REVIEW: Investigations of Photosensitive Proteins by Serial Crystallography
G. K. Selikhanov, M. S. Fando, M. V. Dontsova, and A. G. Gabdulkhakov*
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: azat@vega.protres.ru* To whom correspondence should be addressed.
Received May 21, 2017; Revision received July 7, 2017
This review contains recent data on serial femtosecond X-ray crystallography (SFX), based on a femtosecond X-ray free electron laser, as well as, on the possibilities of its application for studying photosensitive proteins. Development of this method began rather recently, and it has already shown its effectiveness and some unique advantages over conventional X-ray structural analysis. This technology is especially promising for structural studies of membrane proteins and for kinetic studies. The main principle of the method, the possibility of its application in structural biology, its advantages and disadvantages, as well as its prospects for further development are analyzed in this review. Special attention is given to publications in which the SFX method has been used to study photosensitive proteins.
KEY WORDS: X-ray, free electron laser, serial crystallography, photosensitive proteinsDOI: 10.1134/S0006297918140134
Abbreviations: LCP, lipid cubic phase; LSP, lipid sponge phase; PSI(II), photosystem I(II); PYP, photoactive yellow protein; RC, reaction center; SFX, serial femtosecond X-ray analysis; XFEL, X-ray free-electron laser; XRD, X-ray diffraction analysis.
X-ray diffraction analysis (XRD) is one of the most powerful tools for
determination and investigation of atomic structure of macromolecules,
including biopolymers. XRD has been proven effective in studies of
water-soluble (globular) proteins; however, its application to membrane
proteins is still at the stage of development and improvement. The
number of structures of membrane proteins in the protein data bank
(http://www.rcsb.org/) is only about 3% of their total number.
In recent years, a modernized version of X-ray diffraction analysis has been gaining popularity – serial femtosecond X-ray diffraction analysis (SFX), which utilizes free electrons femtosecond laser pulses. This promising technology has several advantages and can solve problems that are beyond the reach of classical XRD, especially in studies of membrane proteins. One of the advantages of SFX in comparison to classical crystallography is the possibility of studying enzymatic reactions in femtosecond periods, provided that a reaction can be triggered in the entire volume of the crystal simultaneously. In this category, photosensitive proteins are the preferred objects of study.
Photosensitive proteins are extremely interesting. They are responsible for such processes as photosynthesis and phototaxis. For the most complete characterization of photosensitive proteins and determination of the mechanism of their functioning, analysis of spatial organization of these molecules is required. However, many photosensitive proteins are integral membrane complexes, and this significantly complicates their crystallization. Nevertheless, quite interesting results have been obtained recently, and this report is devoted to their review.
FREE-ELECTRON LASER TECHNOLOGY
Serial crystallography has long but relatively infrequent usage history. In the past, implementing XRD sometimes required using several isomorphic crystals to obtain a complete set of diffraction data due to rapid crystal destruction under X-rays [1-3]. With increase in power and improvement in focusing of radiation, it became possible to collect data from several positions on a single crystal obtained before the destruction of the sample [4-6], which significantly increased the compatibility of data set fragments. Further software development allowed collection of diffraction data from a variety of crystals of relatively small size that are frozen together on various mounts [7].
The greatest development of the serial method of collecting diffraction data came with the advent of the X-Ray Free Electron Laser (XFEL) technology.
The X-ray free electron laser is a type of laser in which the gain medium consists of high-speed electrons that freely propagate in an undulator (Fig. 1) [8]. The undulator (or “wiggler”) is a device for generating coherent radiation, which is a sequence of short dipole magnets. In such a device, regulation of the parameters of the undulator and electron entry rate can be used to vary the radiation characteristics. Unlike undulators installed on synchrotrons that are several meters in length, those from X-ray lasers are much longer and reach several hundred meters.
Fig. 1. Scheme of the undulator adapted from [11]: 1) position of electron beam entry from the accelerator into the undulator; 2) electron trap; 3) electromagnetic beam generated by the undulator; 4) the system of conjugated dipole magnets.
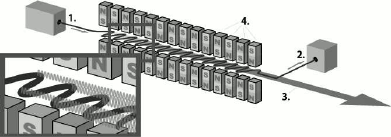
An electron accelerated in the accelerator enters the undulator, where it starts moving along a sinusoidal trajectory. At this point, the particles lose energy, which is converted into a photon flux. To obtain radiation in the X-ray range, electrons must enter the undulator at a speed close to the speed of light. In XFEL, due to the considerable undulator length, electrons are bunched into beams under the influence of Lorentz force. Particles in a beam oscillate synchronously, and their radiation becomes coherent, resulting in an additional increase in electromagnetic radiation intensity and in its pulsed nature [8-10]. The effectiveness of the amplifying effect is directly proportional to the undulator length. XFELs can generate coherent ultrashort (10-100 fs) electromagnetic pulses with peak spectral emission power much higher (up to 1033 photons/s/mm2/mrad2/0.1% bandwidth (BW)) than that of third generation synchrotron sources (up to 1020 photons/s/mm2/mrad2/0.1% BW).
Since 2009, a new XFEL center (European XFEL) is being constructed in Hamburg. The table shows the comparative characteristics of this XFEL, the American LCLS, and the Japanese SACLA. Many studies carried out on LCLS would be conducted much faster on the European XFEL, and it would also be possible to conduct experiments that were previously technologically inaccessible.
Comparative characteristics of LCLS, SACLA, and European XFEL (adapted
from [12])
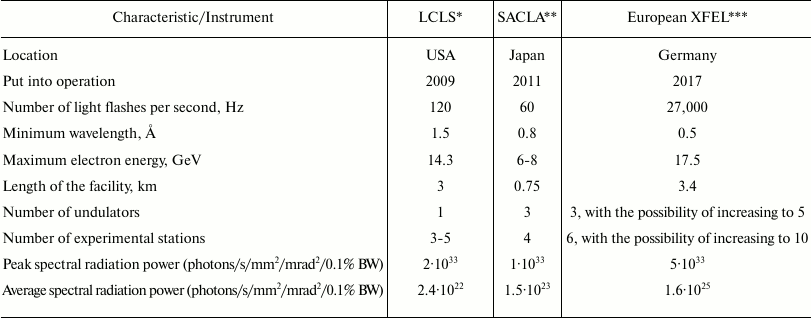
* LCLS, Linac Coherent Light Source.
** SACLA, Spring-8 Angstrom Compact Free Electron Laser.
*** European XFEL, European X-Ray Free-Electron Laser.
THE METHOD OF SERIAL CRYSTALLOGRAPHY AND ITS ADVANTAGES
The physical principle of serial crystallography does not differ from the principle of classical X-ray analysis. This method is also based on interaction of X-rays with molecules whose spatial structure should be determined. The primary photon beam, which is a plane monochromatic electromagnetic wave, hits an object, each electron of which is capable of diffraction; in other words, it is a source of a secondary spherical electromagnetic wave. This radiation is recorded by detectors, resulting in diffraction patterns, which are then processed by software. Then phase problem is solved, and ultimately an electron density map is calculated, on which the spatial structure is based.
One of the main problems of classical XRD is degradation of crystals under radiation. Their destruction occurs due to such factors as heating, destruction of covalent bonds, changes in molecular charges, and formation of free radicals [13]. Due to appearance of free radicals, damage to a sample continues even after irradiation has ceased [14]. For XRD, relatively large three-dimensional crystals are typically used with sides longer than 10 µm [15]. Smaller crystals do not provide a sufficient level of signal to obtain diffraction data; in addition, there are difficulties in centering the beam on microcrystals. Even though modern synchrotrons can generate intense X-ray pulses, crystallographs usually do not use them at their full capacity. There is a certain compromise in the choice of radiation parameters: on one hand, it should not be too powerful, since this will cause rapid crystal destruction, and on the other hand, it should be sufficiently intense to obtain high-resolution diffraction data. To slow sample destruction, cryoprotection is used, but this does not solve the problem completely [13].
Even before XFEL technology became available for use in structural biology, it was suggested that high doses of X-rays delivered in short and high-intensity pulses could be used to obtain diffraction data [16]. Even though a high dose of X-ray radiation destroys the crystal, it is possible to obtain qualitative diffraction data with short pulse duration before damage occurs. Theoretically, the size of the analyzed sample can be reduced to the level of a single molecule under conditions of a corresponding increase in the intensity of the electromagnetic pulse. In this case, spatial structures are solved by using superimposition of diffraction data obtained from thousands of isomorphic crystals.
This approach was applied for the first time in 2006. With the XFEL FLASH located in Hamburg, diffraction data was obtained from a silicon nitride membrane with an image burnt onto it [17]. The pulse duration was 25 fs. The image reconstructed from the diffraction data showed no damage, although the membrane was destroyed after irradiation (Fig. 2). Later, in 2007, X-ray diffraction analysis was carried out for a polystyrene sphere fixed on a silicon nitride membrane with the same setup [18]. The resolving power of the method in this experiment was 60 Å.
