work, the main attention was given to amyloid fibrils built from protofilaments passing along the entire length of the fibril. In our opinion, the interpretation of the EM data of the construction of fibrils/filaments from a series of laterally associated protofibrils could be derived from the interpretation of early studies that used X-ray structural analysis of fibrillar structures, according to which amyloids have cross-β structure. This structure suggests that β-sheets pass along the entire fibril. This model of amyloid fibrils constructed from extended β-sheets formed from β-strands is still used. Practically no attention was given to the work of Inoue et al. [138], when after deciphering the first components of amyloid deposits Glenner [139], who studied amyloid P, showed that amyloidosis is caused by different amyloid proteins. According to the EM data analysis, two types of particles were revealed: (i) pentagonal planar structures 8.5 nm in diameter and 2.5 nm in height with a hole in the center, and (ii) structures formed from such pentagonal particles lying on top of each other in the form of a stack of disk-like, elongated particles. In fact, the results of 1966 were repeated [69].
However, other mechanisms for the formation of amyloid structures already mentioned above have been described. The negative contrasting method for the study of amyloid structures is described in [78]. Samples of Aβ(1-42) peptide at different incubation times were analyzed, and, based on electron microscopy data, it was assumed that fibrils are formed from globular oligomeric particles. A mechanism for the formation of α-synuclein fibrils from homogeneous granules was also proposed [79]. Granules are first formed from monomers, and then the granules are associated with the formation of fibrils whose diameter coincides with the diameter of the granules (a double-concerted fibrillation model). Much attention was given to the study of intermediates along the path of fibril formation [140]: oligomeric dodecamers that interact with each other with the formation of fibrils. It should be noted that in many studies on the mechanism of formation of amyloid structures, it is noted that in the initial stages of fibrillation (most often during the lag period) on EM images, it is possible to observe annular oligomeric particles, which no longer occur in images of mature fibrils. However, most often their absence at late stages of fibril formation is not discussed. It is noteworthy that with sufficiently high-quality images of various fibrils, one can see that they are formed from annular oligomeric particles.
Our experience with various amyloid proteins such as insulin, lispro insulin, and Aβ(1-42) and Aβ(1-40) peptides, their amyloidogenic fragments (Aβ(16-25), Aβ(31-40), Aβ(33-42)), as well as amyloidogenic fragments of the cell wall protein of yeast Bgl2 (GluNB, AspNB) showed that the building block in the formation of fibrils is a ring-like oligomer. Ring-like oligomers interact with each other laterally, often overlapping. At the molecular level, the structures of ring-like oligomers of different proteins/peptides differ [54, 56, 87]. According to our modeling, the structures of ring-like oligomers contain small fragments of β-sheets. Many such fragments can give characteristic reflections for cross-β structure in the X-ray diffraction analysis. However, there are numerous other reflections on the X-ray images of amyloid fibrils. Such reflections can help to show the correct structural organization of amyloid fibrils if one properly interprets them.
Our experience with various amyloidogenic proteins/peptides and analysis of the literature on the mechanism(s) of formation of fibrils shows that not all the data are always taken into account. First, with X-ray structural analysis, the focus is on two reflections, 4.5-4.8 and about 8-12 Å, which indicate the cross-β structural organization of amyloid fibrils. In this case, many other reflections, and they always are present, are not given proper attention. Moreover, most often in the X-ray diffraction patterns there are no clear reflections in the region of about 4.7 and 11 Å; these reflections are indistinct, especially in the case of a normally diffuse reflection at 11 Å (the distance between β-sheets). Most often this is explained by a different way of preparing samples for X-ray analysis or inadequate orientation of fibrils [16, 141]. It is notable that attempts to analyze X-ray analysis data undertaken in the 1980s and 1990s [14, 18, 75, 142] have not been further developed. We think that the general conception of the structure of the amyloid fibril as a polymer built from β-sheets running parallel along the entire axis of the fibril (the β-sheets themselves are formed from β-regions perpendicular to the fibril axis) should be analyzed more carefully. We believe that it is necessary to simulate the diffraction pattern from annular oligomeric structures, with parameters obtained from the EM analysis and using data on the molecular structure of oligomers calculated using theoretical methods. Enough evidence has accumulated indicating that the structure of amyloid fibrils can vary [54, 79, 80, 87]. Proceeding from the generally accepted view on the structural organization of fibrils, it is difficult to imagine the mechanism of fibril formation and the possibility of the existence of a β-sheet passing along the entire fibril (up to 10 or more microns). In addition, it is difficult to explain the polymorphism of fibrils, their branching, and their fragmentation. From our data on fibrillation of various amyloidogenic proteins/peptides, we suggest that the main building block for the formation of fibrils is a ring-like oligomer with the parameters and structure inherent in the corresponding protein/peptide (Fig. 6). The association of such ring-like oligomers in various ways leads to the formation of fibrils of different morphologies.
Fig. 6. X-ray diffraction analysis of Aβ(1-40) and Aβ(1-42) peptides. a) Schematic representation of a typical diffraction pattern for the cross-β structure (8-12 Å, equatorial reflection; 4.5-4.8 Å, meridional reflection). b, c) Diffraction of Aβ(1-40) and Aβ(1-42) synthetic peptides (Sigma-Aldrich, USA) in 50 mM Tris-HCl, pH 7.5 [143]. In addition to the main reflections (4.5-4.8 and 8-12 Å), some other reflections are indicated. d) Schematic representation of the arrangement of ring-like oligomers in amyloid fibrils for Aβ(1-40) and Aβ(1-42) peptides. Equatorial reflections are marked perpendicular to the fibril axis, and meridional reflections are parallel. Ring-like oligomeric structures are short hollow cylinders.
