REVIEW: Analysis of Insulin Analogs and the Strategy of Their Further Development
O. M. Selivanova1, S. Yu. Grishin1,2, A. V. Glyakina1,3, A. S. Sadgyan4, N. I. Ushakova4, and O. V. Galzitskaya1*
1Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: ogalzit@vega.protres.ru2Lomonosov Moscow State University, 119991 Moscow, Russia
3Institute of Mathematical Problems of Biology, Keldysh Institute of Applied Mathematics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia
4Joint-Stock Scientific Production Association Bioran, 119261 Moscow, Russia
* To whom correspondence should be addressed.
Received June 5, 2017; Revision received July 9, 2017
We analyzed the structural properties of the peptide hormone insulin and described the mechanism of its physiological action, as well as effects of insulin in type 1 and 2 diabetes. Recently published data on the development of novel insulin preparations based on combining molecular design and genetic engineering approaches are presented. New strategies for creation of long-acting insulin analogs, the mechanisms of functioning of these analogs and their structure are discussed. Side effects of insulin preparations are described, including amyloidogenesis and possible mitogenic effect. The pathways for development of novel insulin analogs are outlined with regard to the current requirements for therapeutic preparations due to the wider occurrence of diabetes of both types.
KEY WORDS: insulin analogs, diabetes, hyperglycemia, hypoglycemia, glycemic control, insulin fibrilsDOI: 10.1134/S0006297918140122
Abbreviations: αCT, C-terminal domain of insulin receptor α-subunit; BMI, body mass index; ER, endoplasmic reticulum; IR, insulin receptor; IGF-1R, type 1 insulin-like growth factor receptor; IRA, insulin receptor isoform A; IRB, insulin receptor isoform B; L1, leucine-rich repeat domain of the insulin receptor α-subunit; NPH, neutral protamine Hagedorn; PEG, polyethylene glycol; ThT, thioflavin T.
Insulin is a peptide hormone (51 a.a.) produced by the β-cells of
the pancreatic Langerhans islets. An insulin monomer consists of two
polypeptide chains: A (21 a.a.) and B (30 a.a.) connected via two
disulfide bridges formed by cysteine residues at positions A7–B7
and A20–B19. The third intrasubunit S–S bond is formed
between the residues A6 and A11. Although insulin was discovered at the
beginning of the XX century [1], the studies of its
functions have remained relevant up to present time.
Insulin regulates a number of metabolic processes, such as biosynthesis of proteins, fats, and nucleic acids, as well as cell growth and differentiation. However, its key function is regulation of glucose uptake by the cells. Insulin is produced in response to the rise of glucose blood concentration; it binds to the insulin receptor (IR) and activates glucose transporters, mainly Glut4, in fat tissues and cardiac and skeletal muscles. Mobilized transporters are recruited from the intracellular compartments to the plasma membrane, where they facilitate glucose transport into the cell [2]. If insulin production is disrupted, the blood concentration of glucose increases (chronic hyperglycemia), which is the basic diagnostic symptom of type 1 diabetes [3]. Type 2 diabetes develops when the IR signaling is disturbed, even if the hormone is produced in sufficient amounts; it is characterized by the decreased tissue sensitivity to insulin (insulin resistance) [4-6].
Diabetes is classified as a group of metabolic diseases characterized by hyperglycemia caused by defects in the insulin secretion or insulin action [7]. Both reduced cell sensitivity to insulin and inadequate insulin levels in the type 2 and 1 diabetes lead to development of such complications, as vascular diseases, in particular coronary heart disease, cerebrovascular disorders, retinopathy, nephropathy, and neuropathy [8]. Impairments in insulin secretion and functioning can be found in the same patient, which makes revealing the major cause of hyperglycemia rather problematic. The immune response of an organism to its own β-cells in the pancreas can be diagnosed from the presence of autoantibodies against insulin, Langerhans islet cells, tyrosine phosphatases IA2 and IA2β, and glutamate decarboxylase GAD 65. An increased risk of type 1 diabetes development can be revealed by molecular genetic analysis of the HLA–DQB1 gene alleles [9, 10]. As a rule, insulin secretion at the last stage of diabetes is insignificant or lacking at all, which is indicated by low blood plasma levels of the C-peptide. Patients with type 1 diabetes become dependent on insulin administration.
Only 5-10% diabetes patients have type 1 diabetes, while most patients have type 2 diabetes [7]. Although no autoimmune destruction of cells takes place in type 2 diabetes, this type of diabetes can be caused by various other factors. The precise mechanisms of type 2 diabetes are extensively studied [11-15]. Most cases of insulin usage are associated with type 2 diabetes, because it has a higher occurrence.
The efficiency of insulin in diabetes treatment that is related to the ability of this hormone to decrease the blood glucose levels has been demonstrated during decades of insulin use in medical practice. However, at present, novel approaches to the diabetes therapy remain a topical problem, because the number of disease cases continues to rise. Moreover, individual features and preferences of patients should be taken into account during the therapy [16].
The limitations in the use of insulin are related to the necessity of administration of exact doses several times a day in order to maintain physiological levels of glucose in the blood. Besides, the hormone has a narrow therapeutic window associated with the risk of hyperglycemia; it can also cause weight gain that might be dangerous in patients with a high body mass index (BMI) [17, 18].
The above problems that are associated with the properties of native human insulin have promoted the development of its analogs (Fig. 1).
Fig. 1. Structure of insulin and its analogs (modified from [19]).
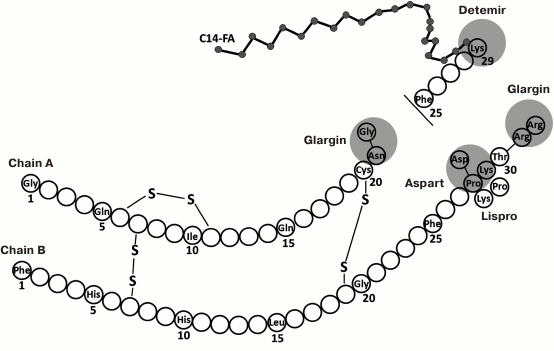
Insulin analogs are synthetically modified molecules that, due to faster or more prolonged action, allow better metabolic control of the blood glucose levels in diabetes [20]. By the present time, different preparations for diabetes therapy have been created that include insulin analogs and mixtures.
Herein, we review existing insulin analogs. There are two main strategies for their development: creation of bolus (short-acting) and basal (long-acting) insulin preparations. These types of insulin analogs can be combined to normalize glucose blood levels and to provide the delivery of the preparations in a form convenient for patients.
The use of insulin as a drug has started almost immediately after its discovery [1]. Since then, numerous biochemical and biomedical studies have been conducted in order to further improve insulin preparations.
BIOSYNTHESIS AND SECRETION OF INSULIN
Insulin biosynthesis starts with the translation of mRNA for its precursor, preproinsulin. The precursor (110 a.a.) is encoded by the INS gene, a single copy of which is located in the short shoulder of chromosome 11 of the human genome [21]. Preproinsulin is synthesized only on polyribosomes associated with the endoplasmic reticulum (ER). The signal peptide (24 a.a.) of preproinsulin is cleaved off by the signal peptidase as the polypeptide is translocated into the ER lumen [22] forming proinsulin. In the ER, proinsulin folds into correct conformation with the formation of three disulfide bonds (B7–A7, B19–A20, A6–A11). Conversion of proinsulin into monomer insulin and C-peptide happens after proinsulin transport from the ER to the Golgi apparatus, where proinsulin is cleaved by peptidases in secretory vesicles, during which the C-peptide (of 31 a.a.) located between fragments B and A is excised from the molecule. Insulin is then stored as a hexamer coordinated by two Zn2+ ions in β-cells of the Langerhans islets in the pancreas [23, 24].
An increase in the glucose blood level acts as a signal for insulin secretion. As a rule, the process starts when the insulin-independent carrier protein Glut2 binds glucose molecules and transports them into β-cells of the Langerhans islets. In β-cells, glucose is phosphorylated by the enzyme glucokinase and eventually converted to pyruvate via glycolysis. Pyruvate is oxidized in the tricarboxylic acid cycle with the formation of ATP. An increase in the ATP/ADP ratio leads to the closing of ATP-dependent potassium channels and depolarization of the plasma membrane. As a result, voltage-dependent potassium channels open and potassium enters the cells. High intracellular potassium concentrations stimulate phospholipase C that cleaves phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-trisphosphate and diacylglycerol. Inositol 1,4,5-trisphosphate is a ligand for the ER receptor proteins responsible for the release of intracellular calcium that results in an increased cytoplasmic Ca2+ concentration. Finally, calcium ions stimulate insulin release from the secretory granules. Besides glucose, other molecules that mediate insulin secretion have been found: nicotinamide adenine dinucleotide phosphate (NADP), glutamate and malonyl-CoA [25, 26], glycerol-3-phosphate [27], and fatty acids [28, 29]. Insulin is released from the cells through exocytosis: a mature secretory granule fuses with the plasma membrane and releases its content to the extracellular space.
STRUCTURAL PROPERTIES AND SURFACE CONTACTS IN THE INSULIN
MOLECULE. BINDING TO THE RECEPTOR
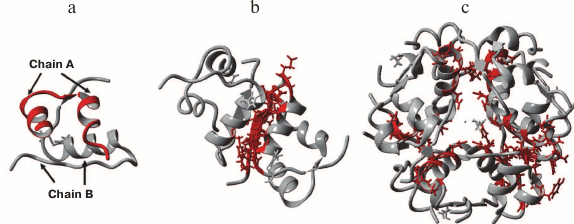
Immediately after insulin hexamers are secreted from the β-cells and diffuse in the blood, a combination of electrostatic repulsion and insulin concentration gradient promotes dissociation of the hexamers into dimers and monomers (only monomers exhibit biological activity). Therefore, hexamers are the storage form of insulin, while monomers are the biologically active form of this hormone (Fig. 2).