Fig. 2. The first experiment on diffraction data collection by a femtosecond laser (adapted from [17]). 1) Scanning electron microscope microphotograph of the membrane before irradiation by the laser. 2) Image reconstructed from diffraction data. 3) Scanning electron microscope microphotograph of the membrane after irradiation by the laser.

With SFX technology, inevitable destruction of the object after its irradiation requires constant replacement of analyzed material in the area of interaction with X-rays. There are several delivery systems. The choice of a system depends largely on the type of analyzed sample. Most experiments utilize a liquid jet, usually a parent solution for growing crystals, which is put into the vacuum compartment of the device where irradiation occurs [19]. The possibility of using a liquid jet for delivering microcrystals is explained by the fact that the intensity of diffraction peaks in this case is much higher than that of the water environment, allowing filtering out of water “noise”. The jet diameter is several hundred nanometers [20].
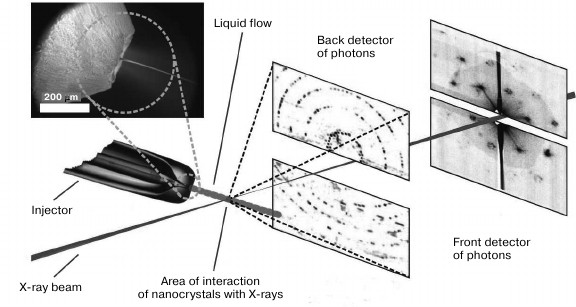
In Fig. 3, a scheme for a setup at Stanford University, California, USA is demonstrated.
Fig. 3. Scheme of the experimental setup for femtosecond serial crystallography (adapted from [26]). Nanocrystals are pulverized in vacuum by a gas-dynamic atomizer. X-rays pass perpendicularly through the resulting jet. Diffraction data is recorded by detectors after each pulse. The front and back detectors is responsible for high and low resolution, respectively.
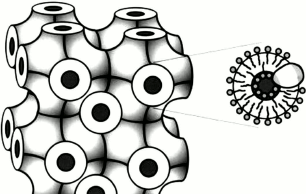
Unlike globular proteins, membrane proteins have extensive hydrophobic surfaces and are located in the lipid bilayer of a cell, causing additional difficulties both for their purification and crystallization, as well as for delivering the resulting crystals to an X-ray beam. To study this kind of samples, various methods can be applied. One method is crystallization and delivery in lipid cubic phase (LCP) [21] (Fig. 4). LCP are lipids, most often monoolein, mixed with water and protein in certain proportions. The LCP matrix enables growth of membrane protein crystals, facilitating molecular packing in which proteins are in contact with each other as well as with hydrophobic and hydrophilic surfaces [22]. Later, LCP was upgraded to obtain lipid sponge phase (LSP) by mixing monoolein with polyethylene glycol or Jeffamine [23, 24]. Unlike LCP, which is a solid, lipid sponge phase is a viscous liquid that like aqueous solutions can be sprayed, creating a stream of microcrystals entering the region of interaction with an X-ray pulse.
Fig. 4. Schematic model of the cubic phase made up of monoolein, water, and a membrane protein [22]. On the left is a matrix consisting of a three-dimensional periodic membrane grid permeated with a system of extended water channels. On the right is an enlarged matrix section showing a lipid bilayer with an integrated membrane protein molecule enveloping the water channel.
Another way of delivering membrane proteins is a formation of liquid bilayer films across an X-ray beam which ensure continuous delivery of membrane proteins to the interaction region [25]. This method is effective for studying two-dimensional protein crystals.
Many molecules that do not form large crystals are nevertheless capable of forming micro- and nanocrystals. They are so small that it is often impossible to detect them with an optical microscope [12]. Nevertheless, due to significant X-ray intensity in XFEL devices, even such small crystals provide a sufficient level of signal amplification for obtaining diffraction data.
To reduce degradation rate, crystals are kept under cryogenic conditions during a standard XRD experiment. A negative consequence of freezing proteins at ultralow temperatures can be disruption of their ability to undergo conformational rearrangements. It was shown that about 35% of side groups of amino acid residues may have a different spatial arrangement under cryogenic conditions than at room temperature [27]. Such changes often affect the structure of catalytic sites necessary for protein functioning. When analyzing by the SFX method, particularly using XFEL, crystals are kept at room temperature, since delivering microcrystals by spraying a jet of parent solution requires its liquid state. This is important, since studying spatial structures of molecules without freezing allows detection of conformational features crucial to the functioning of a protein that might be eliminated at ultralow temperatures. Moreover, keeping proteins under their normal temperature conditions is extremely important for kinetic experiments, since reaction rates usually depend on temperature.
KINETIC STUDIES
Functioning of many proteins is associated with certain conformational rearrangements. For a detailed understanding of their functioning mechanisms, information is needed not only on their spatial organization “at rest”, but also in all their transitional forms that appear because of conformational changes. Among such sensitive methods for studies of molecular dynamics as nuclear magnetic resonance, neutron scattering, ultraviolet and infrared microscopy, X-ray diffraction analysis plays a central role [28].
This is possible because many proteins remain functionally active in their crystal forms. However, to obtain an intermediate state structure, it is required that either this protein state is stable and able to form a crystal, or that conformational rearrangements start simultaneously in all protein molecules forming the crystal without destroying it. This is virtually impossible for all molecules except for photosensitive ones. For example, in the case of enzymes that are activated upon contact with a particular compound, we cannot deliver a substrate molecule to each protein molecule in the crystal simultaneously, and therefore we cannot bring all cells in the crystal to the same conformational state. Molecules of photosensitive proteins can be activated synchronously by irradiating them with light having certain characteristics. That is precisely why kinetic experiments in crystals have mainly been carried out on photosensitive proteins.
Previously, individual stages of the experiment were sufficiently time-separated: a crystal “at rest” was exposed to irradiation, and then a crystal that underwent activation was exposed. Then the resulting structures were compared. Thus, for example, information was received on structural changes occurring under the influence of light in the photosynthetic reaction center of Rhodobacter sphaeroides [29]. One crystal was studied in the dark, and another in the light. The resolution of the resulting structures was 2.2 and 2.6 Å, respectively. This revealed significant changes in the structure of reaction center molecules. However, this method can demonstrate only the initial and the final conformation, which is insufficient for a complete analysis of the protein functioning mechanism.
The modern approach to kinetic X-ray diffraction studies is the “pump and probe” method. This method involves triggering conformational rearrangements of molecules in a crystal by a laser pulse followed by a treatment with an X-ray pulse, which provides the diffraction data necessary for constructing the spatial structure. Pumping and probing must be repeated many times to obtain the necessary amount of data from crystals in their random orientations, and to obtain information with different delays between the excitation and the X-ray pulses to collect kinetic data. The delay value now varies from seconds to nanoseconds [28]. Such experiments are indeed kinetic, since they allow not only the detection of intermediates and their structures, but also their lifetimes and interconversion rates. This approach was used, for example, to obtain data on the intermediate conformations of such photosensitive proteins as photoactive yellow protein [30, 31] and bacterial photosynthetic reaction center [32].
One of the main limitations of the kinetic XRD method is the minimum time of X-ray irradiation required to obtain the needed diffraction data. Before the appearance of powerful synchrotron radiation sources, the duration of irradiation was longer than the lifetime of intermediates. With the advent of third-generation synchrotrons, it became possible to “catch” transitional conformations of molecules. For this, the Laue method is used, in which diffraction data is obtained by exposing a fixed crystal to powerful polychromatic X-ray radiation, which differs from the standard XRD method, where a rotating crystal is irradiated by a monochromatic beam [33]. The Laue method reduces the exposure time of X-ray radiation by 3-4 orders of magnitude in comparison with monochromatic experiments, which makes possible collection of microsecond diffraction data.
However, kinetic XRD has the same drawbacks as classical XRD. That is the problem of preparing crystals and preventing their damage during the experiment. Using hard polychromatic X-rays aggravates the situation by the fact that crystals degrade more rapidly, and the diffraction data is therefore of poor quality [28]. Another important drawback of this method is the impossibility of reducing exposure time of a crystal below the threshold value, which is approximately 100 ps and necessary for obtaining diffraction data, which prevents analysis of intermediates with shorter lifetimes [34].
With serial X-ray diffraction analysis, it is also possible to study the kinetics of various processes. For kinetic SFX, the same approach is used – “pumping and probing”. This technology has already shown its effectiveness in kinetic studies of globular proteins. An example of one of the latest successes of the method is achieving a time resolution of 250 fs in the study of ultrafast structural changes that occur following photolysis of the Fe–CO bond in the complex of myoglobin with carbon monoxide [35]. These changes take about 500 fs, which makes kinetic SFX the only method with sufficient resolving power to detect such short-lived intermediate states.