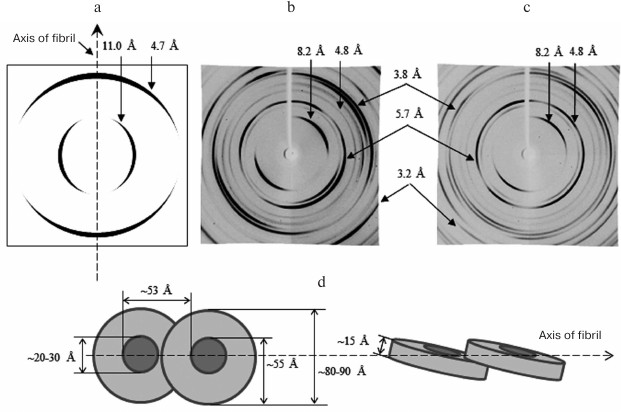
In the literature, there is an opinion that a personal approach should be used when developing therapeutic agents against amyloidosis, since it has been shown that fibrillar formations in different patients can morphologically differ. In this connection, the discovery of a single method for forming all fibrils from oligomeric structures could facilitate the development of generic drugs. However, in our opinion, the main attention should be given not to polymer formations of proteins/peptides in the form of fibrils or even to oligomeric aggregates, but to physiological, genetic, and other reasons leading to destabilization of native protein/peptide molecules and triggering the formation of fibrils.
Acknowledgments
These studies were supported by the Russian Science Foundation (project No. 14-14-00536) and by the Molecular and Cell Biology Program (project No. 01201353567).
REFERENCES
1.Uberti, D., Cenini, G., Bonini, S. A., Barcikowska,
M., Styczynska, M., Szybinska, A., and Memo, M. (2010) Increased CD44
gene expression in lymphocytes derived from Alzheimer’s disease
patients, Neurodegener. Dis., 7, 143-147.
2.Song, J., Wang, S., Tan, M., and Jia, J. (2012)
G1/S checkpoint proteins in peripheral blood lymphocytes are
potentially diagnostic biomarkers for Alzheimer’s disease,
Neurosci. Lett., 526, 144-149.
3.Saha, A. R., Ninkina, N. N., Hanger, D. P.,
Anderton, B. H., Davies, A. M., and Buchman, V. L. (2000) Induction of
neuronal death by alpha-synuclein, Eur. J. Neurosci., 12,
3073-3077.
4.Caughey, B., and Lansbury, P. T. (2003)
Protofibrils, pores, fibrils, and neurodegeneration: separating the
responsible protein aggregates from the innocent bystanders, Annu.
Rev. Neurosci., 26, 267-298.
5.Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R.,
Glabe, C. G., Yang, A., Gallagher, M., and Ashe, K. H. (2006) A
specific amyloid-beta protein assembly in the brain impairs memory,
Nature, 440, 352-357.
6.Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz,
A., Shepardson, N. E., Smith, I., Brett, F. M., Farrell, M. A., Rowan,
M. J., Lemere, C. A., Regan, C. M., Walsh, D. M., Sabatini, B. L., and
Selkoe, D. J. (2008) Amyloid-β protein dimers isolated directly
from Alzheimer’s brains impair synaptic plasticity and memory,
Nat. Med., 14, 837-842.
7.Hardy, J., and Selkoe, D. J. (2002) The amyloid
hypothesis of Alzheimer’s disease: progress and problems on the
road to therapeutics, Science, 297, 353-356.
8.Burns, A., and Iliffe, S. (2009) Alzheimer’s
disease, BMJ, 338, b158.
9.Chartier-Harlin, M. C., Crawford, F., Houlden, H.,
Warren, A., Hughes, D., Fidani, L., Goate, A., Rossor, M., Roques, P.,
and Hardy, J. (1991) Early-onset Alzheimer’s disease caused by
mutations at codon 717 of the beta-amyloid precursor protein gene,
Nature, 353, 844-846.
10.Czlonkowska, A., and Kurkowska-Jastrzebska, I.
(2011) Inflammation and gliosis in neurological diseases –
clinical implications, J. Neuroimmunol., 231, 78-85.
11.Maltsev, A. V., Santockyte, R., Bystryak, S., and
Galzitskaya, O. V. (2014) Activation of neuronal defense mechanisms in
response to pathogenic factors triggering induction of amyloidosis in
Alzheimer’s disease, J. Alzheimer’s Dis., 40,
19-32.
12.Brookmeyer, R., Johnson, E., Ziegler-Graham, K.,
and Arrighi, H. M. (2007) Forecasting the global burden of
Alzheimer’s disease, Alzheimers Dement. J. Alzheimer’s
Assoc., 3, 186-191.
13.Astbury, W. T., Dickinson, S., and Bailey, K.
(1935) The X-ray interpretation of denaturation and the structure of
the seed globulins, Biochem. J., 29, 2351-2360.
14.Inouye, H., Fraser, P. E., and Kirschner, D. A.
(1993) Structure of beta-crystallite assemblies formed by Alzheimer
beta-amyloid protein analogues: analysis by X-ray diffraction,
Biophys. J., 64, 502-519.
15.Malinchik, S. B., Inouye, H., Szumowski, K. E.,
and Kirschner, D. A. (1998) Structural analysis of Alzheimer’s
beta(1-40) amyloid: protofilament assembly of tubular fibrils,
Biophys. J., 74, 537-545.
16.Serpell, L. C., Fraser, P. E., and Sunde, M.
(1999) X-ray fiber diffraction of amyloid fibrils, Methods
Enzymol., 309, 526-536.
17.Sunde, M., and Blake, C. (1997) The structure of
amyloid fibrils by electron microscopy and X-ray diffraction, Adv.
Protein Chem., 50, 123-159.
18.Sunde, M., Serpell, L. C., Bartlam, M., Fraser,
P. E., Pepys, M. B., and Blake, C. C. (1997) Common core structure of
amyloid fibrils by synchrotron X-ray diffraction, J. Mol. Biol.,
273, 729-739.
19.Chiti, F., and Dobson, C. M. (2006) Protein
misfolding, functional amyloid, and human disease, Annu. Rev.
Biochem., 75, 333-366.