Fig. 2. X-ray structure of human recombinant insulin at 0.92 Å resolution: a) monomer; b) dimer; c) hexamer coordinated by two Zn2+ ions (PDB 5E7W, visualization in YASARA).
Insulin monomer consists of chains A and B connected by disulfide bonds. The secondary structure of chain A contains two antiparallel α-helices formed between residues A2-A8 and A13-A19 connected by the fragment A9-A12. The secondary structure of chain B contains β-strands and an α-helix. Amino acid residues B1-B5 form an elongated region; the central α-helix is preceded by a sharp 1→4 turn due to the presence of glycine residue B8. This explains the tight packaging of the α-helix by the 310 type via hydrogen bonds formed by B7, B8, and B9 [30]. Starting from Ser at position B9, hydrogen bonds promote formation of the helical structure of the 1→5 type, and the α-helix is maintained through B19. Then, the 1→4 β-turn is formed by the hydrogen bonds between B19 carbonyl oxygen and B22 amide hydrogen and between B20 carbonyl oxygen and B23 amide hydrogen. Residues B23-B30 form the β-strand. PheB24 and TyrB26 interact with LeuB11 and LeuB15 of the chain B α-helix. Cysteine residues at each end of the chain B helical segment stabilize the native structure of insulin by forming disulfide bonds with chain A (A7–B7 and A20–B19).
Chain B can have two different conformations in a crystal [30]. In the T conformation, the central α-helix runs from B9 through B19, while the N-terminus (B1-B8) is unfolded. In the R conformation, the α-helix is continuous (B1-B19). The transition from the T conformation to the R conformation was studied in a solution of insulin hexamers; the changes in the content of α-helices were registered using circular dichroism [31] and 2D-NMR [32, 33] methods. It was shown that the T6→R6 transition occurs in the presence of phenols and some other cyclic alcohols [34] including cyclohexanol [35]. Addition of phenol ligands induces a conformational transition of the N-terminus in chain B not only in native insulin, but also in its analog insulin lispro (LysB28, ProB29) [36]. The monomeric forms of insulin acquire the R-like conformation in a solution. The secondary structure of both A and B chains determines the affinity of insulin to its receptor, so it was suggested that the T→R transition is important for the binding and activation of IRs.
IR is a homodimer, each subunit of which consists of two (α and β) polypeptide chains. The extracellular α-subunit binds insulin, while the transmembrane β-subunit contains a tyrosine kinase intracellular domain [37]. Insulin monomer binds to the site formed by two α-subunits. This site includes the L1 (leucine-rich repeat domain) of one α-subunit and the carboxyterminal (αCT) domain of the other subunit. The binding of insulin leads to the repositioning of the helix in the αCT domain on the L1 surface in such a way that it interacts, on the one hand, with insulin and, and on the other hand, with L1. The C-terminus of chain B shifts relatively to the other parts of the molecule after binding with αCT [38, 39]. PheB24 plays the role of an anchor in the nonpolar pocket formed by the L1 surface, αCT, and the central α-helix of chain B [40]. It is believed that only three regions close to the insulin molecule surface are responsible for the interaction with the IR because the residues in all the three regions are evolutionary conserved: the N-terminus of chain A (GlyA1-IleA2-ValA3-GluA4), the C-terminus of chain A (TyrA19-CysA20-AsnA21), and the C-terminus of chain B (GlyB23-PheB24-PheB25-TyrB26) [41]. Peptides with mutations in these regions have lower affinity (1-5% of the normal) to the IR. Patients with Leu substitution for Phe in position B25 (Chicago insulin), Ser substitution for Phe in position B24 (Los Angeles insulin), and Leu substitution for Val in position A3 (Wakayama insulin) suffer from type 2 diabetes [42-46].
Insulin binding to the receptor was reduced by 30%, when the N-terminal nitrogen of chain A was acetylated, which shows the importance of free positively charged amine group in A1 [33]. Removal of Gly in A1 decreased insulin affinity to the receptor by 15%, demonstrating that the salt bridge formed by GlyA1 and the C-terminus of chain B is essential for the correct positioning of the peptide [47]. It should be noted that the overall conformation might be more important than the presence of certain amino acid residues. For example, the replacement of GlyA1 with L-amino acids (Ala, Val, Leu, Pro, Trp, Lys, or Glu) reduces insulin binding to the receptor by 2-20%, while replacement of the same GlyA1 with D-amino acids (D-Phe, D-Leu, D-Trp, D-Ala, D-Lys, or D-Glu) does not affect the biological activity of insulin analogs [48-51]. Mutations impairing the folding of the chain A α-helix (as determined by a decreased molar ellipticity of circular dichroism) also result in reduced biological activity [52].
The structure of chain B is especially well studied. It was demonstrated that the first four residues of chain B do not affect the activity of insulin binding to the receptor, while the removal of HisB5 leads to its considerable reduction [52, 53]. LeuB6 is of critical importance because deletion of this amino acid residue results in 99% loss of affinity to the IR [54]. Substitution of His with Asp in B10 in a synthetic insulin analog causes a 500% increase in its affinity to the IR [55]. However, because HisB10 is essential for insulin biosynthesis, its substitution with AspB10 prevents insulin formation from proinsulin, which leads to an elevated concentration of the latter [56, 57]. The C-terminal fragment of chain B contains evolutionary conserved residues involved in the receptor binding (GlyB23, PheB24, PheB25, and TyrB26). In particular, PheB24 forms hydrogen bonds critical for dimerization. The conformation of PheB25 is different in the two molecules forming the dimer, i.e., this residue is important for the generation of insulin native structure [47]. Substitution of PheB24 with methionine or cyclohexylalanine decreases the time of disassembling of insulin analog hexamers [40].
According to the crystallography data, the C-terminus of chain B is essential for the dimer formation. The amino acids at positions B8, B9, B12, B13, B16, B23-B28 form the contacts between the insulin molecules. Essential amino acids, whose substitution results in a significant decrease in the capacity of insulin to form dimers, were identified by mutagenesis [58]. Sequential truncation of the insulin molecule from the C-terminus of chain B showed that ProB28 is critical for the insulin self-association in a solution. Removal of ProB28, its substitution, or, especially, inversion of amino acids in positions B28 and B29 (Lys-Pro) almost completely eliminate the capacity of insulin for the dimer formation, but does not affect its ability to form hexamers [59]. It should be noted that in all commercial pharmaceutical preparations, insulin is in hexamer form [60].
INSULIN ANALOGS
Elucidation of the insulin structure and identification of most important regions responsible for the formation of dimers and hexamers and binding to the IR significantly promoted the development of insulin analogs. Because insulin is active in its monomeric form, creation of insulin analogs with a modified structure should lead to prolongation of their action and a change in the onset of insulin effects in the body.
Basal and bolus analogs of insulin. The aim of insulin replacement therapy is the maximal imitation of physiologic insulin secretion, when small amounts of insulin are discharged continuously (the so-called basal or background secretion). Rapid secretion of insulin by β-cells in response to food intake is called bolus secretion [61]. Insulin therapy is recognized as an efficient way to prevent macro- and microvascular complications in patients suffering from type 1 diabetes. To minimize degenerative changes in organs, patients with diabetes should maintain the glucose blood levels that would correspond to normal insulin secretion [62]. At the same time, regular administration of insulin causes a number of adverse side effects, such as an increased risk of pronounced hypoglycemia, weight gain, and changes in the action profile of insulin [16, 63]. When insulin is injected intravenously, its hexamers almost immediately dissociate into monomers; the monomers interact with IRs in the target tissues and decrease the glucose levels almost instantaneously. When injected subcutaneously, insulin hexamers should dissociate into monomers at the site of injection prior to getting into the bloodstream. This delay in the onset of insulin action depends on the injection site and causes variability in the insulin action profile and discrepancy between the time of insulin administration and food intake. Patients are required to take an insulin dose 15-30 min before the meals, which is problematic in everyday life [64]. To improve the control of glucose blood levels, insulin preparations with different time courses of action have been developed (Table 1).
Table 1. Characteristics of insulin
preparations [65-68]
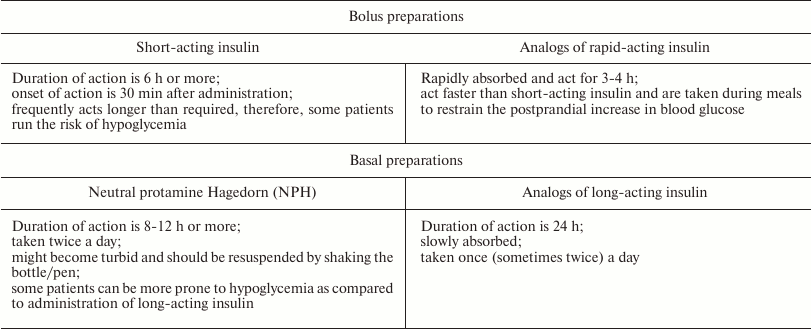
Weakening of contacts between the monomers in the insulin dimer allowed to obtain short/rapid-acting analogs (bolus insulins), such as insulin lispro (Humalog®), insulin aspart (NovoRapid®), and insulin glulisine (Apidra®). The onset of action of these analogs is within 5-15 min after subcutaneous administration; the peak of action is within 1-2 h; and the duration of action does not exceed 4 h. Therefore, these preparations are designed for effective control of hyperglycemia immediately after meals.
The slowing down the dissociation of insulin hexamers into monomers led to the development of long/ultralong-acting preparations, e.g., insulin glargin (Lantus®) and insulin detemir (Levemir®). The unique property of these preparations is the absence of peak increase in the insulin blood level and the ability to maintain stable insulin concentration for 24 h. In other words, these insulin preparations almost completely supplement the basal secretion of insulin typical of healthy subjects. The low absorption rate of long-acting insulin analogs allows administering them once a day. The onset of their action is on average 1 h after subcutaneous injection, and the duration of action may reach 24 h (Fig. 3).
Fig. 3. Pharmacokinetic profiles of insulin analogs (modified from [19]).