In addition to the possibility of analyzing short-lived intermediates, kinetic SFX has a number of other advantages: (i) proteins within micro- and nanocrystals can be more homogenously activated than within large ones; (ii) there is no necessity to obtain data on protein structure both at rest and in the excited state from a single crystal, which is often required for carrying out kinetic XRD; (iii) kinetic SFX is more adapted to obtaining data from crystals of various morphology than classical XRD and is applicable for crystals with comparatively weak reflecting properties [36].
DISADVANTAGES OF THE SFX METHOD
Serial X-ray analysis method certainly has its drawbacks. One of them is high consumption of protein crystals during an experiment compared to classical XRD, since only a small share of them is affected by X-ray pulses and most are skipped and do not interact with the radiation. For example, in a study on the polyhedrin structure of the capsid of the CPV17 virus, the hit rate was only 4% of the total number of crystals [37]. To carry out an experiment, 632 µl of suspension with concentration of approximately 3·109 microcrystals per liter was required. Given that, 144,803 images were taken, of which only 5787 were suitable for further use. Such high losses are characteristic of SFX, where crystals are delivered in a liquid jet.
To decrease the needed volume of a sample, delivery methods are being developed that use crystallographic grids with small cells, which are applied similarly to a crystallographic loop into which large crystals are placed [38]. However, with this type of delivery, crystals are extracted from the mother liquor, which can result in their damage and deterioration of quality.
Another way to reduce the amount of protein needed for analysis is increasing the frequency of X-ray pulses. X-ray free-electron lasers currently used for SFX have pulse frequencies of 60 Hz in the Japanese SACLA and 120 Hz in the American LCLS. The commissioned European XFEL have a pulse frequency of 27,000 Hz, which should significantly increase the efficiency of utilizing crystals in this setup compared to other XFELs.
Other disadvantages of this method are high requirements of computer memory necessary to store sorted diffraction data, as well as problems with the analysis of suspensions containing several crystalline forms of microcrystals.
Moreover, there might be an intensity limit for X-ray radiation in the SFX method above which the crystal will be destroyed before obtaining of diffraction data is complete [39]. XFELs that currently exist cannot be used to test this proposition in practice because they are not sufficiently powerful.
EXAMPLES OF SERIAL CRYSTALLOGRAPHY APPLICATION FOR STUDYING
PHOTOSENSITIVE PROTEINS
Photosystem I. Photosystem I (PSI) is a plastocyanin:ferredoxin oxidoreductase. It is a large multipigment membrane protein complex located in chloroplasts and essential for photosynthesis in plants, algae, and cyanobacteria [40]. PSI converts solar energy into a flow of electrons. During light absorption an electron is transferred in the electron transport chain, first to plastocyanin, and then to ferredoxin [41]. The quantum yield of this process is extremely high, approaching 100% [42]. Analysis of the crystal structure of PSI in the thermophilic cyanobacterium Synechococcus elongatus revealed that the complete functional complex in these organisms is a trimer. One monomer consists of 12 proteins and 127 cofactors, which include 96 chlorophylls, two phylloquinones, three Fe4S4 clusters, 22 carotenoids, four lipids, and the presence of one Ca2+ ion and 201 water molecules is supposed [43]. The complete trimeric complex has a molecular mass of about 1 MDa.
In 2011, Synechococcus elongatus PSI became the first photosensitive membrane complex on which the SFX method using XFEL was applied [26]. The analysis was conducted at room temperature. The microcrystals were delivered by a liquid jet, and the X-ray pulse duration was 70 fs. Analysis of the data provided a structure with 8.5 Å resolution. Despite the low resolution, this result was quite impressive, since the size of the microcrystals was 0.2 to 2 µm and, given the size of the protein complex itself, the amplification of the diffraction signal from crystals of that size was comparatively small.
In 2012, kinetics of PSI were studied using the SFX method [44]. Using a setup with a synchronized excitation laser and XFEL, crystals of PSI complex with ferredoxin were analyzed with post-activation delays of 5 and 10 µs. Data on photoinduced changes in crystals was obtained that correlated with microsecond kinetics of electron transfer from PSI to ferredoxin. It should be noted that after electron transfer the complex decays, resulting in significant rearrangements in the crystal and its subsequent destruction. This was the first example of kinetic SFX application for irreversible photochemical reactions.
Photoactive yellow protein (PYP). PYP is a small globular protein that has a photosensor function in purple photosynthetic bacteria [45].
The p-coumaric acid chromophore in PYP absorbs light in the blue region of the spectrum. The chromophore is bound to a cysteine residue (Cys69) by a thioether bond [40, 45]. In the dark, the chromophore is in a deprotonated trans-isomeric form. After absorption of a photon, it transits into an excited state, and the protein enters an photocycle that includes many intermediates. The main photochemical event controlling the entry into the photocycle is the isomerization of the chromophore from trans- to cis-conformation, which occurs along the torsion angle of the C2=C3 bond. PYP is the ideal model for studying spatial rearrangements necessary for a protein to function in response to an external signal. Due to the protein environment, the majority of photobiological systems are characterized by high speed and efficiency of chromophore isomerization. The rate of this process is measured in femto- and picoseconds, and the intermediates are short-lived, which is a problem for determining their structure, and therefore for studying the reaction mechanism [46].
Serial kinetic X-ray diffraction analysis was first applied to PYP in 2014 [47]. Previous studies using kinetic XRD with the Laue method revealed six intermediates in the photocycle: IT, ICT, pR1, pR2, pB1, and pB2 (Fig. 5).
Fig. 5. A simplified scheme of the photocycle of photoactive yellow protein (adapted from [47]). The cycle demonstrated here was obtained with data from kinetic XRD. pG, the protein state in the dark; pG*, the excited protein state after absorption of a 450-nm photon. Crystal structures of relatively long-lived intermediates IT, ICT, pR1, pR2, pB1, and pB2 are known.
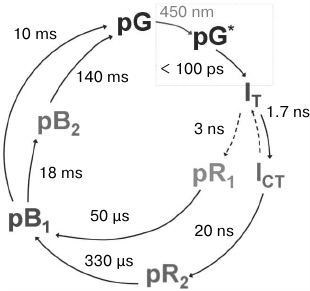
The strongest features are expressed in electron density difference maps, given the population of the pR1 and pR2 states, since the sulfur belonging to amino acid residue Cys69, to which the chromophore is covalently bound, significantly changes its spatial arrangement within both these intermediates. This occurs in a time interval between 200 ns and 100 µs. To detect these changes in the experiment, a delay time of 1 µs was chosen.
The second data set was collected with a delay of 10 ns, during which a population of three different intermediates was observed. Maximum resolution was 1.6 Å. The data obtained by kinetic XRD with Laue diffraction gave very similar difference maps.
In a later study, structural dynamics of cis-trans-isomerization in PYP at the earliest stages of the photocycle was studied by kinetic SFX [40]. Many structures were obtained with a delay after activation from 100 fs to 3 ps; the duration of the X-ray pulse was 40 fs, and a resolution of 1.6 Å was achieved.
Photosynthetic reaction center (RC) of purple bacteria. Photosynthetic RCs are macromolecular complexes that consist of proteins, pigments, and other cofactors that take part in the conversion of light energy into chemical energy during photosynthesis [41]. Energy can be received by an RC during direct excitation of its own pigments, but more often energy is transferred from light-harvesting complexes.
Bacterial RCs are interesting in that their architecture is similar to that of the central part of photosystem II (PSII), and therefore they have been used as structural and functional models for studying the more complex PSII for many years. The bacterial photosynthetic RC of Blastochloris viridis was the first membrane protein whose spatial structure was obtained by XRD in 1985 [48]. This photoreaction center is a four-subunit membrane protein with a molecular mass of approximately 135 kDa. Four bacteriochlorophyll molecules (two of which form a special pair), two molecules of bacteriopheophytins, two molecules of quinones, one carotenoid, one Fe2+, and four hemes act as its cofactors.
In contrast to the case of purple bacterial RCs, PSI is able to be crystallized in solution with low ionic strength (9 mM MgSO4) [49]. However, such conditions are rare for membrane proteins. Routinely, high salt concentrations (50-300 mM) with the addition of polyethylene glycol (10-35% and more) are used for their crystallization [24]. These solutions are not suitable for crystal delivery by a liquid jet in SFX devices due to their high viscosity and the risk of formation of salt crystals that can block the injector. In 2012, the SFX method was used for B. viridis RCs [24]. Their structure was determined at 8.2 Å resolution. The technique of crystallization in lipid sponge phase (LSP) was just then adapted and applied for the first time for the delivery of microcrystals by a liquid jet. LSP can be safely used for microcrystal delivery, and it is now used for many membrane proteins.