20.Cohen, S. I. A., Linse, S., Luheshi, L. M.,
Hellstrand, E., White, D. A., Rajah, L., Otzen, D. E., Vendruscolo, M.,
Dobson, C. M., and Knowles, T. P. J. (2013) Proliferation of
amyloid-β42 aggregates occurs through a secondary nucleation
mechanism, Proc. Natl. Acad. Sci. USA, 110,
9758-9763.
21.Meisl, G., Yang, X., Hellstrand, E., Frohm, B.,
Kirkegaard, J. B., Cohen, S. I. A., Dobson, C. M., Linse, S., and
Knowles, T. P. J. (2014) Differences in nucleation behavior underlie
the contrasting aggregation kinetics of the Aβ40 and Aβ42
peptides, Proc. Natl. Acad. Sci. USA, 111, 9384-9389.
22.Dovidchenko, N. V., Finkelstein, A. V., and
Galzitskaya, O. V. (2014) How to determine the size of folding nuclei
of protofibrils from the concentration dependence of the rate and
lag-time of aggregation. I. Modeling the amyloid protofibril formation,
J. Phys. Chem. B, 118, 1189-1197.
23.Dovidchenko, N. V., and Galzitskaya, O. V. (2015)
Computational approaches to identification of aggregation sites and the
mechanism of amyloid growth, Adv. Exp. Med. Biol., 855,
213-239.
24.Dovidchenko, N. V., Glyakina, A. V., Selivanova,
O. M., Grigorashvili, E. I., Suvorina, M. Y., Dzhus, U. F., Mikhailina,
A. O., Shiliaev, N. G., Marchenkov, V. V., Surin, A. K., and
Galzitskaya, O. V. (2016) One of the possible mechanisms of amyloid
fibrils formation based on the sizes of primary and secondary folding
nuclei of Aβ40 and Aβ42, J. Struct. Biol., 194,
404-414.
25.Tanzi, R. E., Gusella, J. F., Watkins, P. C.,
Bruns, G. A., St. George-Hyslop, P., Van Keuren, M. L., Patterson, D.,
Pagan, S., Kurnit, D. M., and Neve, R. L. (1987) Amyloid beta protein
gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer
locus, Science, 235, 880-884.
26.Goate, A., Chartier-Harlin, M. C., Mullan, M.,
Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving,
N., and James, L. (1991) Segregation of a missense mutation in the
amyloid precursor protein gene with familial Alzheimer’s disease,
Nature, 349, 704-706.
27.Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum,
J. M., Masters, C. L., Grzeschik, K. H., Multhaup, G., Beyreuther, K.,
and Muller-Hill, B. (1987) The precursor of Alzheimer’s disease
amyloid A4 protein resembles a cell-surface receptor, Nature,
325, 733-736.
28.Eschenko, N. D. (2004) Biochemistry of Mental
and Nervous Diseases [in Russian], SPbSU, St. Petersburg.
29.Stanton, L. R., and Coctzee, R. H. (2004)
Down’s syndrome and dementia, Adv. Psych. Treatment,
10, 50-58.
30.Molinari, M., Eriksson, K. K., Calanca, V.,
Galli, C., Cresswell, P., Michalak, M., and Helenius, A. (2004)
Contrasting functions of calreticulin and calnexin in glycoprotein
folding and ER quality control, Mol. Cell, 13,
125-135.
31.Van Nostrand, W. E., Melchor, J. P., Cho, H. S.,
Greenberg, S. M., and Rebeck, G. W. (2001) Pathogenic effects of D23N
Iowa mutant amyloid beta-protein, J. Biol. Chem., 276,
32860-32866.
32.Nilsberth, C., Westlind-Danielsson, A., Eckman,
C. B., Condron, M. M., Axelman, K., Forsell, C., Stenh, C., Luthman,
J., Teplow, D. B., Younkin, S. G., Naslund, J., and Lannfelt, L. (2001)
The “Arctic” APP mutation (E693G) causes Alzheimer’s
disease by enhanced Abeta protofibril formation, Nat. Neurosci.,
4, 887-893.
33.Priller, C., Bauer, T., Mitteregger, G., Krebs,
B., Kretzschmar, H. A., and Herms, J. (2006) Synapse formation and
function is modulated by the amyloid precursor protein, J. Neurosci.
Off. J. Soc. Neurosci., 26, 7212-7221.
34.Mal’tsev, A. V., and Galzitskaya, O. (2010)
Formation and participation of nonamyloid in the pathogenesis of
Alzheimer’s disease and other amyloidogenic diseases, Biomed.
Khim., 56, 624-638.
35.Hefter, D., and Draguhn, A. (2017) APP as a
protective factor in acute neuronal insults, Front. Mol.
Neurosci., 10, 22.
36.Pardossi-Piquard, R., Petit, A., Kawarai, T.,
Sunyach, C., Alves da Costa, C., Vincent, B., Ring, S., D’Adamio,
L., Shen, J., Muller, U., St. George Hyslop, P., and Checler, F. (2005)
Presenilin-dependent transcriptional control of the Abeta-degrading
enzyme neprilysin by intracellular domains of betaAPP and APLP,
Neuron, 46, 541-554.
37.Belyaev, N. D., Nalivaeva, N. N., Makova, N. Z.,
and Turner, A. J. (2009) Neprilysin gene expression requires binding of
the amyloid precursor protein intracellular domain to its promoter:
implications for Alzheimer’s disease, EMBO Rep.,
10, 94-100.
38.Fraering, P. C. (2007) Structural and functional
determinants of gamma-secretase, an intramembrane protease implicated
in Alzheimer’s disease, Curr. Genomics, 8,
531-549.
39.Citron, M., Westaway, D., Xia, W., Carlson, G.,
Diehl, T., Levesque, G., Johnson-Wood, K., Lee, M., Seubert, P., Davis,
A., Kholodenko, D., Motter, R., Sherrington, R., Perry, B., Yao, H.,
Strome, R., Lieberburg, I., Rommens, J., Kim, S., Schenk, D., Fraser,
P., St George Hyslop, P., and Selkoe, D. J. (1997) Mutant presenilins
of Alzheimer’s disease increase production of 42-residue amyloid
beta-protein in both transfected cells and transgenic mice, Nat.