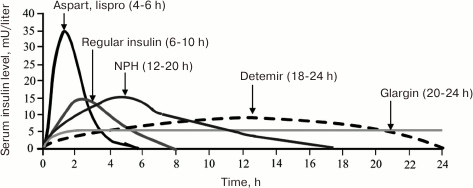
Therefore, combined administration of ultrashort- and long-acting insulin analogs might completely imitate the two-phase physiological secretion of insulin in healthy organisms. The use of these preparations is associated with a lower risk of hypoglycemia, which is the most hazardous complication of insulin therapy.
Humulin® was the first insulin preparation produced with the use of recombinant DNA technology [69]. Different insulins have been designed since then: neutral protamine Hagedorn (NPH), rapid-acting analogs, basal analogs, and premixed insulin analogs from such companies as Eli Lilly (USA), Novo Nordisk (Denmark), and Sanofi (France) [70].
Production and application of insulin analogs. The first medical insulin preparations had been produced from animal pancreatic extracts. However, the amino acid sequences of bovine and porcine insulins differ from that of human insulin (in the porcine insulin in the B chain, instead of the threonine in the B30 position, alanine is found; in the bovine insulin in addition to this substitution, in the A chain, instead of the threonine in the A8 position, alanine is found, and in the A10 position, instead of isoleucine, valine is found), therefore animal insulin preparations caused adverse side effects in humans (e.g., allergy). Besides, due to the limited amount of source materials, animal insulins could not satisfy the requirements for the insulin preparations.
The invention of recombinant DNA technologies allowed for the first time the large-scale production of recombinant insulin identical to the human insulin as well as production of its analogs with designed properties and predetermined duration of action.
Production of insulins with different duration of action is of great importance, because combination of such insulins in one preparation makes it possible for diabetes patients to administer the drug directly before the meal and to conveniently decrease the number of injections.
In the developed countries and Europe, almost all diabetes patients in need of insulin therapy are treated with genetic engineered insulin analogs. In Russia, 100% children with diabetes and requiring insulin therapy receive genetically engineered analogs of human insulin as the most efficient and safe preparations.
The most important feature of the Russian insulin market is that for more than twenty years it has been controlled by foreign manufacturers with three pharmaceutical companies being the leaders in this field: Novo Nordisk, Eli Lilly, and Sanofi [71] (Table 2).
Table 2. Percentage of producers of insulin
and its analogs sold in the Russian Federation in 2015-2016 according
to the Klifar-Goszakupki Data Analysis System

Up to the present, the criterion of the Russian Ministry of Industrial Trade for a local preparation has been the recognition of the packing stage, which is sufficient for a foreign preparation to get a license for its distribution in Russia. The most popular imported insulin preparations in Russia are Actrapid HM and Protaphane HM (Novo Nordisk) and Humulin regular and Humulin NPH (Eli Lilly). These preparations are analogs of Insuran P and Insuran NPH (Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences), respectively.
In 2014, the patent terms for Levemir® and NovoLog® (active components are detemir and aspart, respectively) from Novo Nordisk and for Humalog® (insulin lispro) from Eli Lilly expired, which allowed other companies to start manufacturing analogous insulin products, the so-called biosimilars (Table 3).
Table 3. Main current insulin biosimilars
[16, 72]
The characteristics of biosimilars are analogous to those of the original preparations. To get a permission from the regulating authorities to produce biosimilars, the manufacturing company should prove that the biosimilar is safe, efficient, and similar to the original drug and provide identification of the patent status of the original biopharmaceutical preparation. A biosimilar may not be identical to the original preparation, but should exhibit similar therapeutic action. The terms of patent defense and exclusive access to the market should be terminated in the area of patent action.
Biosimilars include commercially available analogs of glargine, such as Basalog® (Biocon Ltd., Bangalore, India) and Basalin® (Gan & Lee, Beijing, China), and premixed insulin compositions Gensulin® (Bioton S.A., Warsaw, Poland), Insugen® (Biocon Ltd, Bangalore, India), Wosulin® (Wockhardt Ltd, Mumbai, India), and Biosulin® (MJ Biopharm Pvt Ltd, Mumbai, India) [72].
Since 2000, the number of genetically engineered preparations of human insulin that have been developed and registered in the Russian Federation has grown essentially. The procedure for the confirmation of safety and efficiency of genetically engineered human insulin preparations registered in the Russian Federation does not require the proof that these preparations have characteristics analogous to the original ones. So strictly speaking, these preparations are not bioanalogs. At the same time, they cannot be recognized as original medical preparations because their preregistration studies have been significantly reduced.
To confirm that novel preparations of genetically engineered human insulin and its analogs represent bioanalogs of the original preparations, it is necessary to follow earlier developed recommendations [73-75], as well as use the new ones.
In addition, the emerging willingness to comply with the European standards on the confirmation of the bioanalogous nature of medical preparations will allow the development of scientific and methodical procedures, as well as the program for preclinical and clinical studies of genetically engineered bioanalogs of human insulin and its analogs, and will lead to new approaches to the evaluation of the results of these studies in order to estimate the benefits vs. risks of the developed drugs.
Strategies and results of insulin analog design. From the theoretical point of view, the differences in the action profiles of insulin analogs make it possible for the patients with type 1 diabetes to dynamically control glucose blood levels in order to avoid hyper- and hypoglycemia. However, such control requires imitation of the activity of healthy pancreas, i.e., maintenance of basal insulin levels with peak increases in the hormone concentration during meals [76, 77].
The first designed insulin analog was rapid-acting insulin lispro that was released on the market in 1996. As described above, the action of insulin lispro is based on the weakening of self-association of insulin molecules by inversion of amino acid residues ProB28 and LysB29 [78]. The Pro–Lys inversion changes the local conformation of the chain B C-terminus by eliminating two critical hydrophobic interactions, which results in the weakening of the two terminal hydrogen bonds in the β-sheet and insulin dimer destabilization [79]. Phenol ligands induce conformational changes by binding to specific sites on the insulin hexamer and promoting transition of eight N-terminal residues in chain B from the elongated conformation to the α-helix [80]. Another classical short- and ultrashort insulin analogs are aspart and glulisine, which dissociate into dimers and monomers faster than regular human insulin. Therefore, they are more rapidly absorbed, but have a shorter duration of action [81-83].
During the first hour after administration, the pharmacokinetic profile of the lispro plasma concentration is two times higher than that of human insulin. Insulin lispro also reaches its maximal concentration two times faster than regular insulin. Three to four hours after injection, the concentration of rapid-acting insulins drops to 20% of the maximum, while regular insulin is still absorbed at the injection site [84]. Insulin lispro more efficiently lowers the glucose level 0-4 h after the meal, which makes it possible to administer it within 15 min after beginning of the meal. Unlike regular human insulin, the time for the onset of the insulin lispro effect depends less on the site of injection. However, it is recommended to inject rapid-acting insulin analogs into the abdominal wall, because absorption in the deltoid and quadriceps muscles proceeds slower than in the abdominal cavity and results in prolongation of the action of both regular insulin and insulin lispro, as evidenced by pharmacokinetic and pharmacodynamic data [66].
The other essential component of the glucose level control in insulin-dependent diabetes is the use of long-acting insulin analogs to substitute the basal insulin in order to reduce the risk of nocturnal hypoglycemia that could be caused by administration of human insulin preparations [85, 86]. Neutral protamine Hagedorn (NPH), as well as insulins lente and ultra-lente [67, 87], were the first preparations used to maintain the basal hormone level. NPH is obtained by adding protamine and Zn2+ to insulin. This procedure results in the formation of heterogeneous suspensions, which slightly increases the duration of the insulin action. However, the reproducibility of the action profiles of such insulin preparations remains low, which in part might be related to the necessity to resuspend insulin in a vial before administration. The differences in the duration of action of these insulin preparations can be explained by the stochastic release of hexamers from the aggregates [88].
The strategy based on diminishing the solubility of injected preparation at the injection site has proven to be more effective for designing basal insulin preparations. For example, modifications in insulin glargin led to the shift in the molecule isoelectric point in order to significantly reduce its solubility at physiological pH, which makes injected insulin glargin much less soluble [68, 89, 90]. The increase in the isoelectric point was achieved by introducing two Arg residues at positions B31 and B32; additionally, Gly was introduced at A21 to provide the chemical stability of the resulting preparation. It was found that both human insulin and its analogs were equally efficient in the glucose level control, but the analogs counteracted the effects of hypoglycemia better, especially at night. However, insulin glargin was found to cause weight gain in diabetes patients [91]. The strategy used for developing glargin has been rejected by now because of the inflammatory response occurring at the site of injection [16].
An alternative approach for developing long-acting insulin analogs is acetylation of LysB29 with a fatty acid residue, which leads to the reversible hormone binding to the blood serum albumin and prolongation of its action. This strategy was used to develop insulins detemir [92] and degludec [93, 94]. Insulin degludec (Tresiba, Novo Nordisk) is human insulin in which LysB29 is modified with a palmitic acid residue and ThrB30 is deleted (de-B30). The medical preparation of insulin degludec contains phenol and zinc; in the presence of these components, it exists in a stable dihexameric form, where the fatty acids are positioned between the hexamers and interact with PheB1 residues in each chain [95]. The duration of action of insulin degludec is determined mainly by its oligomeric structure, whereas the binding to the serum albumin provides for the amortization effect and decreases variability of the action profile due to more uniform uptake into the bloodstream [93]. The clinical assessment of insulin degludec demonstrated that decreased levels of glucose are maintained for up to 42 h with simultaneous reduction of the episodes of nocturnal hypoglycemia, as compared to the use of insulin glargin [96].