Development of the SFX technique and improvement of crystal quality resulted in improved resolution of the B. viridis RC structure to 3.5 Å in 2013 [50]. This increased resolution revealed several differences between the structures obtained by the XRD and SFX methods. Comparing the data on the covalent thioether bond between the N-terminal cysteine of the RC C-subunit and the diacylglycerol molecule is indicative (Fig. 6).
Fig. 6. Disruption of a covalent thioether bond in RC of Blastochloris viridis that is sensitive to X-rays (adapted from [50]). 1) Electron density map obtained from XRD data at 100 K using X-rays with power 4.4 MGy. 2) Electron density map obtained from XRD data at 100 K using X-rays with power 77 MGy. 3) Electron density map obtained from SFX data at room temperature using X-rays with power 33 MGy.
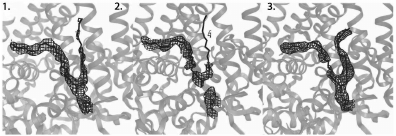
As seen in Fig. 6, the continuous electron density demonstrated on the first panel is significantly disturbed on the second one, which is a result of thioether bond destruction by the powerful X-ray radiation. The third panel does not show a break in the covalent bond, although the radiation power is sufficient for this, which demonstrates the ability of the SFX method to provide completely intact diffraction data.
Photosystem II. Photosystem II, like PSI, is a protein–pigment complex. It is located in the thylakoid membrane of chloroplasts in the form of a dimer, each monomer of which contains 19 to 31 protein subunits depending on the species [51]. PSII is distinguished by its ability to oxidize water with the formation of molecular oxygen, and it is the only biological system capable of doing this [41].
A unique part of PSII is the manganese cluster, also known as the water-oxidizing complex. It consists of four manganese atoms in different oxidation states, from 3+ to 5+, five oxygen atoms linking them, and one calcium atom. In 1970, a five-stage cycle was proposed for the functioning of the water-oxidizing complex [52]. The stages are designated as S0 to S4, where the index reflects the number of oxidative equivalents accumulated in the complex.
For several decades, various groups of researchers tried to determine the spatial structure of the components that comprise the PSII complex. In 2001, Zouni et al. determined the 3.8 Å resolution spatial structure of PSII from the cyanobacterium Synechococcus elongatus by X-ray diffraction analysis [53]. Subsequently, the quality of the PSII structure and the limit of its resolution have been improved gradually for several years [54-58].
In 2011, the structure of PSII from the cyanobacterium Thermosynechococcus vulcanus was obtained by XRD with 1.9 Å resolution. This revealed the structure of the Mn4CaO5 cluster and its protein environment, and a more detailed mechanism for its functioning was suggested [59]. Unfortunately, this and other structures obtained by XRD turned out to be not entirely reliable, since the state of manganese can be modified under the action of X-ray radiation. Approximately 25% of the ions in the Mn(III/IV) oxidation state turn into the Mn(II) oxidation state [60]. This process significantly changes the distances between atoms and deforms cluster structure [60-62]. Therefore, even at cryogenic temperatures that prevent formation of a native structure to begin with, application of classical X-ray analysis for PSII has limitations.
Serial crystallography with the use of XFEL was applied for the first time for PSII in 2012 [63]. This method solved the problem of manganese ion reduction and allowed analysis at room temperature. The diffraction data obtained from microcrystals of PSII of the thermophilic cyanobacterium Thermosynechococcus elongatus was used to reconstruct the structure with 6.5 Å resolution. In 2013, with optimization of techniques of microcrystallization and microcrystal delivery, the resolution was improved to 5.7 Å [64, 65].
The structure of PSII of the cyanobacterium T. vulcanus with significantly higher quality was obtained in 2014 [66]. A resolution of 1.95 Å was achieved, allowing study of the structure of the manganese cluster and its comparison with the structure obtained using a synchrotron. However, in contrast to previous SFX experiments, this one was carried out on large crystals at 100 K. Therefore, even though no photoinduced damage was observed in the structure, since a free-electron X-ray laser was used, it was nevertheless obtained under temperature conditions unnatural for the protein.
An example of successful application of the SFX method was a 2016 study in which the structure of PSII in the S1 stage in the dark state and two PSII structures in the S3 stage were obtained, one in the presence of ammonia molecules [67]. Diffraction data was collected at room temperature, using the kinetic SFX approach, for the transition to the S3 state. Ammonia was used as an analog of water to study its binding sites in the manganese cluster. Resolution of 3.0 Å was achieved for the S1 stage, 2.8 Å for the S3 stage with ammonia, and 2.25 Å for the S3 stage without ammonia. When compared with the structure obtained under cryogenic conditions [66], differences in the spatial arrangement of PSII monomers relative to each other and to the transmembrane α-helices within the monomers were revealed. In addition, some differences in bond lengths around the pigments involved in the energy transfer process were found. It is assumed that most of these changes are due precisely to the difference in temperatures during the recordings. The data on PSII in the S3 stage served as a source of new information on the structural elements important for the functioning of the photosystem. More information on the mechanism of functioning of the Mn4CaO5 cluster was obtained in a study in 2017 [68]. Previously unknown photoactivated structural changes in the water-oxidizing complex were found, and the maximum resolution of the intermediate structures was 2.35 Å.
Bacteriorhodopsin. Bacteriorhodopsin is an archaean protein that occurs in members of the Halobacterium genus and participates in photosynthesis. Bacteriorhodopsin photosynthesis is more primitive than chlorophyll photosynthesis since the protein is a proton pump that, by using light energy, transfers protons from the cell to the surrounding media through the membrane. The resulting proton gradient is further used in its conversion to chemical energy [69].
Bacteriorhodopsin is an integral membrane protein. Its characteristic feature is that it participates in the formation of specific sites in membranes called purple membranes. Purple membranes are two-dimensional crystalline layers that can occupy up to 50% of surface area of archaean cells. Bacteriorhodopsin forms repetitive elements organized in chains. Each chain has seven transmembrane α-helices, each chain containing one retinal molecule. Retinal is a chromophore that is linked to bacteriorhodopsin by a Schiff base [69, 70].
Because bacteriorhodopsin in archaean membranes forms two-dimensional crystals on its own, it is a convenient model object for X-ray diffraction studies. The method of serial X-ray diffraction analysis using XFEL was first applied to this protein in 2013 [71]. Purple membranes were isolated from H. salinarum, then, using detergents, small bacteriorhodopsin plates were obtained. A suspension of two-dimensional crystals containing sucrose was placed on a thin silicon nitride membrane, forming a window of a silicon chip placed in vacuum. The crystals were not cryopreserved, and sucrose prevented their dehydration. The use of fixed targets for X-ray pulses allowed study with the use of only a few micrograms of two-dimensional crystals. The maximum resolution of the diffraction data was 8 Å. Later, the technique was optimized, providing data with 7 Å resolution [72].
It should be noted that the shape of a two-dimensional crystal is most natural for many membrane proteins, since this state is similar to their location in cellular membranes [73]. In addition, the conformation of membrane proteins in two-dimensional crystals is supposed to be similar to their in vivo conformation, and potential structural changes in molecules are not as limited as they may be in three-dimensional crystals, which is especially important for studying the mechanisms of protein functioning [74].
Bacterial phytochrome. Phytochromes are a family of photoreceptors that take part in photosensor reactions of plants, fungi, and bacteria. These proteins detect light and trigger intracellular signal cascades responsible for the organism’s reaction to light. In bacteria, they often function as histidine kinases in two-component signaling systems and can be responsible, for example, for control of carotenoid synthesis [75, 76].
All phytochromes have a similar architecture. They have three conservative domains, PAS, GAF, and PHY, and a tetrapyrrole bilin chromophore. The chromophore is covalently bound by a thioether bond to a cysteine residue in the PAS domain in bacteria or in the GAF domain in cyanobacteria and plants. Bacterial phytochromes contain biliverdin as a chromophore [76]. Absorption of light by the chromophore results in several consecutive structural changes in the protein, which lead to a shift in the absorption spectrum of these molecules from the red to the far-red region. The details of the isomerization reaction and the sequence of structural changes are still not fully understood.
In 2016, the crystal structure of the chromophore-binding domain (PAS-GAF) of the bacterium Deinococcus radiodurans was obtained by serial X-ray diffraction analysis using XFEL at room temperature, attaining resolution of 2.1 Å [77]. For comparison, another PAS-GAF crystal structure was obtained with resolution 1.35 Å using conventional X-ray diffraction analysis under cryogenic conditions. With both XRD methods, similar conditions were used for crystallization, and the procedures of protein isolation and purification were identical. In this case, both directly grown microcrystals and crushed large crystals were used for serial analysis. To deliver microcrystals, a jet of the parent solution was used, and to deliver split crystals, their mixture with a viscous substance was used. In both cases, the possibility of obtaining qualitative diffraction data was demonstrated.