Med., 3, 67-72.
40.Sun, L., Zhou, R., Yang, G., and Shi, Y. (2017)
Analysis of 138 pathogenic mutations in presenilin-1 on the in
vitro production of Aβ42 and Aβ40 peptides by
γ-secretase, Proc. Natl. Acad. Sci. USA, 114,
E476-E485.
41.Vetrivel, K. S., Cheng, H., Lin, W., Sakurai, T.,
Li, T., Nukina, N., Wong, P. C., Xu, H., and Thinakaran, G. (2004)
Association of gamma-secretase with lipid rafts in post-Golgi and
endosome membranes, J. Biol. Chem., 279, 44945-44954.
42.Paschkowsky, S., Hamze, M., Oestereich, F., and
Munter, L. M. (2016) Alternative processing of the amyloid precursor
protein family by rhomboid protease RHBDL4, J. Biol. Chem.,
291, 21903-21912.
43.Willem, M., Tahirovic, S., Busche, M. A.,
Ovsepian, S. V., Chafai, M., Kootar, S., Hornburg, D., Evans, L. D. B.,
Moore, S., Daria, A., Hampel, H., Muller, V., Giudici, C., Nuscher, B.,
Wenninger-Weinzierl, A., Kremmer, E., Heneka, M. T., Thal, D. R.,
Giedraitis, V., Lannfelt, L., Müller, U., Livesey, F. J.,
Meissner, F., Herms, J., Konnerth, A., Marie, H., and Haass, C. (2015)
η-Secretase processing of APP inhibits neuronal activity in the
hippocampus, Nature, 526, 443-447.
44.Nussbaum, J. M., Seward, M. E., and Bloom, G. S.
(2013) Alzheimer’s disease: a tale of two prions, Prion,
7, 14-19.
45.Marcelli, S., Corbo, M., Iannuzzi, F., Negri, L.,
Blandini, F., Nistico, R., and Feligioni, M. (2017) The involvement of
post-translational modifications in Alzheimer’s disease, Curr.
Alzheimer Res., doi: 10.2174/1567205014666170505095109.
46.Guntupalli, S., Jang, S. E., Zhu, T., Huganir, R.
L., Widagdo, J., and Anggono, V. (2017) GluA1 subunit ubiquitination
mediates amyloid-β-induced loss of surface
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)
receptors, J. Biol. Chem., 292, 8186-8194.
47.Girvan, P., Miyake, T., Teng, X., Branch, T., and
Ying, L. (2016) Kinetics of the interactions between copper and
amyloid-β with FAD mutations and phosphorylation at the
N-terminus, ChemBioChem, 17, 1732-1737.
48.Ahmed, M., Davis, J., Aucoin, D., Sato, T.,
Ahuja, S., Aimoto, S., Elliott, J. I., Van Nostrand, W. E., and Smith,
S. O. (2010) Structural conversion of neurotoxic amyloid-beta(1-42)
oligomers to fibrils, Nat. Struct. Mol. Biol., 17,
561-567.
49.Yu, L., Edalji, R., Harlan, J. E., Holzman, T.
F., Lopez, A. P., Labkovsky, B., Hillen, H., Barghorn, S., Ebert, U.,
Richardson, P. L., Miesbauer, L., Solomon, L., Bartley, D., Walter, K.,
Johnson, R. W., Hajduk, P. J., and Olejniczak, E. T. (2009) Structural
characterization of a soluble amyloid beta-peptide oligomer,
Biochemistry (Moscow), 48, 1870-1877.
50.Bitan, G., Kirkitadze, M. D., Lomakin, A.,
Vollers, S. S., Benedek, G. B., and Teplow, D. B. (2003) Amyloid
beta-protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize
through distinct pathways, Proc. Natl. Acad. Sci. USA,
100, 330-335.
51.Bernstein, S. L., Dupuis, N. F., Lazo, N. D.,
Wyttenbach, T., Condron, M. M., Bitan, G., Teplow, D. B., Shea, J.-E.,
Ruotolo, B. T., Robinson, C. V., and Bowers, M. T. (2009)
Amyloid-β protein oligomerization and the importance of tetramers
and dodecamers in the etiology of Alzheimer’s disease, Nat.
Chem., 1, 326-331.
52.Garai, K., and Frieden, C. (2013) Quantitative
analysis of the time course of Aβ oligomerization and subsequent
growth steps using tetramethylrhodamine-labeled Aβ, Proc. Natl.
Acad. Sci. USA, 110, 3321-3326.
53.Jeong, J. S., Ansaloni, A., Mezzenga, R.,
Lashuel, H. A., and Dietler, G. (2013) Novel mechanistic insight into
the molecular basis of amyloid polymorphism and secondary nucleation
during amyloid formation, J. Mol. Biol., 425,
1765-1781.
54.Selivanova, O. M., Surin, A. K., Marchenkov, V.
V., Dzhus, U. F., Grigorashvili, E. I., Suvorina, M. Y., Glyakina, A.
V., Dovidchenko, N. V., and Galzitskaya, O. V. (2016) The mechanism
underlying amyloid polymorphism is opened for Alzheimer’s disease
amyloid-β peptide, J. Alzheimer’s Dis., 54,
821-830.
55.Selivanova, O. M., Suvorina, M. Y., Dovidchenko,
N. V., Eliseeva, I. A., Surin, A. K., Finkelstein, A. V., Schmatchenko,
V. V., and Galzitskaya, O. V. (2014) How to determine the size of
folding nuclei of protofibrils from the concentration dependence of the
rate and lag-time of aggregation. II. Experimental application for
insulin and LysPro insulin: aggregation morphology, kinetics, and sizes
of nuclei, J. Phys. Chem. B, 118, 1198-1206.
56.Selivanova, O. M., Suvorina, M. Y., Surin, A. K.,
Dovidchenko, N. V., and Galzitskaya, O. V. (2017) Insulin and lispro
insulin: what is common and different in their behavior? Curr.