Another trend in the development of basal insulins is creation of hepatoselective analogs with a higher potential for attenuation of glucose levels and lower risk of hypoglycemia and body weight growth than those of regular insulin [97]. For example, insulin PEGLispro (Eli Lilly) was obtained by attaching polyethylene glycol (PEG) to the insulin molecule [98]. This modification increases the hydrodynamic radius of the molecule, which, on one hand, allows insulin PEGLispro monomers to penetrate via liver capillaries but, on the other hand, prevents penetration via capillaries of other tissues, such as fat tissue or muscles. According to pharmacokinetic studies, the half-time of insulin PEGLispro elimination was prolonged to 2-3 days. No difference was observed in the frequency of hypoglycemia in patients taking insulins PEGLispro and glargine, although the frequency of nocturnal hypoglycemia in the first group was lower [99]. Regrettably, these studies were terminated because clinical trials showed that insulin PEGLispro impairs liver functions; in particular, higher levels of enzymes alanine aminotransferase and aspartate aminotransferase were observed [100].
An important direction in the development of insulin preparations is design of analogs specific to particular isoforms of the IR, since it was suggested that some insulin analogs can promote activation of the IGF-1 receptor, which might result in the tumor growth stimulation [101, 102]. Insulin binds with a high affinity (Kd ~ 5·10–11 M) to the IR and with lower (but still noticeable) affinity (Kd ~ 10–8 M) – to IGF-1R (type 1 insulin-like growth factor receptor) [33]. The functions and tissue expression of the IR isoforms IRA and IRB differ. The physiological metabolic effect of insulin is mediated by IRB, while excessive hormone biding to IRA and IGF-1R might cause pronounced mitogenic effect. In vitro models demonstrated that short-acting insulin analogs bind to IRA, IRB, and IGF-1R, similarly to insulin. On the contrary, long-acting analogs, such as insulins glargin and detemir, preferably bind to IGF-1R and to a lesser extent – to IR. Therefore, insulin analogs predominantly activate the ERK kinase signaling pathway, while regular insulin activates AKT kinase [20].
The receptor-binding activity of the hormone can be enhanced by the following modifications: removal of residues B27-B30 at the C-end of chain B together with amidation of the C-terminal residue in position B26 and inclusion of N-methylated amino acid or D-enantiomer. However, methylation of TyrB26 or its substitution with N-methylated alanine decreases the receptor-binding activity in the same way as bulk amino acids do it in truncated insulin analogs N-MePheB26 and N-MeTyrB26 [103].
The novel strategies for the design of basal insulin preparations include primarily three directions: (i) insulin binding to glucose-responsive polymers for controlled release of the hormone [62], (ii) rearrangement of insulin hexamers [104], and (iii) pH-dependent binding of zinc ions by created His-X3-His sites (“histidine staples”) on the hexamer surface [105, 106].
Hence, insulin analogs can be differentiated by the profile of their action in the following way:
1) short- and ultrashort acting analogs that imitate the postprandial insulin secretion;
2) long-acting (peakless) analogs designed to imitate a stable (basal) profile of insulin secretion by healthy pancreas.
At present, the basal-bolus therapy is used because it is more flexible than other regimes [107-110]. As to the intermediate-acting insulins, the widely used long-acting insulin preparations have made them irrelevant.
FIBRILLATION OF INSULIN PREPARATIONS
Insulin possesses a high propensity for aggregation and formation of fibrils (fibrillation). This disposition for polymerization, as well as the possibility to obtain reproducible kinetic data, explains the attention that insulin has received as a model object for studying fibril formation. When searching for different insulin monomeric forms, researchers have also investigated insulin capacity for aggregation and fibrils formation.
Fibrillation is undesirable in insulin therapy. Regular non-mutant insulin used for injections is prone to fibrillation under the action of various external factors. Insulin preparations often spontaneously polymerize during storage and transportation. There is also a problem of insulin-derived amyloidosis upon frequent insulin injections.
Aggregation of insulin into insoluble fibrils may cause numerous complications, such as an attenuated therapeutic effect of insulin, clogging of insulin delivery systems, or activation of immune response. Fibrillation might affect the glucose level control; besides, it raises the cost of treatment and decreases its efficiency [111].
Identification of regions in the insulin molecule, which are responsible for the aggregation and stability, might promote the development of novel insulin analogs for better regulation of glucose blood levels and reduce the propensity of insulin for fibril formation by decreasing the probability of its self-association [112]. The early stages of fibrillation are of particular interest [113].
It is believed that the key role in the growth of insulin fibrils belongs to the conformational shift of the chain B C-terminus that exposes a domain containing amino acid residues A2, A3, B11, and B15. In its turn, this protruding domain interacts with another hydrophobic domain containing residues A13, B6, B14, B17, and B18 [114]. Substitution of ThrA8 with His slows down insulin fibrillation [115].
Disulfide bridges also play an important role in insulin fibrillation. A disruption of the A7–B7 disulfide bond induces unfolding of the insulin molecule and promotes fibrillation more than disruption of the intrachain A6–A11 disulfide bond [116]. Apparently, this is related to a low propensity for fibrillation of single-chain insulin analogs [117].
Although the kinetics of fibril formation in native insulin has been actively studied [118-120], the data on fibrillation of insulin analogs are scarce. As has been mentioned, at present there are insulin analogs with different time of action, such as rapid-acting insulins (lispro, aspart, etc.) and long-acting insulins (glargin, degludec, etc.). It has been shown that in most insulin analogs, formation of fibrils occurs slower than in regular insulin [115].
Fibrillation of rapid-acting insulin analogs has been studied in detail. Insulins lispro, aspart, and glulisine have a longer lag-period and a lower fibrillation rate, as compared to regular insulin. However, the data on the differences between fibrillation rates of these analogs from different studies are contradictory [121, 122]. As a rule, the most attention in the design of rapid-acting insulins is focused on amino acid residues involved in the dimer formation, but not in hexamerization. Amino acid residues responsible for interaction with the IR are also taken into account (Table 4).
Table 4. Amino acid residues involved in
dimer and hexamer formation and binding to IR [58,
104, 114, 123]

For our studies, we used recombinant insulin and insulin lispro (both preparations were kindly provided by the Bioran Scientific & Production Corporation, Russian Federation). As mentioned above, insulin lispro is a rapid-acting insulin analog and differs from the regular human insulin by the inversion of chain B amino acid residues at positions 28 and 29 (inversion of ProB28(Lys) and LysB29(Pro)). These residues are involved in dimerization of insulin molecules and at the same time do not interfere with the formation of insulin hexamers.
The key research methods in studying fibrillary structures are fluorescence spectroscopy (binding of thioflavin T (ThT)), electron microscopy (fibril formation), and X-ray analysis (the presence of specific reflexions).
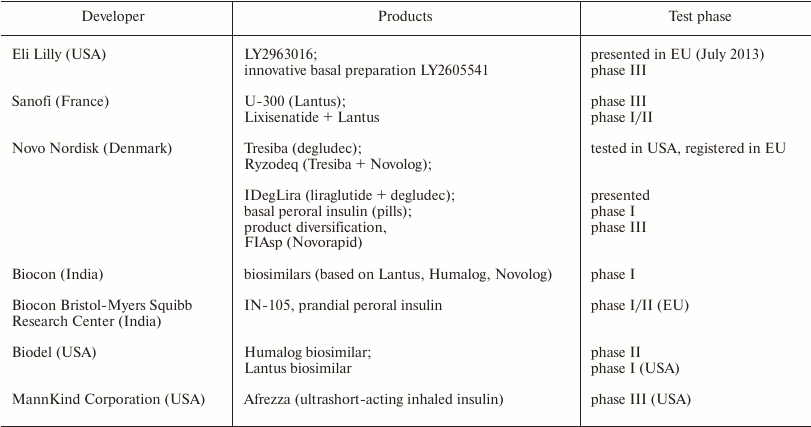
We have found that the fibrillation rates of human recombinant insulin and insulin lispro are different [124]. As shown by fluorescence spectroscopy, the lag period for the recombinant insulin is about 8-10 h (2 mg/ml; 20% acetic acid, pH 2.0, 140 mM NaCl; 37°C). Active polymerization of insulin fibrils occurs after 10-12 h of incubation under the same conditions; formation of large clusters of fibrils happens after 24 h of incubation. The lag period for insulin lispro is 5 h longer than for the recombinant insulin (under the same conditions). However, according to the electron microscopy data, the morphology of fibrils in recombinant insulin and insulin lispro is identical. In both cases, mature fibrils are 10 to 12 µm long, and the diameter of the thinnest single fibrils is about 6-7 nm (Figs. 4 and 5).
Fig. 4. Electron microscopy of recombinant zinc-free human insulin in 20% acetic acid (pH 2.0), 140 mM NaCl after 24 h incubation at 37°C (2 mg/ml) (modified and adapted from [123]). a) Observation field; b) fragments of fibrils with the smallest diameter; c and d) single fibrils under highest magnification: c) packing of ring oligomers in a ring-to-ring manner, top view (two images); d) side view.
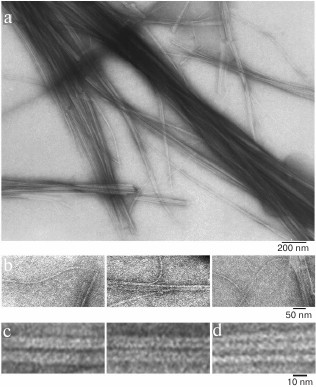
Fig. 5. Electron microscopy images of recombinant insulin lispro in 20% acetic acid (pH 2.0), 140 mM NaCl after 24 h incubation at 37°C (2 mg/ml) (modified and adapted from [123]). a) Observation field; b) fragments of fibrils with the smallest diameter; c and d) single fibrils under highest magnification: c) packing of ring oligomers in a ring-to-ring manner, top view (two images); d) side view.
An increase in the incubation time resulted in a considerable polymorphism of mature fibrils that interact laterally with each other with the formation of ribbons of different widths, twisted ribbons, and bundles of different diameter. When the incubation time was prolonged even more, large clusters of insulin fibrils were formed [125, 126].