Comparison of the crystal structures of the PAS-GAF fragment demonstrated that the crystal structures obtained using both SFX and classical XRD are very similar, and the differences are mainly found in unstructured regions, while virtually no differences for atoms of the main chain and for the chromophore were observed. No defects were observed in the crystal structure obtained at room temperature.
The resolution of 2.1 Å, obtained in this study using the SFX method, is sufficient for studying isomerization reactions in a chromophore, changes in spatial arrangement of water molecules, and movements of side groups of amino acid residues. To obtain this data, researchers plan to apply the kinetic SFX method to bacteriophytochrome.
Analyzing spatial organization of protein molecules has become an integral part of many modern biological studies. The database of protein structures is being updated daily, and the largest contribution to this process is made by X-ray crystallography using synchrotron radiation. With all its advantages, this method has serious limitations that prevent its universal use for all proteins.
In this regard, the method of serial X-ray diffraction analysis using XFEL is being actively developed. It has capabilities that allow studies of kinetic processes, particularly those of photosensitive proteins, to a previously unreachable level.
Therefore, we hope that in the future SFX will move from the category of experimental developments to the category of routine methods of structural biology. Then studies will be much quicker and less labor-intensive, and their results will become more reliable and of higher quality.
Acknowledgments
This study was supported by the program of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology and Postgenomic Technologies” and the Russian Foundation for Basic Research (grants Nos. 17-00-00207 and 17-44-500828).
REFERENCES
1.Skarzynski, T., Mistry, A., Wonacott, A.,
Hutchinson, S. E., Kelly, V. A., and Duncan, K. (1996) Structure of
UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential
for the synthesis of bacterial peptidoglycan, complexed with substrate
UDP-N-acetylglucosamine and the drug fosfomycin, Structure,
4, 1465-1474.
2.Liu, J., Tse, A. G. D., Chang, H.-C., Liu, J.-H.,
Wang, J., Hussey, R. E., Chishti, Y., Rheinhold, B., Spoerl, R.,
Nathenson, S. G., Sacchettini, J. C., and Reinherz, E. L. (1996)
Crystallization of a deglycosylated T cell receptor (TCR) complexed
with an anti-TCR Fab fragment, J. Biol. Chem., 271,
33639-33646.
3.Wikoff, W. R., Tsai, C. J., Wang, G., Baker, T. S.,
and Johnson, J. E. (1997) The structure of cucumber mosaic virus:
cryoelectron microscopy, X-ray crystallography, and sequence analysis,
Virology, 232, 91-97.
4.Wenzel, S., Martins, B. M., Rosch, P., and Wohrl,
B. M. (2010) Crystal structure of the human transcription elongation
factor DSIF hSpt4 subunit in complex with the hSpt5 dimerization
interface, Biochem. J., 425, 373-380.
5.Hamiaux, C., Stanley, D., Greenwood, D. R., Baker,
E. N., and Newcomb, R. D. (2009) Crystal structure of Epiphyas
postvittana takeout 1 with bound ubiquinone supports a role as
ligand carriers for takeout proteins in insects, J. Biol. Chem.,
284, 3496-3503.
6.Gabdulkhakov, A. G., Dontsova, M. V., and Saenger,
W. (2011) Three-dimensional structure of photosystem II from
Thermosynechococcus elongates in complex with terbutryn,
Crystallogr. Rep., 56, 1054-1059.
7.Gabadinho, J., Beteva, A., Guijarro, M.,
Rey-Bakaikoa, V., Spruce, D., Bowler, M. W., Brockhauser, S., Flot, D.,
Gordon, E. J., Hall, D. R., Lavault, B., McCarthy, A. A., McCarthy, J.,
Mitchell, E., Monaco, S., Mueller-Dieckmann, C., Nurizzo, D., Ravelli,
R. B. G., Thibault, X., Walsh, M. A., Leonard, G. A., and McSweeney, S.
M. (2010) MxCuBE: a synchrotron beamline control environment customized
for macromolecular crystallography experiments, J. Synchrotron
Radiat., 17, 700-707.
8.Huang, Z., and Kim, K.-J. (2007) Review of X-ray
free-electron laser theory, Phys. Rev. Spec. Top. Accel. Beams,
10, 034801-034826.
9.Margaritondo, G., and Rebernik Ribic, P. (2011) A
simplified description of X-ray free-electron lasers, J. Synchrotron
Radiat., 18, 101-108.
10.Struder, L., Epp, S., Rolles, D., Hartmann, R.,
Holl, P., Lutz, G., Soltau, H., Eckart, R., Reich, C., Heinzinger, K.,
Thamm, C., Rudenko, A., Krasniqi, F., Kuhnel, K.-U., Bauer, C.,
Schroter, C.-D., Moshammer, R., Techert, S., Miessner, D., Porro, M.,
Halker, O., Meidinger, N., Kimmel, N., Andritschke, R., Schopper, F.,
Weidenspointner, G., Ziegler, A., Pietschner, D., Herrmann, S.,
Pietsch, U., Walenta, A., Leitenberger, W., Bostedt, C., Moller, T.,
Rupp, D., Adolph, M., Graafsma, H., Hirsemann, H., Gartner, K.,
Richter, R., Foucar, L., Shoeman, R. L., Schlichting, I., and Ullrich,
J. (2010) Large-format, high-speed, X-ray pnCCDs combined with electron
and ion imaging spectrometers in a multipurpose chamber for experiments
at 4th generation light sources, Nucl. Instrum. Methods Phys. Res.
Sect. Accel. Spectrometers Detect. Assoc. Equip., 614,
483-496.
11.Carroll, D. (2011) Overview of high energy
lasers: past, present, and future, in 42nd AIAA Plasmadynamics and
Lasers Conf. in conjunction with the 18th Int. Conf. on MHD Energy
Conversion (ICMHD), p. 3102.
12.European XFEL – Overview – In
comparison
(https://www.xfel.eu/facility/comparison/index_eng.html).
13.Massover, W. H. (2007) Radiation damage to
protein specimens from electron beam imaging and diffraction: a
mini-review of anti-damage approaches, with special reference to
synchrotron X-ray crystallography, J. Synchrotron Radiat.,
14, 116-127.
14.Smyth, M. S. (2000) X-ray crystallography,
Mol. Pathol., 53, 8-14.
15.Holton, J. M., and Frankel, K. A. (2010) The
minimum crystal size needed for a complete diffraction data set,
Acta Crystallogr. D Biol. Crystallogr., 66, 393-408.
16.Neutze, R., Wouts, R., Van der Spoel, D.,
Weckert, E., and Hajdu, J. (2000) Potential for biomolecular imaging
with femtosecond X-ray pulses, Nature, 406, 752-757.
17.Chapman, H. N., Barty, A., Bogan, M. J., Boutet,
S., Frank, M., Hau-Riege, S. P., Marchesini, S., Woods, B. W., Bajt,
S., Benner, W. H., London, R. A., Plonjes, E., Kuhlmann, M., Treusch,
R., Dusterer, S., Tschentscher, T., Schneider, J. R., Spiller, E.,
Moller, T., Bostedt, C., Hoener, M., Shapiro, D. A., Hodgson, K. O.,
Van der Spoel, D., Burmeister, F., Bergh, M., Caleman, C., Huldt, G.,
Seibert, M. M., Maia, F. R. N. C., Lee, R. W., Szoke, A., Timneanu, N.,
and Hajdu, J. (2006) Femtosecond diffractive imaging with a soft-X-ray
free-electron laser, Nat. Phys., 2, 839-843.
18.Chapman, H. N., Hau-Riege, S. P., Bogan, M. J.,
Bajt, S., Barty, A., Boutet, S., Marchesini, S., Frank, M., Woods, B.
W., Benner, W. H., London, R. A., Rohner, U., Szoke, A., Spiller, E.,
Moller, T., Bostedt, C., Shapiro, D. A., Kuhlmann, M., Treusch, R.,
Plonjes, E., Burmeister, F., Bergh, M., Caleman, C., Huldt, G.,
Seibert, M. M., and Hajdu, J. (2007) Femtosecond time-delay X-ray
holography, Nature, 448, 676-679.
19.Weierstall, U., Spence, J. C. H., and Doak, R. B.
(2012) Injector for scattering measurements on fully solvated
biospecies, Rev. Sci. Instrum., 83, 035108-035120.
20.Deponte, D. P., Mckeown, J. T., Weierstall, U.,
Doak, R. B., and Spence, J. C. H. (2011) Towards ETEM serial
crystallography: electron diffraction from liquid jets,
Ultramicroscopy, 111, 824-827.