Protein Pept. Sci., 18, 57-64.
57.Walti, M. A., Ravotti, F., Arai, H., Glabe, C.
G., Wall, J. S., Bockmann, A., Guntert, P., Meier, B. H., and Riek, R.
(2016) Atomic-resolution structure of a disease-relevant Aβ(1-42)
amyloid fibril, Proc. Natl. Acad. Sci. USA, doi:
10.1073/pnas.1600749113.
58.Lu, J.-X., Qiang, W., Yau, W.-M., Schwieters, C.
D., Meredith, S. C., and Tycko, R. (2013) Molecular structure of
β-amyloid fibrils in Alzheimer’s disease brain tissue,
Cell, 154, 1257-1268.
59.Sgourakis, N. G., Merced-Serrano, M., Boutsidis,
C., Drineas, P., Du, Z., Wang, C., and Garcia, A. E. (2011)
Atomic-level characterization of the ensemble of the Aβ(1-42)
monomer in water using unbiased molecular dynamics simulations and
spectral algorithms, J. Mol. Biol., 405, 570-583.
60.Cho, K. R., Huang, Y., Yu, S., Yin, S., Plomp,
M., Qiu, S. R., Lakshminarayanan, R., Moradian-Oldak, J., Sy, M.-S.,
and De Yoreo, J. J. (2011) A multistage pathway for human prion protein
aggregation in vitro: from multimeric seeds to β-oligomers
and nonfibrillar structures, J. Am. Chem. Soc., 133,
8586-8593.
61.Bieschke, J., Zhang, Q., Powers, E. T., Lerner,
R. A., and Kelly, J. W. (2005) Oxidative metabolites accelerate
Alzheimer’s amyloidogenesis by a two-step mechanism, eliminating
the requirement for nucleation, Biochemistry (Moscow),
44, 4977-4983.
62.Suvorina, M. Y., Selivanova, O. M.,
Grigorashvili, E. I., Nikulin, A. D., Marchenkov, V. V., Surin, A. K.,
and Galzitskaya, O. V. (2015) Studies of polymorphism of
amyloid-β42 peptide from different suppliers, J.
Alzheimer’s Dis., 47, 583-593.
63.Derrick, J. S., and Lim, M. H. (2015) Tools of
the trade: investigations into design strategies of small molecules to
target components in Alzheimer’s disease, ChemBioChem,
16, 887-898.
64.Cohen, A. S., and Calkins, E. (1959) Electron
microscopic observations on a fibrous component in amyloid of diverse
origins, Nature, 183, 1202-1203.
65.Gueft, B., and Ghidoni, J. J. (1963) The site of
formation and ultrastructure of amyloid, Am. J. Pathol.,
43, 837-854.
66.Terry, R. D., Gonatas, N. K., and Weiss, M.
(1964) Ultrastructural studies in Alzheimer’s presenile dementia,
Am. J. Pathol., 44, 269-297.
67.Shirahama, T., and Cohen, A. S. (1967) Fine
structure of the glomerulus in human and experimental renal
amyloidosis, Am. J. Pathol., 51, 869-911.
68.Shirahama, T., and Cohen, A. S. (1965) Structure
of amyloid fibrils after negative staining and high-resolution electron
microscopy, Nature, 206, 737-738.
69.Benditt, E. P., and Eriksen, N. (1966) Amyloid.
3. A protein related to the subunit structure of human amyloid fibrils,
Proc. Natl. Acad. Sci. USA, 55, 308-316.
70.Glenner, G. G., and Bladen, H. A. (1966)
Purification and reconstitution of the periodic fibril and unit
structure of human amyloid, Science, 154, 271-272.
71.Shirahama, T., and Cohen, A. S. (1967)
High-resolution electron microscopic analysis of the amyloid fibril,
J. Cell Biol., 33, 679-708.
72.Shirahama, T., and Cohen, A. S. (1967)
Reconstitution of amyloid fibrils from alkaline extracts, J. Cell
Biol., 35, 459-464.
73.Pras, M., Schubert, M., Zucker-Franklin, D.,
Rimon, A., and Franklin, E. C. (1968) The characterization of soluble
amyloid prepared in water, J. Clin. Invest., 47,
924-933.
74.Glenner, G. G., Eanes, E. D., Bladen, H. A.,
Linke, R. P., and Termine, J. D. (1974) Beta-pleated sheet fibrils. A
comparison of native amyloid with synthetic protein fibrils, J.
Histochem. Cytochem. Off. J. Histochem. Soc., 22,
1141-1158.
75.Kirschner, D. A., Inouye, H., Duffy, L. K.,
Sinclair, A., Lind, M., and Selkoe, D. J. (1987) Synthetic peptide
homologous to beta protein from Alzheimer’s disease forms
amyloid-like fibrils in vitro, Proc. Natl. Acad. Sci.
USA, 84, 6953-6957.
76.Goldsbury, C. S., Wirtz, S., Muller, S. A.,
Sunderji, S., Wicki, P., Aebi, U., and Frey, P. (2000) Studies on the
in vitro assembly of a beta 1-40: implications for the search
for a beta fibril formation inhibitors, J. Struct. Biol.,
130, 217-231.
77.Goldsbury, C., Frey, P., Olivieri, V., Aebi, U.,
and Muller, S. A. (2005) Multiple assembly pathways underlie
amyloid-beta fibril polymorphisms, J. Mol. Biol., 352,
282-298.
78.Nielsen, E. H., Nybo, M., and Svehag, S. E.
(1999) Electron microscopy of prefibrillar structures and amyloid
fibrils, Methods Enzymol., 309, 491-496.
79.Bhak, G., Lee, J.-H., Hahn, J.-S., and Paik, S.
R. (2009) Granular assembly of alpha-synuclein leading to the
accelerated amyloid fibril formation with shear stress, PLoS
One, 4, e4177.
80.Hill, S. E., Robinson, J., Matthews, G., and
Muschol, M. (2009) Amyloid protofibrils of lysozyme nucleate and grow
via oligomer fusion, Biophys. J., 96, 3781-3790.