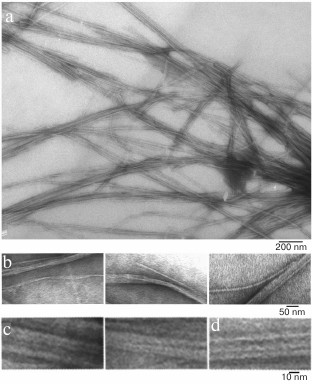
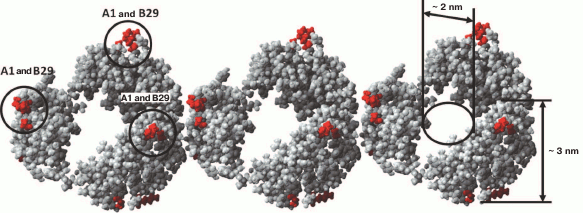
According to a simplified scheme, fibrillation proceeds as follows: destabilized monomers → oligomers → mature fibrils → fibril aggregates of different morphology. As demonstrated by electron microscopy, oligomer particles are formed at the early stages of fibrillation. Formation of fibrils and their lengthening triggers gradual disappearance of oligomer particles. This transition from oligomers to fibrils is the most puzzling stage of fibrillation. When observed under high magnification (Figs. 4c and 5c), mature single fibrils appear to be formed by ring-like oligomers with a diameter coinciding with the diameter of oligomers at the initial stage of fibrillation. When such a fibril is placed with its ring oligomers on a support (formvar film), it is possible to estimate the size of a ring oligomer. For both insulin preparations, the outer diameter of ring-like oligomers (fibril diameter) is about 6-7 nm, and the inner diameter is about 2 nm. The ring-like oligomers in a fibril are either positioned in a ring-to-ring fashion by touching their lateral sides or slightly overlap each other like dominoes. If the ring-like oligomers in a fibril are adsorbed on the Formvar film by their lateral sides, it is possible to estimate the height of a ring-like oligomer, which is approximately 3-4 nm. It should be noted that in ribbons and bundles, single fibrils are associated laterally, so that butt sides of ring-like oligomers interact with each other. Therefore, we can only see the side surfaces of oligomers, their height being ~3 nm. This height corresponds to the diameter of single fibrils in ribbons and bundles [126]. The X-ray images of fibrillar insulin show the existence of two reflexions characteristic of cross-β structure: meridional (4.8 Å) and equatorial (10.7 Å). The equatorial reflexion is very diffuse; in addition, there is also a 30 Å reflexion that has been ascribed to the distance between laterally positioned fibrils. These data correlate well with the earlier obtained results [127, 128]. The cross-β structure implies that a fibril consists of β-sheets positioned at a distance of 9-11 Å from each other and parallel to the fibril axis, and the β-sheet itself is formed by β-strands located at a distance of 4.6-4.8 Å and positioned perpendicularly to the fibril axis [129]. Consequently, a question arises on how the 10-15-µm-long fibrils with a diameter of 6-10 nm are formed by β-sheets positioned along the whole fibril axis. In our opinion, upon insulin fibrillation the main building block is a ring-like oligomer. The diffraction pattern obtained for insulin may evidence that ring-like oligomers contain small β-sheets, which is the reason for a diffraction pattern characteristic of the cross-β structure. It should be noted in this connection that as early as in 1953, Koltun et al. demonstrated using X-ray analysis that fibrils can include insulin molecules in their normal folded state [130]. Based on the data obtained, we suggest that the main building block in the formation of insulin fibrils is a ring-like oligomer, i.e. a hexamer (Figs. 6 and 7). This model of fibrillation might explain the polymorphism of fibrils and their breakage, branching, and surface roughness (due to irregular association of ring-like oligomers). It should be also noted that hexamers involved in the fibril formation are structurally different from the zinc-coordinated hexamers in the crystal. This is indirectly indicated by the fact that no fibrillation takes place upon cross-linking of residues A1 and B29 [114]. The formed bond prevents structural rearrangement of the monomer, thereby eliminating the possibility of acquiring the conformation required for the fibril formation. Moreover, it also prevents binding of the modified insulin monomer to the IR [131].
Fig. 6. The mechanism of amyloid fibril formation by human insulin and insulin lispro (modified and adapted from [123]). a) Destabilized monomers; b) ring oligomers; c) differently oriented fragments of single fibrils; d) ribbons (lateral association of fibrils); e) bundles of fibrils.
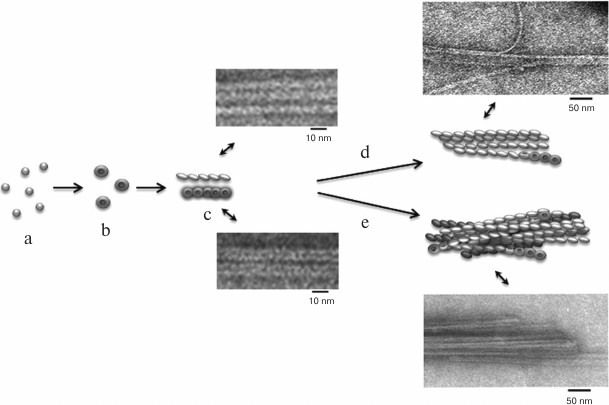
Fig. 7. Model of packing of three insulin hexamers (designed using the YASARA program). Dimer insulin (PDB code 5E7W) was used as the minimal structural unit. Amino acid residues GlyA1 and LysB29 important for the fibrillation are highlighted.
The requirements for insulin replacement therapy for the treatment of diabetes have promoted considerate interest in the development of insulin structural analogs. The studies abroad and in Russia have resulted in the creation of various bolus and basal insulin preparations. Nevertheless, the control of glucose blood levels without the risk of hypoglycemia, as well as insulin delivery in a form convenient for the patients, still remain the major strategic aims in diabetes therapy. The following should be taken into account when developing new insulin analogs for therapeutic purposes.
1. The hexamer form of insulin is more stable; therefore, amino acid substitutions should not involve residues responsible for hexamer formation.
2. When substituting residues involved in insulin molecule dimerization, it should be taken into consideration that some of these residues are responsible for the molecule binding to the IR.
3. Since the monomeric form of insulin is more prone to fibrillation, insulin mutants should be studied for their propensity for fibril formation.
In general, the design of receptor-selective insulin analogs with decreased propensity for amyloidogenesis is of great importance, because it will allow the maintenance of normal glucose blood levels without adverse side effects associated with the mitogenic action and fibrillation of insulin preparations.
Acknowledgments
The study was supported by the Molecular and Cell Biology Program (01201353567).
REFERENCES
1.Banting, F. G., and Best, C. J. (1922) The internal
secretion of the pancreas, Reprinted in 1972, Vol. 80, to mark 50th
anniversary of the discovery, J. Lab. Clin. Med., 7,
251-266.
2.De Meyts, P. (2004) Insulin and its receptor:
structure, function and evolution, Bioessays, 26,
1351-1362.
3.Cabrera, S. M., Chen, Y. G., Hagopian, W. A., and
Hessner, M. J. (2016) Blood-based signatures in type 1 diabetes,
Diabetologia, 59, 414-425.
4.Edelman, S., and Pettus, J. (2014) Challenges
associated with insulin therapy in type 2 diabetes mellitus, Am. J.
Med., 127, 11-16.
5.Tkachuk, V. A., and Vorotnikov, A. V. (2014)
Molecular mechanisms of development of insulin resistance, Saharnii
Diabet, 2, 29-40.
6.Titov, V. N. (2012) Phylogenesis, etiology and
pathogenesis of insulin resistance. Differences from type ii diabetes
mellitus, Vestnik RAMN, 4, 65-73.
7.American Diabetes Association (2012) Diagnosis and
classification of diabetes mellitus, Diabetes Care, 35
(Suppl. 1), 64-71.
8.Chin, J. A., and Sumpio, B. E. (2014) Diabetes
mellitus and peripheral vascular disease: diagnosis and management,
Clin. Podiatr. Med. Surg., 31, 11-26.
9.Todd, J. A. (1990) Genetic control of autoimmunity
in type 1 diabetes, Immunol. Today, 11, 122-129.
10.Redondo, M. J., Fain, P. R., and Eisenbarth, G.
S. (2001) Genetics of type 1A diabetes, Recent Prog. Horm. Res.,
56, 69-89.
11.Ohtsubo, K., Chen, M. Z., Olefsky, J. M., and
Marth, J. D. (2011) Pathway to diabetes through attenuation of
pancreatic beta cell glycosylation and glucose transport, Nat.
Med., 17, 1067-1075.
12.Ye, J. (2013) Mechanisms of insulin resistance in
obesity, Front. Med., 7, 14-24.
13.Chakraborty, C., Doss, C. G. P., Bandyopadhyay,
S., and Agoramoorthy, G. (2014) Influence of miRNA in insulin signaling
pathway and insulin resistance: micro-molecules with a major role in
type-2 diabetes, Wiley Interdiscip. Rev. RNA, 5,
697-712.
14.Feng, X., Tang, H., Leng, J., and Jiang, Q.
(2014) Suppressors of cytokine signaling (SOCS) and type 2 diabetes,
Mol. Biol. Rep., 41, 2265-2274.
15.Cheng, K., Andrikopoulos, S., and Gunton, J. E.
(2013) First phase insulin secretion and type 2 diabetes, Curr. Mol.
Med., 13, 126-139.
16.Zaykov, A. N., Mayer, J. P., and DiMarchi, R. D.
(2016) Pursuit of a perfect insulin, Nat. Rev. Drug Discov.,
15, 425-439.
17.Home, P., Riddle, M., Cefalu, W. T., Bailey, C.
J., Bretzel, R. G., Del Prato, S., Leroith, D., Schernthaner, G., Van
Gaal, L., and Raz, I. (2014) Insulin therapy in people with type 2
diabetes: opportunities and challenges? Diabetes Care,
37, 1499-1508.
18.Dzhavakhishvili, T. S., Romantsova, T. I., and
Roik, O. V. (2010) Dynamics of body weight in patients with type 2
diabetes during the first year of insulin therapy, Obes. Metab.,
4, 13-19.
19.Dedov, I. I., Shestakova, M. V., and Moiseev, S.