21.Liu, W., Wacker, D., Wang, C., Abola, E., and
Cherezov, V. (2014) Femtosecond crystallography of membrane proteins in
the lipidic cubic phase, Philos. Trans. R. Soc. B Biol. Sci.,
369, 20130314-20130324.
22.Landau, E. M., and Rosenbusch, J. P. (1996)
Lipidic cubic phases: a novel concept for the crystallization of
membrane proteins, Proc. Natl. Acad. Sci. USA, 93,
14532-14535.
23.Wadsten, P., Wohri, A. B., Snijder, A., Katona,
G., Gardiner, A. T., Cogdell, R. J., Neutze, R., and Engstrom, S.
(2006) Lipidic sponge phase crystallization of membrane proteins, J.
Mol. Biol., 364, 44-53.
24.Johansson, L. C., Arnlund, D., White, T. A.,
Katona, G., DePonte, D. P., Weierstall, U., Doak, R. B., Shoeman, R.
L., Lomb, L., Malmerberg, E., Davidsson, J., Nass, K., Liang, M.,
Andreasson, J., Aquila, A., Bajt, S., Barthelmess, M., Barty, A.,
Bogan, M. J., Bostedt, C., Bozek, J. D., Caleman, C., Coffee, R.,
Coppola, N., Ekeberg, T., Epp, S. W., Erk, B., Fleckenstein, H.,
Foucar, L., Graafsma, H., Gumprecht, L., Hajdu, J., Hampton, C. Y.,
Hartmann, R., Hartmann, A., Hauser, G., Hirsemann, H., Holl, P.,
Hunter, M. S., Kassemeyer, S., Kimmel, N., Kirian, R. A., Maia, F. R.
N. C., Marchesini, S., Martin, A. V., Reich, C., Rolles, D., Rudek, B.,
Rudenko, A., Schlichting, I., Schulz, J., Seibert, M. M., Sierra, R.
G., Soltau, H., Starodub, D., Stellato, F., Stern, S., Struder, L.,
Timneanu, N., Ullrich, J., Wahlgren, W. Y., Wang, X., Weidenspointner,
G., Wunderer, C., Fromme, P., Chapman, H. N., Spence, J. C. H., and
Neutze, R. (2012) Lipidic phase membrane protein serial femtosecond
crystallography, Nat. Methods, 9, 263-265.
25.Beerlink, A., Wilbrandt, P.-J., Ziegler, E.,
Carbone, D., Metzger, T. H., and Salditt, T. (2008) X-ray structure
analysis of free-standing lipid membranes facilitated by micromachined
apertures, Langmuir, 24, 4952-4958.
26.Chapman, H. N., Fromme, P., Barty, A., White, T.
A., Kirian, R. A., Aquila, A., Hunter, M. S., Schulz, J., DePonte, D.
P., Weierstall, U., Doak, R. B., Maia, F. R. N. C., Martin, A. V.,
Schlichting, I., Lomb, L., Coppola, N., Shoeman, R. L., Epp, S. W.,
Hartmann, R., Rolles, D., Rudenko, A., Foucar, L., Kimmel, N.,
Weidenspointner, G., Holl, P., Liang, M., Barthelmess, M., Caleman, C.,
Boutet, S., Bogan, M. J., Krzywinski, J., Bostedt, C., Bajt, S.,
Gumprecht, L., Rudek, B., Erk, B., Schmidt, C., Homke, A., Reich, C.,
Pietschner, D., Struder, L., Hauser, G., Gorke, H., Ullrich, J.,
Herrmann, S., Schaller, G., Schopper, F., Soltau, H., Kuhnel, K.-U.,
Messerschmidt, M., Bozek, J. D., Hau-Riege, S. P., Frank, M., Hampton,
C. Y., Sierra, R. G., Starodub, D., Williams, G. J., Hajdu, J.,
Timneanu, N., Seibert, M. M., Andreasson, J., Rocker, A., Jonsson, O.,
Svenda, M., Stern, S., Nass, K., Andritschke, R., Schroter, C.-D.,
Krasniqi, F., Bott, M., Schmidt, K. E., Wang, X., Grotjohann, I.,
Holton, J. M., Barends, T. R. M., Neutze, R., Marchesini, S., Fromme,
R., Schorb, S., Rupp, D., Adolph, M., Gorkhover, T., Andersson, I.,
Hirsemann, H., Potdevin, G., Graafsma, H., Nilsson, B., and Spence, J.
C. H. (2011) Femtosecond X-ray protein nanocrystallography,
Nature, 470, 73-77.
27.Fraser, J. S., Van den Bedem, H., Samelson, A.
J., Lang, P. T., Holton, J. M., Echols, N., and Alber, T. (2011)
Accessing protein conformational ensembles using room-temperature X-ray
crystallography, Proc. Natl. Acad. Sci. USA, 108,
16247-16252.
28.Bourgeois, D., Schotte, F., Brunori, M., and
Vallone, B. (2007) Time-resolved methods in biophysics. 6.
Time-resolved Laue crystallography as a tool to investigate
photo-activated protein dynamics, Photochem. Photobiol. Sci.,
6, 1047-1056.
29.Stowell, M. H. (1997) Light-induced structural
changes in photosynthetic reaction center: implications for mechanism
of electron–proton transfer, Science, 276,
812-816.
30.Ihee, H., Rajagopal, S., Srajer, V., Pahl, R.,
Anderson, S., Schmidt, M., Schotte, F., Anfinrud, P. A., Wulff, M., and
Moffat, K. (2005) From the cover: visualizing reaction pathways in
photoactive yellow protein from nanoseconds to seconds, Proc. Natl.
Acad. Sci. USA, 102, 7145-7150.
31.Ren, Z., Perman, B., Srajer, V., Teng, T.-Y.,
Pradervand, C., Bourgeois, D., Schotte, F., Ursby, T., Kort, R., Wulff,
M., and Moffat, K. (2001) A molecular movie at 1.8 Å resolution
displays the photocycle of photoactive yellow protein, a eubacterial
blue-light receptor, from nanoseconds to seconds,
Biochemistry,40, 13788-13801.
32.Baxter, R. H. G., Ponomarenko, N., Srajer, V.,
Pahl, R., Moffat, K., and Norris, J. R. (2004) Time-resolved
crystallographic studies of light-induced structural changes in the
photosynthetic reaction center, Proc. Natl. Acad. Sci.
USA, 101, 5982-5987.
33.Moffat, K., Szebenyi, D., and Bilderback, D.
(1984) X-ray Laue diffraction from protein crystals, Science,
223, 1423-1425.
34.Schotte, F. (2003) Watching a protein as it
functions with 150-ps time-resolved X-ray crystallography,
Science, 300, 1944-1947.
35.Barends, T. R. M., Foucar, L., Ardevol, A., Nass,
K., Aquila, A., Botha, S., Doak, R. B., Falahati, K., Hartmann, E.,
Hilpert, M., Heinz, M., Hoffmann, M. C., Kofinger, J., Koglin, J. E.,
Kovacsova, G., Liang, M., Milathianaki, D., Lemke, H. T., Reinstein,
J., Roome, C. M., Shoeman, R. L., Williams, G. J., Burghardt, I.,
Hummer, G., Boutet, S., and Schlichting, I. (2015) Direct observation
of ultrafast collective motions in CO myoglobin upon ligand
dissociation, Science, 350, 445-450.
36.Panneels, V., Wu, W., Tsai, C.-J., Nogly, P.,
Rheinberger, J., Jaeger, K., Cicchetti, G., Gati, C., Kick, L. M.,
Sala, L., Capitani, G., Milne, C., Padeste, C., Pedrini, B., Li, X.-D.,
Standfuss, J., Abela, R., and Schertler, G. (2015) Time-resolved
structural studies with serial crystallography: a new light on retinal
proteins, Struct. Dyn., 2, 041718-041726.
37.Ginn, H. M., Messerschmidt, M., Ji, X., Zhang,
H., Axford, D., Gildea, R. J., Winter, G., Brewster, A. S., Hattne, J.,
Wagner, A., Grimes, J. M., Evans, G., Sauter, N. K., Sutton, G., and
Stuart, D. I. (2015) Structure of CPV17 polyhedrin determined by the
improved analysis of serial femtosecond crystallographic data, Nat.
Commun., 6, 6435-6443.
38.Cohen, A. E., Soltis, S. M., Gonzalez, A.,
Aguila, L., Alonso-Mori, R., Barnes, C. O., Baxter, E. L., Brehmer, W.,
Brewster, A. S., Brunger, A. T., Calero, G., Chang, J. F., Chollet, M.,
Ehrensberger, P., Eriksson, T. L., Feng, Y., Hattne, J., Hedman, B.,
Hollenbeck, M., Holton, J. M., Keable, S., Kobilka, B. K., Kovaleva, E.