81.Quist, A., Doudevski, I., Lin, H., Azimova, R.,
Ng, D., Frangione, B., Kagan, B., Ghiso, J., and Lal, R. (2005) Amyloid
ion channels: a common structural link for protein-misfolding disease,
Proc. Natl. Acad. Sci. USA, 102, 10427-10432.
82.Roychaudhuri, R., Yang, M., Hoshi, M. M., and
Teplow, D. B. (2009) Amyloid beta-protein assembly and
Alzheimer’s disease, J. Biol. Chem., 284,
4749-4753.
83.Kumar, S., and Udgaonkar, J. B. (2009)
Conformational conversion may precede or follow aggregate elongation on
alternative pathways of amyloid protofibril formation, J. Mol.
Biol., 385, 1266-1276.
84.Walsh, D. M., Lomakin, A., Benedek, G. B.,
Condron, M. M., and Teplow, D. B. (1997) Amyloid beta-protein
fibrillogenesis. Detection of a protofibrillar intermediate, J.
Biol. Chem., 272, 22364-22372.
85.Morris, A. M., Watzky, M. A., and Finke, R. G.
(2009) Protein aggregation kinetics, mechanism, and curve-fitting: a
review of the literature, Biochim. Biophys. Acta, 1794,
375-397.
86.Grigorashvili, E. I., Selivanova, O. M.,
Dovidchenko, N. V., Dzhus, U. F., Mikhailina, A. O., Suvorina, M. Y.,
Marchenkov, V. V., Surin, A. K., and Galzitskaya, O. V. (2016)
Determination of size of folding nuclei of fibrils formed from
recombinant Aβ(1-40) peptide, Biochemistry (Moscow),
81, 538-547.
87.Selivanova, O. M., Glyakina, A. V., Gorbunova, E.
Y., Mustaeva, L. G., Suvorina, M. Y., Grigorashvili, E. I., Nikulin, A.
D., Dovidchenko, N. V., Rekstina, V. V., Kalebina, T. S., Surin, A. K.,
and Galzitskaya, O. V. (2016) Structural model of amyloid fibrils for
amyloidogenic peptide from Bgl2p-glucantransferase of S.
cerevisiae cell wall and its modifying analog. New morphology of
amyloid fibrils, Biochim. Biophys. Acta, 1864,
1489-1499.
88.Selivanova, O. M., Gorbunova, E. Y., Mustaeva, L.
G., Grigorashvili, E. I., Suvorina, M. Y., Surin, A. K., and
Galzitskaya, O. V. (2016) Peptide Aβ(16-25) forms nanofilms in the
process of its aggregation, Biochemistry (Moscow), 81,
755-761.
89.Galzitskaya, O. V., and Selivanova, O. M. (2017)
Rosetta stone for amyloid fibrils: the key role of ring-like oligomers
in amyloidogenesis, J. Alzheime’rs Dis., 59,
785-795.
90.Terry, R. D., Masliah, E., Salmon, D. P.,
Butters, N., DeTeresa, R., Hill, R., Hansen, L. A., and Katzman, R.
(1991) Physical basis of cognitive alterations in Alzheimer’s
disease: synapse loss is the major correlate of cognitive impairment,
Ann. Neurol., 30, 572-580.
91.Ferreira, S. T., Vieira, M. N. N., and De Felice,
F. G. (2007) Soluble protein oligomers as emerging toxins in
Alzheimer’s and other amyloid diseases, IUBMB Life,
59, 332-345.
92.Sakono, M., and Zako, T. (2010) Amyloid
oligomers: formation and toxicity of Aβ oligomers: formation of
toxic Aβ oligomers, FEBS J., 277, 1348-1358.
93.Stroud, J. C., Liu, C., Teng, P. K., and
Eisenberg, D. (2012) Toxic fibrillar oligomers of amyloid-β have
cross-β structure, Proc. Natl. Acad. Sci. USA, 109,
7717-7722.
94.Friedrich, R. P., Tepper, K., Ronicke, R., Soom,
M., Westermann, M., Reymann, K., Kaether, C., and Fandrich, M. (2010)
Mechanism of amyloid plaque formation suggests an intracellular basis
of A pathogenicity, Proc. Natl. Acad. Sci. USA,
107, 1942-1947.
95.Gouras, G. K., Tampellini, D., Takahashi, R. H.,
and Capetillo-Zarate, E. (2010) Intraneuronal β-amyloid
accumulation and synapse pathology in Alzheimer’s disease,
Acta Neuropathol. (Berl.), 119, 523-541.
96.Canter, R. G., Penney, J., and Tsai, L.-H. (2016)
The road to restoring neural circuits for the treatment of
Alzheimer’s disease, Nature, 539, 187-196.
97.Mattson, M. P. (2004) Pathways towards and away
from Alzheimer’s disease, Nature, 430, 631-639.
98.Kayed, R., and Lasagna-Reeves, C. A. (2013)
Molecular mechanisms of amyloid oligomers toxicity, J.
Alzheimer’s Dis., 33, Suppl. 1, S67-78.
99.Fu, L., Sun, Y., Guo, Y., Chen, Y., Yu, B.,
Zhang, H., Wu, J., Yu, X., Kong, W., and Wu, H. (2017) Comparison of
neurotoxicity of different aggregated forms of Aβ40, Aβ42 and
Aβ43 in cell cultures, J. Pept. Sci. Off. Publ. Eur. Pept.
Soc., 23, 245-251.
100.Connelly, L., Jang, H., Arce, F. T., Capone,
R., Kotler, S. A., Ramachandran, S., Kagan, B. L., Nussinov, R., and
Lal, R. (2012) Atomic force microscopy and MD simulations reveal
pore-like structures of all-D-enantiomer of Alzheimer’s
β-amyloid peptide: relevance to the ion channel mechanism of AD
pathology, J. Phys. Chem. B, 116, 1728-1735.