V. (2005) Analogues of insulin, Klin. Farmakol. Ter., 14,
49-55.
20.Vigneri, R., Squatrito, S., and Sciacca, L.
(2010) Insulin and its analogs: actions via insulin and IGF receptors,
Acta Diabetol., 47, 271-278.
21.Bell, G. I., Pictet, R. L., Rutter, W. J.,
Cordell, B., Tischer, E., and Goodman, H. M. (1980) Sequence of the
human insulin gene, Nature, 284, 26-32.
22.Alarcón, C., Leahy, J. L., Schuppin, G.
T., and Rhodes, C. J. (1995) Increased secretory demand rather than a
defect in the proinsulin conversion mechanism causes
hyperproinsulinemia in a glucose-infusion rat model of
non-insulin-dependent diabetes mellitus, J. Clin. Invest.,
95, 1032-1039.
23.Greider, M. H., Howell, S. L., and Lacy, P. E.
(1969) Isolation and properties of secretory granules from rat islets
of Langerhans. II. Ultrastructure of the beta granule, J. Cell
Biol., 41, 162-166.
24.Michael, J., Carroll, R., Swift, H. H., and
Steiner, D. F. (1987) Studies on the molecular organization of rat
insulin secretory granules, J. Biol. Chem., 262,
16531-16535.
25.Chang, T. W., and Goldberg, A. L. (1978) The
metabolic fates of amino acids and the formation of glutamine in
skeletal muscle, J. Biol. Chem., 253, 3685-3693.
26.Eto, K., Tsubamoto, Y., Terauchi, Y., Sugiyama,
T., Kishimoto, T., Takahashi, N., Yamauchi, N., Kubota, N., Murayama,
S., Aizawa, T., Akanuma, Y., Aizawa, S., Kasai, H., Yazaki, Y., and
Kadowaki, T. (1999) Role of NADH shuttle system in glucose-induced
activation of mitochondrial metabolism and insulin secretion,
Science, 283, 981-985.
27.Bender, K., Newsholme, P., Brennan, L., and
Maechler, P. (2006) The importance of redox shuttles to pancreatic
beta-cell energy metabolism and function, Biochem. Soc. Trans.,
34, 811-814.
28.Nolan, C. J., Leahy, J. L., Delghingaro-Augusto,
V., Moibi, J., Soni, K., Peyot, M. L., Fortier, M., Guay, C.,
Lamontagne, J., Barbeau, A., Przybytkowski, E., Joly, E., Masiello, P.,
Wang, S., Mitchell, G. A., and Prentki, M. (2006) Beta cell
compensation for insulin resistance in Zucker fatty rats: increased
lipolysis and fatty acid signalling, Diabetologia, 49,
2120-2130.
29.Prentki, M., Joly, E., El-Assaad, W., and Roduit,
R. (2002) Malonyl-CoA signaling, lipid partitioning, and
glucolipotoxicity: role in beta-cell adaptation and failure in the
etiology of diabetes, Diabetes, 51 (Suppl. 3),
405-413.
30.Baker, E. N., Blundell, T. L., Cutfield, J. F.,
Cutfield, S. M., Dodson, E. J., Dodson, G. G., Hodgkin, D. M., Hubbard,
R. E., Isaacs, N. W., and Reynolds, C. D. (1988) The structure of 2Zn
pig insulin crystals at 1.5 Å resolution, Philos. Trans. R.
Soc. Lond. B Biol. Sci., 319, 369-456.
31.Wood, S. P., Blundell, T. L., Wollmer, A.,
Lazarus, N. R., and Neville, R. W. (1975) The relation of conformation
and association of insulin to receptor binding; X-ray and
circular-dichroism studies on bovine and hystricomorph insulins,
Eur. J. Biochem., 55, 531-542.
32.Williamson, K. L., and Williams, R. J. (1979)
Conformational analysis by nuclear magnetic resonance: insulin,
Biochemistry, 18, 5966-5972.
33.Ramesh, V., and Bradbury, J. H. (1987)
1H NMR studies of insulin: histidine residues, metal
binding, and dissociation in alkaline solution, Arch. Biochem.
Biophys., 258, 112-122.
34.Wollmer, A., Rannefeld, B., Johansen, B. R.,
Hejnaes, K. R., Balschmidt, P., and Hansen, F. B. (1987)
Phenol-promoted structural transformation of insulin in solution,
Biol. Chem. Hoppe. Seyler., 368, 903-911.
35.Pittman, I., and Tager, H. S. (1995) A
spectroscopic investigation of the conformational dynamics of insulin
in solution, Biochemistry, 34, 10578-10590.
36.Bakaysa, D. L., Radziuk, J., Havel, H. A.,
Brader, M. L., Li, S., Dodd, S. W., Beals, J. M., Pekar, A. H., and
Brems, D. N. (1996) Physicochemical basis for the rapid time-action of
LysB28ProB29-insulin: dissociation of a protein–ligand complex,
Protein Sci., 5, 2521-2531.
37.De Meyts, P., and Whittaker, J. (2002) Structural
biology of insulin and IGF1 receptors: implications for drug design,
Nat. Rev. Drug Discov., 1, 769-783.
38.Menting, J. G., Whittaker, J., Margetts, M. B.,
Whittaker, L. J., Kong, G. K., Smith, B. J., Watson, C. J., Zakova, L.,
Kletvíkova, E., Jiracek, J., Chan, S. J., Steiner, D. F.,
Dodson, G. G., Brzozowski, A. M., Weiss, M. A., Ward, C. W., and
Lawrence, M. C. (2013) How insulin engages its primary binding site on
the insulin receptor, Nature, 493, 241-245.
39.Menting, J. G., Yang, Y., Chan, S. J., Phillips,
N. B., Smith, B. J., Whittaker, J., Wickramasinghe, N. P., Whittaker,
L. J., Pandyarajan, V., Wan, Z. L., Yadav, S. P., Carroll, J. M.,
Strokes, N., Roberts, C. T., Jr., Ismail-Beigi, F., Milewski, W.,
Steiner, D. F., Chauhan, V. S., Ward, C. W., Weiss, M. A., and
Lawrence, M. C. (2014) Protective hinge in insulin opens to enable its
receptor engagement, Proc. Natl. Acad. Sci. USA, 111,
3395-3404.
40.Pandyarajan, V., Smith, B. J., Phillips, N. B.,
Whittaker, L., Cox, G. P., Wickramasinghe, N., Menting, J. G., Wan, Z.
L., Whittaker, J., Ismail-Beigi, F., Lawrence, M. C., and Weiss, M. A.
(2014) Aromatic anchor at an invariant hormone–receptor
interface, J. Biol. Chem., 289, 34709-34727.
41.Pullen, R. A., Lindsay, D. G., Wood, S. P.,
Tickle, I. J., Blundell, T. L., Wollmer, A., Krail, G., Brandenburg,
D., Zahn, H., Gliemann, J., and Gammeltoft, S. (1976) Receptor-binding
region of insulin, Nature, 259, 369-373.
42.Kwok, S. C., Steiner, D. F., Rubenstein, A. H.,
and Tager, H. S. (1983) Identification of a point mutation in the human
insulin gene giving rise to a structurally abnormal insulin (insulin
Chicago), Diabetes, 32, 872-875.
43.Shoelson, S., Haneda, M., Blix, P., Nanjo, A.,
Sanke, T., Inouye, K., Steiner, D., Rubenstein, A., and Tager, H.
(1983) Three mutant insulins in man, Nature, 302,
540-543.
44.Shoelson, S., Fickova, M., Haneda, M., Nahum, A.,
Musso, G., Kaiser, E. T., Rubenstein, A. H., and Tager, H. (1983)
Identification of a mutant human insulin predicted to contain a
serine-for-phenylalanine substitution, Proc. Natl. Acad. Sci.
USA, 80, 7390-7394.
45.Kobayashi, M., Ohgaku, S., Iwasaki, M., Maegawa,
H., Shigeta, Y., and Inouye, K. (1982) Supernormal insulin:
[D-PheB24]-insulin with increased affinity for insulin receptors,
Biochem. Biophys. Res. Commun., 107, 329-336.
46.Nanjo, K., Sanke, T., Miyano, M., Okai, K., Sowa,
R., Kondo, M., Nishimura, S., Iwo, K., Miyamura, K., and Given, B. D.
(1986) Diabetes due to secretion of a structurally abnormal insulin
(insulin Wakayama). Clinical and functional characteristics of [LeuA3]
insulin, J. Clin. Invest., 77, 514-519.
47.Blundell, T. L., Cutfield, J. F., Cutfield, S.
M., Dodson, E. J., Dodson, G. G., Hodgkin, D. C., and Mercola, D. A.
(1972) Three-dimensional atomic structure of insulin and its
relationship to activity, Diabetes, 21, 492-505.
48.Cosmatos, A., Cheng, K., Okada, Y., and
Katsoyannis, P. G. (1978) The chemical synthesis and biological
evaluation of [1-L-alanine-A]-and [1-D-alanine-A] insulins, J. Biol.
Chem., 253, 6586-6590.
49.Geiger, R., Geisen, K., Summ, H. D., and Langer,
D. (1975) (A1-D-alanine) insulin, Hoppe. Seylers. Z. Physiol.
Chem., 356, 1635-1649.
50.Geiger, R., Geisen, K., and Summ, H. D. (1982)
Exchange of A1-glycine in bovine insulin with L- and D-tryptophan,
Hoppe. Seylers. Z. Physiol. Chem., 363, 1231-1239.
51.Nakagawa, S. H., and Tager, H. S. (1989)
Perturbation of insulin–receptor interactions by intramolecular
hormone cross-linking. Analysis of relative movement among residues A1,
B1, and B29, J. Biol. Chem., 264, 272-279.
52.Ogawa, H., Burke, G. T., Chanley, J. D., and
Katsoyannis, P. G. (1987) Effect of N-methylation of selected peptide
bonds on the biological activity of insulin.