G., Kruse, A. C., Lemke, H. T., Lin, G., Lyubimov, A. Y., Manglik, A.,
Mathews, I. I., McPhillips, S. E., Nelson, S., Peters, J. W., Sauter,
N. K., Smith, C. A., Song, J., Stevenson, H. P., Tsai, Y.,
Uervirojnangkoorn, M., Vinetsky, V., Wakatsuki, S., Weis, W. I.,
Zadvornyy, O. A., Zeldin, O. B., Zhu, D., and Hodgson, K. O. (2014)
Goniometer-based femtosecond crystallography with X-ray free electron
lasers, Proc. Natl. Acad. Sci. USA, 111,
17122-17127.
39.Lunin, V. Y., Grum-Grzhimailo, A. N., Gryzlova,
E. V., Sinitsyn, D. O., Petrova, T. E., Lunina, N. L., Balabaev, N. K.,
Tereshkina, K. B., Stepanov, A. S., and Krupyanskii, Y. F. (2015)
Efficient calculation of diffracted intensities in the case of
nonstationary scattering by biological macromolecules under XFEL
pulses, Acta Crystallogr. D Biol. Crystallogr., 71,
293-303.
40.Pande, K., Hutchison, C. D. M., Groenhof, G.,
Aquila, A., Robinson, J. S., Tenboer, J., Basu, S., Boutet, S.,
DePonte, D. P., Liang, M., White, T. A., Zatsepin, N. A., Yefanov, O.,
Morozov, D., Oberthuer, D., Gati, C., Subramanian, G., James, D., Zhao,
Y., Koralek, J., Brayshaw, J., Kupitz, C., Conrad, C., Roy-Chowdhury,
S., Coe, J. D., Metz, M., Xavier, P. L., Grant, T. D., Koglin, J. E.,
Ketawala, G., Fromme, R., Rajer, V., Henning, R., Spence, J. C. H.,
Ourmazd, A., Schwander, P., Weierstall, U., Frank, M., Fromme, P.,
Barty, A., Chapman, H. N., Moffat, K., Van Thor, J. J., and Schmidt, M.
(2016) Femtosecond structural dynamics drives the
trans/cis isomerization in photoactive yellow protein,
Science, 352, 725-729.
41.Blankenship, R. E. (2014) Molecular Mechanisms
of Photosynthesis, Wiley/Blackwell, Chichester, West Sussex, p.
296.
42.Nelson, N. (2009) Plant photosystem
I – the most efficient nano-photochemical machine, J.
Nanosci. Nanotechnol., 9, 1709-1713.
43.Jordan, P., Fromme, P., Witt, H. T., Klukas, O.,
Saenger, W., and Krauß, N. (2001) Three-dimensional structure of
cyanobacterial photosystem I at 2.5 Å resolution, Nature,
411, 909-917.
44.Aquila, A., Hunter, M. S., Doak, R. B., Kirian,
R. A., Fromme, P., White, T. A., Andreasson, J., Arnlund, D., Bajt, S.,
Barends, T. R. M., Barthelmess, M., Bogan, M. J., Bostedt, C., Bottin,
H., Bozek, J. D., Caleman, C., Coppola, N., Davidsson, J., DePonte, D.
P., Elser, V., Epp, S. W., Erk, B., Fleckenstein, H., Foucar, L.,
Frank, M., Fromme, R., Graafsma, H., Grotjohann, I., Gumprecht, L.,
Hajdu, J., Hampton, C. Y., Hartmann, A., Hartmann, R., Hau-Riege, S.,
Hauser, G., Hirsemann, H., Holl, P., Holton, J. M., Homke, A.,
Johansson, L., Kimmel, N., Kassemeyer, S., Krasniqi, F., Kuhnel, K.-U.,
Liang, M., Lomb, L., Malmerberg, E., Marchesini, S., Martin, A. V.,
Maia, F. R. N. C., Messerschmidt, M., Nass, K., Reich, C., Neutze, R.,
Rolles, D., Rudek, B., Rudenko, A., Schlichting, I., Schmidt, C.,
Schmidt, K. E., Schulz, J., Seibert, M. M., Shoeman, R. L., Sierra, R.,
Soltau, H., Starodub, D., Stellato, F., Stern, S., Struder, L.,
Timneanu, N., Ullrich, J., Wang, X., Williams, G. J., Weidenspointner,
G., Weierstall, U., Wunderer, C., Barty, A., Spence, J. C. H., and
Chapman, H. N. (2012) Time-resolved protein nanocrystallography using
an X-ray free-electron laser, Opt. Express, 20,
2706-2716.
45.Imamoto, Y., and Kataoka, M. (2007) Structure and
photoreaction of photoactive yellow protein, a structural prototype of
the PAS domain superfamily, Photochem. Photobiol., 83,
40-49.
46.Moffat, K. (1989) Time-resolved macromolecular
crystallography, Annu. Rev. Biophys. Biophys. Chem., 18,
309-332.
47.Tenboer, J., Basu, S., Zatsepin, N., Pande, K.,
Milathianaki, D., Frank, M., Hunter, M., Boutet, S., Williams, G. J.,
Koglin, J. E., Oberthuer, D., Heymann, M., Kupitz, C., Conrad, C., Coe,
J., Roy-Chowdhury, S., Weierstall, U., James, D., Wang, D., Grant, T.,
Barty, A., Yefanov, O., Scales, J., Gati, C., Seuring, C., Srajer, V.,
Henning, R., Schwander, P., Fromme, R., Ourmazd, A., Moffat, K., Van
Thor, J. J., Spence, J. C. H., Fromme, P., Chapman, H. N., and Schmidt,
M. (2014) Time-resolved serial crystallography captures high-resolution
intermediates of photoactive yellow protein, Science,
346, 1242-1246.
48.Deisenhofer, J., Epp, O., Miki, K., Huber, R.,
and Michel, H. (1985) Structure of the protein subunits in the
photosynthetic reaction center of Rhodopseudomonas
viridis at 3 Å resolution, Nature, 318,
618-624.
49.Fromme, P., and Witt, H. T. (1998) Improved
isolation and crystallization of photosystem I for structural analysis,
Biochim. Biophys. Acta, 1365, 175-184.
50.Johansson, L. C., Arnlund, D., Katona, G., White,
T. A., Barty, A., DePonte, D. P., Shoeman, R. L., Wickstrand, C.,
Sharma, A., Williams, G. J., Aquila, A., Bogan, M. J., Caleman, C.,
Davidsson, J., Doak, R. B., Frank, M., Fromme, R., Galli, L.,
Grotjohann, I., Hunter, M. S., Kassemeyer, S., Kirian, R. A., Kupitz,
C., Liang, M., Lomb, L., Malmerberg, E., Martin, A. V., Messerschmidt,
M., Nass, K., Redecke, L., Seibert, M. M., Sjohamn, J., Steinbrener,
J., Stellato, F., Wang, D., Wahlgren, W. Y., Weierstall, U.,
Westenhoff, S., Zatsepin, N. A., Boutet, S., Spence, J. C. H.,
Schlichting, I., Chapman, H. N., Fromme, P., and Neutze, R. (2013)
Structure of a photosynthetic reaction center determined by serial
femtosecond crystallography, Nat. Commun., 4,
2911-2917.
51.Vinyard, D. J., Ananyev, G. M., and Charles
Dismukes, G. (2013) Photosystem II: the reaction center of oxygenic
photosynthesis, Annu. Rev. Biochem., 82, 577-606.
52.Kok, B., Forbush, B., and McGloin, M. (1970)
Cooperation of charges in photosynthetic O2 evolution. I. A
linear four-step mechanism, Photochem. Photobiol., 11,
457-475.
53.Zouni, A., Witt, H.-T., Kern, J., Fromme, P.,
Krauss, N., Saenger, W., and Orth, P. (2001) Crystal structure of
photosystem II from Synechococcus elongatus at 3.8
Å resolution, Nature, 409, 739-743.
54.Guskov, A., Kern, J., Gabdulkhakov, A., Broser,
M., Zouni, A., and Saenger, W. (2009) Cyanobacterial photosystem II at
2.9-Å resolution and the role of quinones, lipids, channels and
chloride, Nat. Struct. Mol. Biol., 16, 334-342.
55.Loll, B., Kern, J., Saenger, W., Zouni, A., and
Biesiadka, J. (2005) Towards complete cofactor arrangement in the 3.0
Å resolution structure of photosystem II, Nature,
438, 1040-1044.
56.Ferreira, K. N. (2004) Architecture of the
photosynthetic oxygen-evolving center, Science, 303,
1831-1838.
57.Biesiadka, J., Loll, B., Kern, J., Irrgang,
K.-D., and Zouni, A. (2004) Crystal structure of cyanobacterial
photosystem II at 3.2 Å resolution: a closer look at the
Mn-cluster, Phys. Chem. Chem. Phys., 6, 4733-4736.