101.Soscia, S. J., Kirby, J. E., Washicosky, K. J.,
Tucker, S. M., Ingelsson, M., Hyman, B., Burton, M. A., Goldstein, L.
E., Duong, S., Tanzi, R. E., and Moir, R. D. (2010) The
Alzheimer’s disease-associated amyloid beta-protein is an
antimicrobial peptide, PLoS One, 5, e9505.
102.Clare, D. K., Vasishtan, D., Stagg, S., Quispe,
J., Farr, G. W., Topf, M., Horwich, A. L., and Saibil, H. R. (2012)
ATP-triggered conformational changes delineate substrate-binding and
-folding mechanics of the GroEL chaperonin, Cell, 149,
113-123.
103.Koike-Takeshita, A., Arakawa, T., Taguchi, H.,
and Shimamura, T. (2014) Crystal structure of a symmetric
football-shaped GroEL:GroES2-ATP14 complex determined at 3.8 Å
reveals rearrangement between two GroEL rings, J. Mol. Biol.,
426, 3634-3641.
104.Horwich, A. L. (2011) Protein folding in the
cell: an inside story, Nat. Med., 17, 1211-1216.
105.Marchenkov, V. V., Sokolovskii, I. V., Kotova,
N. V., Galzitskaya, O. V., Bochkareva, E. S., Girshovich, A. S., and
Semisotnov, G. V. (2004) The interaction of the GroEL chaperone with
early kinetic intermediates of renaturing proteins inhibits the
formation of their native structure, Biofizika, 49,
987-994.
106.Yagi-Utsumi, M., Kunihara, T., Nakamura, T.,
Uekusa, Y., Makabe, K., Kuwajima, K., and Kato, K. (2013) NMR
characterization of the interaction of GroEL with amyloid β as a
model ligand, FEBS Lett., 587, 1605-1609.
107.Fukui, N., Araki, K., Hongo, K., Mizobata, T.,
and Kawata, Y. (2016) Modulating the effects of the bacterial
chaperonin GroEL on fibrillogenic polypeptides through modification of
domain hinge architecture, J. Biol. Chem., 291,
25217-25226.
108.Haslbeck, M., Peschek, J., Buchner, J., and
Weinkauf, S. (2016) Structure and function of α-crystallins:
traversing from in vitro to in vivo, Biochim. Biophys.
Acta, 1860, 149-166.
109.Shammas, S. L., Waudby, C. A., Wang, S., Buell,
A. K., Knowles, T. P. J., Ecroyd, H., Welland, M. E., Carver, J. A.,
Dobson, C. M., and Meehan, S. (2011) Binding of the molecular chaperone
αB-crystallin to Aβ amyloid fibrils inhibits fibril
elongation, Biophys. J., 101, 1681-1689.
110.Dehle, F. C., Ecroyd, H., Musgrave, I. F., and
Carver, J. A. (2010) αB-Crystallin inhibits the cell toxicity
associated with amyloid fibril formation by κ-casein and the
amyloid-β peptide, Cell Stress Chaperones, 15,
1013-1026.
111.Bonito-Oliva, A., Barbash, S., Sakmar, T. P.,
and Graham, W. V. (2017) Nucleobindin 1 binds to multiple types of
pre-fibrillar amyloid and inhibits fibrillization, Sci. Rep.,
7, 42880.
112.Soper-Hopper, M. T., Eschweiler, J. D., and
Ruotolo, B. T. (2017) Ion mobility-mass spectrometry reveals a
dipeptide that acts as a molecular chaperone for amyloid β, ACS
Chem. Biol., 12, 1113-1120.
113.Romeo, M., Stravalaci, M., Beeg, M., Rossi, A.,
Fiordaliso, F., Corbelli, A., Salmona, M., Gobbi, M., Cagnotto, A., and
Diomede, L. (2017) Humanin specifically interacts with amyloid-β
oligomers and counteracts their in vivo toxicity, J.
Alzheimer’s Dis., 57, 857-871.
114.Li, J., Zhu, M., Manning-Bog, A. B., Di Monte,
D. A., and Fink, A. L. (2004) Dopamine and L-DOPA disaggregate amyloid
fibrils: implications for Parkinson’s and Alzheimer’s
disease, FASEB J., 18, 962-964.
115.Eanes, E. D., and Glenner, G. G. (1968) X-ray
diffraction studies on amyloid filaments, J. Histochem.
Cytochem., 16, 673-677.
116.Petkova, A. T., Buntkowsky, G., Dyda, F.,
Leapman, R. D., Yau, W.-M., and Tycko, R. (2004) Solid state NMR
reveals a pH-dependent antiparallel beta-sheet registry in fibrils
formed by a beta-amyloid peptide, J. Mol. Biol., 335,
247-260.
117.Petkova, A. T., Ishii, Y., Balbach, J. J.,
Antzutkin, O. N., Leapman, R. D., Delaglio, F., and Tycko, R. (2002) A
structural model for Alzheimer’s beta-amyloid fibrils based on
experimental constraints from solid state NMR, Proc. Natl. Acad.
Sci. USA, 99, 16742-16747.
118.Paravastu, A. K., Leapman, R. D., Yau, W.-M.,
and Tycko, R. (2008) Molecular structural basis for polymorphism in
Alzheimer’s beta-amyloid fibrils, Proc. Natl. Acad. Sci.
USA, 105, 18349-18354.
119.Schmidt, M., Rohou, A., Lasker, K., Yadav, J.
K., Schiene-Fischer, C., Fandrich, M., and Grigorieff, N. (2015)
Peptide dimer structure in an Aβ(1-42) fibril visualized with
cryo-EM, Proc. Natl. Acad. Sci. USA, 112,
11858-11863.
120.Zhang, R., Hu, X., Khant, H., Ludtke, S. J.,
Chiu, W., Schmid, M. F., Frieden, C., and Lee, J.-M. (2009)
Interprotofilament interactions between Alzheimer’s Abeta1-42
peptides in amyloid fibrils revealed by cryo-EM, Proc. Natl. Acad.
Sci. USA, 106, 4653-4658.