[2-N-methylisoleucine-A]insulin and [3-N-methylvaline-A]insulin,
Int. J. Pept. Protein Res., 30, 460-473.
53.Schwartz, G., and Katsoyannis, P. G. (1978)
Synthesis of des(tetrapeptide B(1-4)) and des(pentapeptide B(1-5))
human insulins. Two biologically active analogues, Biochemistry,
17, 4550-4556.
54.Nakagawa, S. H., and Tager, H. S. (1991)
Implications of invariant residue LeuB6 in insulin–receptor
interactions, J. Biol. Chem., 266, 11502-11509.
55.Schwartz, G. P., Burke, G. T., and Katsoyannis,
P. G. (1987) A superactive insulin: [B10-aspartic acid]insulin(human),
Proc. Natl. Acad. Sci. USA, 84, 6408-6411.
56.Chan, S. J., Seino, S., Gruppuso, P. A.,
Schwartz, R., and Steiner, D. F. (1987) A mutation in the B chain
coding region is associated with impaired proinsulin conversion in a
family with hyperproinsulinemia, Proc. Natl. Acad. Sci. USA,
84, 2194-2197.
57.Gruppuso, P. A., Gorden, P., Kahn, C. R.,
Cornblath, M., Zeller, W. P., and Schwartz, R. (1984) Familial
hyperproinsulinemia due to a proposed defect in conversion of
proinsulin to insulin, N. Engl. J. Med., 311,
629-634.
58.Brange, J. Ribel, U., Hansen, J. F., Dodson, G.,
Hansen, M. T., Havelund, S., Melberg, S. G., Norris, F., Norris, K.,
and Snel, L. (1988) Monomeric insulins obtained by protein engineering
and their medical implications, Nature, 333, 679-682.
59.Brems, D. N., Alter, L. A., Beckage, M. J.,
Chance, R. E., DiMarchi, R. D., Green, L. K., Long, H. B., Pekar, A.
H., Shields, J. E., and Frank, B. H. (1992) Altering the association
properties of insulin by amino acid replacement, Protein Eng.,
5, 527-533.
60.Blundell, T. L., Cutfield, J. F., Cutfield, S.
M., Dodson, E. J., Dodson, G. G., Hodgkin, D. C., and Mercola, D. A.
(1972) Three-dimensional atomic structure of insulin and its
relationship to activity, Diabetes, 21, 492-505.
61.Nathan, D. M., Genuth, S., Lachin, J., Cleary,
P., Crofford, O., Davis, M., Rand, L., and Siebert, C. (1993) The
effect of intensive treatment of diabetes on the development and
progression of long-term complications in insulin-dependent diabetes
mellitus, N. Engl. J. Med., 329, 977-986.
62.Chou, H. S., Larsson, M., Hsiao, M. H., Chen, Y.
C., Roding, M., Nyden, M., and Liu, D. M. (2016) Injectable
insulin-lysozyme-loaded nanogels with enzymatically-controlled
degradation and release for basal insulin treatment: in vitro
characterization and in vivo observation, J. Control.
Release, 224, 33-42.
63.Ionova, T. I., Odin, V. I., Nikitina, T. P.,
Kurbatova, K. A., and Shablovskaya, N. E. (2013) Quality of life and
problems posed by hypoglycemia in type 2 diabetes mellitus during oral
hypoglycemic therapy, Klin. Med., 9, 34-40.
64.Hirsch, I. B., Farkas-Hirsch, R., and Skyler, J.
S. (1990) Intensive insulin therapy for treatment of type I diabetes,
Diabetes Care, 13, 1265-1283.
65.Gusarov, D. A., Gusarova, V. D., Bayramashvili,
D. I., and Mironov, A. F. (2008) Human insulin and its pharmaceutical
analogs, Biomed. Khim., 54, 624-642.
66.Home, P. D. (2012) The pharmacokinetics and
pharmacodynamics of rapid-acting insulin analogues and their clinical
consequences, Diabetes Obes. Metab., 14, 780-788.
67.Oakley, W., Hill, D., and Oakley, N. (1966)
Combined use of regular and crystalline protamine (NPH) insulins in the
treatment of severe diabetes, Diabetes, 15, 219-222.
68.Rosenstock, J., Schwartz, S. L., Clark, C. M.,
Jr., Park, G. D., Donley, D. W., and Edwards, M. B. (2001) Basal
insulin therapy in type 2 diabetes: 28-week comparison of insulin
glargine (HOE 901) and NPH insulin, Diabetes Care, 24,
631-636.
69.Johnson, I. S. (1983) Human insulin from
recombinant DNA technology, Science, 219, 632-637.
70.Peters, A. L., Pollom, R. D., Zielonka, J. S.,
Carey, M. A., and Edelman, S. V. (2015) Biosimilars and new insulin
versions, Endocr. Pract., 21, 1387-1394.
71.Miroshnikov, A. I. (2009) Recipe for Russian
insulin, Acta Naturae, 3, 18-20.
72.Owens, D. R., Landgraf, W., Schmidt, A., Bretzel,
R. G., and Kuhlmann, M. K. (2012) The emergence of biosimilar insulin
preparations – a cause for concern? Diabetes Technol.
Ther., 14, 989-996.
73.Habriev, R. U. (2005) in Manual on
Experimental (Preclinical) Study of New Pharmacological Substances
(Habriev, R. U., ed.) [in Russian], Meditsina, Moscow.
74.Mironov, A. N. (2012) in Manual on Preclinical
Trials of Medicines, Part I (Mironov, A. N., ed.) [in Russian],
Grif i K, Moscow.
75.Mironov, A. N. (2013) in Manual on Expertise
of Medicines, Part V. I. (Mironov, A. N., ed.) [in Russian], Grif i
K, Moscow.
76.Atkinson, M. A., Eisenbarth, G. S., and Michels,
A. W. (2014) Type 1 diabetes, Lancet, 383, 69-82.
77.Kuraeva, T. L. (2010) Analogues of insulin in
achieving compensation and improving the quality of life of children
and adolescents with type 1 diabetes mellitus, Saharnii Diabet,
3, 147-152.
78.Howey, D. C., Bowsher, R. R., Brunelle, R. L.,
and Woodworth, J. R. (1994) [Lys(B28), Pro(B29)]-human insulin. A
rapidly absorbed analogue of human insulin, Diabetes, 43,
396-402.
79.Ciszak, E., Beals, J. M., Frank, B. H., Baker, J.
C., Carter, N. D., and Smith, G. D. (1995) Role of C-terminal B-chain
residues in insulin assembly: the structure of hexameric
LysB28ProB29-human insulin, Structure, 3, 615-622.
80.Bakaysa, D. L., Radziuk, J., Havel, H. A.,
Brader, M. L., Li, S., Dodd, S. W., Beals, J. M., Pekar, A. H., and
Brems, D. N. (1996) Physicochemical basis for the rapid time-action of
Lys B28 Pro B29-insulin: dissociation of a protein–ligand
complex, Protein Sci., 5, 2521-2531.
81.Becker, R. H. A., Frick, A. D., Burger, F.,
Potgieter, J. H., and Scholtz, H. (2005) Insulin glulisine, a new
rapid-acting insulin analogue, displays a rapid time-action profile in
obese non-diabetic subjects, Exp. Clin. Endocrinol. Diabetes,
113, 435-443.
82.Dreyer, M., Prager, R, Robinson, A., Busch, K.,
Ellis, G., Souhami, E., and Van Leendert, R. (2005) Efficacy and safety
of insulin glulisine in patients with type 1 diabetes, Horm. Metab.
Res., 37, 702-707.
83.Pavlova, M. G. (2008) Apidra (insulin glulisin)
in the treatment of type 1 diabetes mellitus, Saharnii Diabet,
2, 65-68.
84.Heinemann, L., Heise, T., Wahl, L. C., Trautmann,
M. E., Ampudia, J., Starke, A. A., and Berger, M. (1996) Prandial
glycaemia after a carbohydrate-rich meal in type I diabetic patients:
using the rapid acting insulin analogue [Lys(B28), Pro(B29)] human
insulin, Diabet. Med., 13, 625-629.
85.Owens, D. R., Matfin, G., and Monnier, L. (2014)
Basal insulin analogues in the management of diabetes mellitus: what
progress have we made? Diabetes Metab. Res. Rev., 30,
104-119.
86.Zinman, B. (2013) Newer insulin analogs: advances
in basal insulin replacement, Diabetes Obes. Metab., 15
(Suppl. 1), 6-10.
87.Hallas-Moller, K. (1956) The lente insulins,
Diabetes, 5, 7-14.
88.Heise, T., Nosek, L., Ronn, B. B., Endahl, L.,
Heinemann, L., Kapitza, C., and Draeger, E. (2004) Lower within-subject
variability of insulin detemir in comparison to NPH insulin and insulin
glargine in people with type 1 diabetes, Diabetes, 53,
1614-1620.
89.Peterkova, V. A., Kuraeva, T. L., and Titovich,
E. V. (2003) Lantus (insulin glargine): real benefits and perspectives
of use in pediatrics, Saharnii Diabet, 3, 26-28.
90.Klimontov, V. V., and Myakina, N. E. (2014)
Insulin glargine: pharmacokinetic and pharmacodynamic bases of clinical
effect, Saharnii Diabet, 4, 99-107.
91.Migdalis, I. N. (2011) Insulin analogs versus
human insulin in type 2 diabetes, Diabetes Res. Clin. Pract.,
93 (Suppl. 1), 102-104.
92.Swinnen, S. G., Simon, A. C., Holleman, F.,
Hoekstra, J. B., and Devries, J. H. (2011) Insulin detemir versus
insulin glargine for type 2 diabetes mellitus, Cochrane Database
Syst. Rev., 7, 63-83.
93.Jonassen, I., Havelund, S., Hoeg-Jensen, T.,
Steensgaard, D. B., Wahlund, P. O., and Ribel, U. (2012) Design of the
novel protraction mechanism of insulin degludec, an ultra-long-acting
basal insulin, Pharm. Res., 29, 2104-2114.