121.Xiao, Y., Ma, B., McElheny, D., Parthasarathy,
S., Long, F., Hoshi, M., Nussinov, R., and Ishii, Y. (2015)
Aβ(1-42) fibril structure illuminates self-recognition and
replication of amyloid in Alzheimer’s disease, Nat. Struct.
Mol. Biol., 22, 499-505.
122.Colvin, M. T., Silvers, R., Ni, Q. Z., Can, T.
V., Sergeyev, I., Rosay, M., Donovan, K. J., Michael, B., Wall, J.,
Linse, S., and Griffin, R. G. (2016) Atomic resolution structure of
monomorphic Aβ42 amyloid fibrils, J. Am. Chem.
Soc., 138, 9663-9674.
123.Waugh, D. F. (1948) Regeneration of insulin
from insulin fibrils by the action of alkali, J. Am. Chem. Soc.,
70, 1850-1857.
124.Waugh, D. F. (1947) A comparison of the
regeneration products of fibrous insulin with native insulin, Fed.
Proc., 6, 223.
125.Elliott, A., Ambrose, E. J., and Robinson, C.
(1950) Chain configurations in natured and denatured insulin: evidence
from infrared spectra, Nature, 166, 194.
126.Koltun, W. L., Waugh, D. F., and Bear, R. S.
(1954) An X-ray diffraction investigation of selected types of insulin
fibrils, J. Am. Chem. Soc., 76, 413-417.
127.Reithel, F. J. (1963) The dissociation and
association of protein structures, Adv. Protein Chem.,
18, 123-226.
128.Beaven, G. H., Gratzer, W. B., and Davies, H.
G. (1969) Formation and structure of gels and fibrils from glucagon,
Eur. J. Biochem., 11, 37-42.
129.Burke, M. J., and Rougvie, M. A. (1972)
Cross-protein structures. I. Insulin fibrils, Biochemistry
(Moscow), 11, 2435-2439.
130.Geddes, A. J., Parker, K. D., Atkins, E. D.,
and Beighton, E. (1968) “Cross-beta” conformation in
proteins, J. Mol. Biol., 32, 343-358.
131.Lim, K. H., Dasari, A. K. R., Hung, I., Gan,
Z., Kelly, J. W., Wright, P. E., and Wemmer, D. E. (2016) Solid-state
NMR studies reveal native-like β-sheet structures in transthyretin
amyloid, Biochemistry (Moscow), 55, 5272-5278.
132.Economou, N. J., Giammona, M. J., Do, T. D.,
Zheng, X., Teplow, D. B., Buratto, S. K., and Bowers, M. T. (2016)
Amyloid β-protein assembly and Alzheimer’s disease:
dodecamers of Aβ42, but not of Aβ40, seed fibril formation,
J. Am. Chem. Soc., 138, 1772-1775.
133.Tran, J., Chang, D., Hsu, F., Wang, H., and
Guo, Z. (2017) Cross-seeding between Aβ40 and Aβ42 in
Alzheimer’s disease, FEBS Lett., 591, 177-185.
134.Pauwels, K., Williams, T. L., Morris, K. L.,
Jonckheere, W., Vandersteen, A., Kelly, G., Schymkowitz, J., Rousseau,
F., Pastore, A., Serpell, L. C., and Broersen, K. (2012) Structural
basis for increased toxicity of pathological Aβ42 : Aβ40
ratios in Alzheimer’s disease, J. Biol. Chem., 287,
5650-5660.
135.Cukalevski, R., Yang, X., Meisl, G., Weininger,
U., Bernfur, K., Frohm, B., Knowles, T. P. J., and Linse, S. (2015) The
Aβ40 and Aβ42 peptides self-assemble into separate
homomolecular fibrils in binary mixtures but cross-react during primary
nucleation, Chem. Sci., 6, 4215-4233.
136.Pauling, L., and Corey, R. B. (1953) Two
rippled-sheet configurations of polypeptide chains, and a note about
the pleated sheets, Proc. Natl. Acad. Sci. USA, 39,
253-256.
137.Glenner, G. G., Keiser, H. R., Bladen, H. A.,
Cuatrecasas, P., Eanes, E. D., Ram, J. S., Kanfer, J. N., and DeLellis,
R. A. (1968) Amyloid. VI. A comparison of two morphologic components of
human amyloid deposits, J. Histochem. Cytochem., 16,
633-644.
138.Inoue, S., Skinner, M., Leblond, C. P.,
Shirahama, T., and Cohen, A. S. (1986) Isolation of the amyloid P
component from the Engelbreth–Holm–Swarm (EHS) tumor of the
mouse, Biochem. Biophys. Res. Commun., 134, 995-999.
139.Glenner, G. G., Terry, W., Harada, M., Isersky,
C., and Page, D. (1971) Amyloid fibril proteins: proof of homology with
immunoglobulin light chains by sequence analyses, Science,
172, 1150-1151.
140.Dean, D. N., Das, P. K., Rana, P., Burg, F.,
Levites, Y., Morgan, S. E., Ghosh, P., and Rangachari, V. (2017)
Strain-specific fibril propagation by an Aβ dodecamer, Sci.
Rep., 7, 40787.
141.Makin, O. S., and Serpell, L. C. (2005) X-ray
diffraction studies of amyloid structure, Methods Mol. Biol.,
299, 67-80.
142.Kirschner, D. A., Abraham, C., and Selkoe, D.
J. (1986) X-ray diffraction from intraneuronal paired helical filaments
and extraneuronal amyloid fibers in Alzheimer’s disease indicates
cross-beta conformation, Proc. Natl. Acad. Sci. USA, 83,
503-507.
143.Selivanova, O. M., Grigorashvili, E. I.,
Suvorina, M. Y., Dzhus, U. F., Nikulin, A. D., Marchenkov, V. V.,
Surin, A. K., and Galzitskaya, O. V. (2016) X-ray diffraction and
electron microscopy data for amyloid formation of Αβ40 and
Αβ42, Data Brief, 8, 108-113.