94.Dedov, I. I., and Shestakova, M. V. (2014)
Insulin degludec – a new analogue of insulin super-long-acting,
Saharnii Diabet, 2, 91-104.
95.Whittingham, J. L., Havelund, S., and Jonassen,
I. (1997) Crystal structure of a prolonged-acting insulin with
albumin-binding properties, Biochemistry, 36,
2826-2831.
96.Sorli, C., Warren, M., Oyer, D., Mersebach, H.,
Johansen, T., and Gough, S. C. (2013) Elderly patients with diabetes
experience a lower rate of nocturnal hypoglycaemia with insulin
degludec than with insulin glargine: a meta-analysis of phase IIIa
trials, Drugs Aging, 30, 1009-1018.
97.Henry, R. R., Mudaliar, S., Ciaraldi, T. P.,
Armstrong, D. A., Burke, P., Pettus, J., Garhyan, P., Choi, S. L.,
Jacober, S. J., Knadler, M. P., Lam, E. C., Prince, M. J., Bose, N.,
Porksen, N., Sinha, V. P., and Linnebjerg, H. (2014) Basal insulin
peglispro demonstrates preferential hepatic versus peripheral action
relative to insulin glargine in healthy subjects, Diabetes Care,
37, 2609-2615.
98.Mathieu, C., Gillard, P., and Benhalima, K.
(2017) Insulin analogues in type 1 diabetes mellitus: getting better
all the time, Nat. Rev. Endocrinol., 13, 385-399.
99.Bergenstal, R. M., Rosenstock, J., Bastyr, E. J.,
Prince, M. J., Qu, Y., and Jacober, S. J. (2014) Lower glucose
variability and hypoglycemia measured by continuous glucose monitoring
with novel long-acting insulin LY2605541 versus insulin glargine,
Diabetes Care, 37, 659-665.
100.Caparrotta, T. M., and Evans, M. (2014)
PEGylated insulin Lispro, (LY2605541) – a new basal insulin
analogue, Diabetes Obes. Metab., 16, 388-395.
101.Ciaraldi, T. P., and Sasaoka, T. (2011) Review
on the in vitro interaction of insulin glargine with the
insulin/insulin-like growth factor system: potential implications for
metabolic and mitogenic activities, Horm. Metab. Res.,
43, 1-10.
102.Monnier, L., Colette, C., and Owens, D. (2013)
Basal insulin analogs: from pathophysiology to therapy. What we see,
know, and try to comprehend? Diabetes Metab., 39,
468-476.
103.Kurapkat, G., Siedentop, M., Gattner, H. G.,
Hagelstein, M., Brandenburg, D., Grotzinger, J., and Wollmer, A. (1999)
The solution structure of a superpotent β-chain-shortened
single-replacement insulin analogue, Protein Sci., 8,
499-508.
104.Jiracek, J., Zakova, L., Antolikova, E.,
Watson, C. J., Turkenburg, J. P., Dodson, G. G., and Brzozowski, A. M.
(2010) Implications for the active form of human insulin based on the
structural convergence of highly active hormone analogues, Proc.
Natl. Acad. Sci. USA, 107, 1966-1970.
105.Phillips, N. B., Wan, Z. L., Whittaker, L., Hu,
S. Q., Huang, K., Hua, Q. X., Whittaker, J., Ismail-Beigi, F., and
Weiss, M. A. (2010) Supramolecular protein engineering: design of
zinc-stapled insulin hexamers as a long acting depot, J. Biol.
Chem., 285, 11755-11759.
106.Berenson, D. F., Weiss, A. R., Wan, Z., and
Weiss, M. A. (2011) Insulin analogs for the treatment of diabetes
mellitus: therapeutic applications of protein engineering, Ann. N.
Y. Acad. Sci., 1243, 40-54.
107.Morishita, H. (2015) Premixed insulin and
intermediate-acting insulin, Nihon Rinsho, 73,
453-457.
108.Arinina, E. E., and Rashid, M. A. (2012)
Clinical and economic benefits of using human insulin analogues,
Pharmacoeconomics, 5, 41-46.
109.Dedov, I. I., and Shestakova, M. V. (2014)
Insulin degludec/insulin aspart – the first combined preparation
of basal and prandial insulin analogues, Saharnii Diabet,
4, 108-119.
110.Rodionova, T. I., and Orlova, M. M. (2014)
Assessment of efficiency of application of various analogues of insulin
in treatment of diabetes type 2, Saratov J. Med. Sci. Res.,
10, 461-464.
111.Nilsson, M. R. (2016) Insulin amyloid at
injection sites of patients with diabetes, Amyloid, 23,
139-147.
112.Berhanu, W. M., and Masunov, A. E. (2012)
Controlling the aggregation and rate of release in order to improve
insulin formulation: molecular dynamics study of full-length insulin
amyloid oligomer models, J. Mol. Model., 18,
1129-1142.
113.Amdursky, N., Gazit, E., and Rosenman, G.
(2012) Formation of low-dimensional crystalline nucleus region during
insulin amyloidogenesis process, Biochem. Biophys. Res. Commun.,
419, 232-237.
114.Brange, J., Andersen, L., Laursen, E. D., Meyn,
G., and Rasmussen, E. (1997) Toward understanding insulin fibrillation,
J. Pharm. Sci., 86, 517-525.
115.Yang, Y., Petkova, A., Huang, K., Xu, B., Hua,
Q. X., Ye, I. J., Chu, Y. C., Hu, S. Q., Phillips, N. B., Whittaker,
J., Ismail-Beigi, F., Mackin, R. B., Katsoyannis, P. G., Tycko, R., and
Weiss, M. A. (2010) An Achilles’ heel in an amyloidogenic protein
and its repair: insulin fibrillation and therapeutic design, J.
Biol. Chem., 285, 10806-10821.
116.Li, Y., Gonga, H., Sunb, Y., Yana, J., Chenga,
B., Zhanga, X., Huang, J., Yua, M., Guoa, Y., Zhengb, L., and Huanga,
K. (2012) Dissecting the role of disulfide bonds on the amyloid
formation of insulin, Biochem. Biophys. Res. Commun.,
423, 373-378.
117.Huang, K., Maiti, N. C., Phillips, N. B.,
Carey, P. R., and Weiss, M. A. (2006) Structure-specific effects of
protein topology on cross-beta assembly: studies of insulin
fibrillation, Biochemistry, 45, 10278-10293.
118.Fodera, V., Librizzi, F., Groenning, M., Van de
Weert, M., and Leone, M. (2008) Secondary nucleation and accessible
surface in insulin amyloid fibril formation, J. Phys. Chem. B,
112, 3853-3858.
119.Groenning, M., Frokjaer, S., and Vestergaard,
B. (2009) Formation mechanism of insulin fibrils and structural aspects
of the insulin fibrillation process, Curr. Protein Pept. Sci.,
10, 509-528.
120.Ahmad, A., Uversky, V. N., Hong, D., and Fink,
A. L. (2005) Early events in the fibrillation of monomeric insulin,
J. Biol. Chem., 280, 42669-42675.
121.Zhou, C., Qi, W., Lewis, E. N., and Carpenter,
J. F. (2016) Characterization of sizes of aggregates of insulin analogs
and the conformations of the constituent protein molecules: a
concomitant dynamic light scattering and raman spectroscopy study,
J. Pharm. Sci., 105, 551-558.
122.Woods, R. J., Alarcon, J., McVey, E., and
Pettis, R. J. (2012) Intrinsic fibrillation of fast-acting insulin
analogs, J. Diabetes Sci. Technol., 6, 265-276.
123.Blundell, T., Dodson G., Hodgkin, D., and
Mercola, D. (1972) Insulin: the structure in the crystal and its
reflection in chemistry and biology, Adv. Protein Chem.,
26, 279-402.
124.Selivanova, O. M., Suvorina, M. Y., Surin, A.
K., Dovidchenko, N. V., and Galzitskaya, O. V. (2017) Insulin and
lispro insulin: what is common and different in their behavior?
Curr. Protein Pept. Sci., 18, 57-64.
125.Selivanova, O. M., and Galzitskaya, O. V.
(2012) Structural polymorphism and possible pathways of amyloid fibril
formation on the example of insulin protein, Biochemistry
(Moscow), 77, 1237-1247.
126.Selivanova, O. M., Suvorina, M. Y.,
Dovidchenko, N. V., Eliseeva, I. A., Surin, A. K., Finkelstein, A. V.,
Schmatchenko, V. V., and Galzitskaya, O. V. (2014) How to determine the
size of folding nuclei of protofibrils from the concentration
dependence of the rate and lag-time of aggregation. II. Experimental
application for insulin and Lispro insulin: aggregation morphology,
kinetics, and sizes of nuclei, J. Phys. Chem. B, 118,
1198-1206.
127.Burke, M. J., and Rougvie, M. A. (1972)
Cross-β protein structures. I. Insulin fibrils,
Biochemistry, 11, 2435-2439.
128.Bouchard, M., Zurdo, J., Nettleton, E. J.,
Dobson, C. M., and Robinson, C. V. (2000) Formation of insulin amyloid
fibrils followed by FTIR simultaneously with CD and electron
microscopy, Protein Sci., 9, 1960-1967.
129.Jimenez, J. L., Nettleton, E. J., Bouchard, M.,
Robinson, C. V., Dobson, C. M., and Saibil, H. R. (2002) The
protofilament structure of insulin amyloid fibrils, Proc. Natl.
Acad. Sci. USA, 99, 9196-9201.
130.Koltun, W. L., Waugh, D. F., and Bear, R. S.
(1954) An X-ray diffraction investigation of selected types of insulin
fibrils, J. Am. Chem. Soc., 76, 413-417.
131.Winocour, P. H., Mitchell, W. S., Gush, R. J.,
Taylor, L. J., and Baker, R. D. (1988) Altered hand skin blood flow in
type 1 (insulin-dependent) diabetes mellitus, Diabet. Med.,
5, 861-866.