58.Kamiya, N., and Shen, J.-R. (2003) Crystal
structure of oxygen-evolving photosystem II from
Thermosynechococcus vulcanus at 3.7-Å resolution,
Proc. Natl. Acad. Sci. USA, 100, 98-103.
59.Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya,
N. (2011) Crystal structure of oxygen-evolving photosystem II at a
resolution of 1.9 Å, Nature, 473, 55-60.
60.Yano, J., Kern, J., Irrgang, K.-D., Latimer, M.
J., Bergmann, U., Glatzel, P., Pushkar, Y., Biesiadka, J., Loll, B.,
Sauer, K., Messinger, J., Zouni, A., and Yachandra, V. K. (2005) X-ray
damage to the Mn4Ca complex in single crystals of
photosystem II: a case study for metalloprotein crystallography,
Proc. Natl. Acad. Sci. USA, 102, 12047-12052.
61.Glockner, C., Kern, J., Broser, M., Zouni, A.,
Yachandra, V., and Yano, J. (2013) Structural changes of the
oxygen-evolving complex in photosystem II during the catalytic cycle,
J. Biol. Chem., 288, 22607-22620.
62.Grabolle, M., Haumann, M., Muller, C., Liebisch,
P., and Dau, H. (2006) Rapid loss of structural motifs in the manganese
complex of oxygenic photosynthesis by X-ray irradiation at 10-300 K,
J. Biol. Chem., 281, 4580-4588.
63.Kern, J., Alonso-Mori, R., Hellmich, J., Tran,
R., Hattne, J., Laksmono, H., Glockner, C., Echols, N., Sierra, R. G.,
Sellberg, J., Lassalle-Kaiser, B., Gildea, R. J., Glatzel, P.,
Grosse-Kunstleve, R. W., Latimer, M. J., McQueen, T. A., DiFiore, D.,
Fry, A. R., Messerschmidt, M., Miahnahri, A., Schafer, D. W., Seibert,
M. M., Sokaras, D., Weng, T.-C., Zwart, P. H., White, W. E., Adams, P.
D., Bogan, M. J., Boutet, S., Williams, G. J., Messinger, J., Sauter,
N. K., Zouni, A., Bergmann, U., Yano, J., and Yachandra, V. K. (2012)
Room temperature femtosecond X-ray diffraction of photosystem II
microcrystals, Proc. Natl. Acad. Sci. USA, 109,
9721-9726.
64.Kern, J., Alonso-Mori, R., Tran, R., Hattne, J.,
Gildea, R. J., Echols, N., Glockner, C., Hellmich, J., Laksmono, H.,
Sierra, R. G., Lassalle-Kaiser, B., Koroidov, S., Lampe, A., Han, G.,
Gul, S., DiFiore, D., Milathianaki, D., Fry, A. R., Miahnahri, A.,
Schafer, D. W., Messerschmidt, M., Seibert, M. M., Koglin, J. E.,
Sokaras, D., Weng, T.-C., Sellberg, J., Latimer, M. J.,
Grosse-Kunstleve, R. W., Zwart, P. H., White, W. E., Glatzel, P.,
Adams, P. D., Bogan, M. J., Williams, G. J., Boutet, S., Messinger, J.,
Zouni, A., Sauter, N. K., Yachandra, V. K., Bergmann, U., and Yano, J.
(2013) Simultaneous femtosecond X-ray spectroscopy and diffraction of
photosystem II at room temperature, Science, 340,
491-495.
65.Gabdulkhakov, A., and Dontsova, M. (2013)
Structural studies on photosystem II of cyanobacteria, Biochemistry
(Moscow), 78, 1524-1538.
66.Suga, M., Akita, F., Hirata, K., Ueno, G.,
Murakami, H., Nakajima, Y., Shimizu, T., Yamashita, K., Yamamoto, M.,
Ago, H., and Shen, J.-R. (2014) Native structure of photosystem II at
1.95 Å resolution viewed by femtosecond X-ray pulses,
Nature, 517, 99-103.
67.Young, I. D., Ibrahim, M., Chatterjee, R., Gul,
S., Fuller, F. D., Koroidov, S., Brewster, A. S., Tran, R.,
Alonso-Mori, R., Kroll, T., Michels-Clark, T., Laksmono, H., Sierra, R.
G., Stan, C. A., Hussein, R., Zhang, M., Douthit, L., Kubin, M., de
Lichtenberg, C., Vo Pham, L., Nilsson, H., Cheah, M. H., Shevela, D.,
Saracini, C., Bean, M. A., Seuffert, I., Sokaras, D., Weng, T.-C.,
Pastor, E., Weninger, C., Fransson, T., Lassalle, L., Brauer, P.,
Aller, P., Docker, P. T., Andi, B., Orville, A. M., Glownia, J. M.,
Nelson, S., Sikorski, M., Zhu, D., Hunter, M. S., Lane, T. J., Aquila,
A., Koglin, J. E., Robinson, J., Liang, M., Boutet, S., Lyubimov, A.
Y., Uervirojnangkoorn, M., Moriarty, N. W., Liebschner, D., Afonine, P.
V., Waterman, D. G., Evans, G., Wernet, P., Dobbek, H., Weis, W. I.,
Brunger, A. T., Zwart, P. H., Adams, P. D., Zouni, A., Messinger, J.,
Bergmann, U., Sauter, N. K., Kern, J., Yachandra, V. K., and Yano, J.
(2016) Structure of photosystem II and substrate binding at room
temperature, Nature, 540, 453-457.
68.Suga, M., Akita, F., Sugahara, M., Kubo, M.,
Nakajima, Y., Nakane, T., Yamashita, K., Umena, Y., Nakabayashi, M.,
Yamane, T., Nakano, T., Suzuki, M., Masuda, T., Inoue, S., Kimura, T.,
Nomura, T., Yonekura, S., Yu, L.-J., Sakamoto, T., Motomura, T., Chen,
J.-H., Kato, Y., Noguchi, T., Tono, K., Joti, Y., Kameshima, T.,
Hatsui, T., Nango, E., Tanaka, R., Naitow, H., Matsuura, Y., Yamashita,
A., Yamamoto, M., Nureki, O., Yabashi, M., Ishikawa, T., Iwata, S., and
Shen, J.-R. (2017) Light-induced structural changes and the site of O=O
bond formation in PSII caught by XFEL, Nature, 543,
131-135.
69.Voet, D., and Voet, J. G. (2011)
Biochemistry, John Wiley & Sons.
70.Hucho, F. (1986) Neurochemistry: Fundamentals
and Concepts, VCH, Weinheim.
71.Frank, M., Carlson, D. B., Hunter, M. S.,
Williams, G. J., Messerschmidt, M., Zatsepin, N. A., Barty, A., Benner,
W. H., Chu, K., Graf, A. T., Hau-Riege, S. P., Kirian, R. A., Padeste,
C., Pardini, T., Pedrini, B., Segelke, B., Seibert, M. M., Spence, J.
C. H., Tsai, C.-J., Lane, S. M., Li, X.-D., Schertler, G., Boutet, S.,
Coleman, M., and Evans, J. E. (2014) Femtosecond X-ray diffraction from
two-dimensional protein crystals, IUCrJ, 1, 95-100.
72.Pedrini, B., Tsai, C.-J., Capitani, G., Padeste,
C., Hunter, M. S., Zatsepin, N. A., Barty, A., Benner, W. H., Boutet,
S., Feld, G. K., Hau-Riege, S. P., Kirian, R. A., Kupitz, C.,
Messerschmitt, M., Ogren, J. I., Pardini, T., Segelke, B., Williams, G.
J., Spence, J. C. H., Abela, R., Coleman, M., Evans, J. E., Schertler,
G. F. X., Frank, M., and Li, X.-D. (2014) 7 Å resolution in
protein two-dimensional-crystal X-ray diffraction at Linac Coherent
Light Source, Philos. Trans. R. Soc. B Biol. Sci., 369,
20130500-20130505.
73.Fujiyoshi, Y. (2011) Electron crystallography for
structural and functional studies of membrane proteins,
Microscopy, 60, S149-S159.
74.Srivastava, S. K., Gayathri, S., Manjasetty, B.
A., and Gopal, B. (2012) Analysis of conformational variation in
macromolecular structural models, PLoS One, 7,
e39993.
75.Davis, S. J. (1999) Bacteriophytochromes:
phytochrome-like photoreceptors from nonphotosynthetic eubacteria,
Science, 286, 2517-2520.
76.Auldridge, M. E., and Forest, K. T. (2011)
Bacterial phytochromes: more than meets the light, Crit. Rev.
Biochem. Mol. Biol., 46, 67-88.
77.Edlund, P., Takala, H., Claesson, E., Henry, L.,
Dods, R., Lehtivuori, H., Panman, M., Pande, K., White, T., Nakane, T.,
Berntsson, O., Gustavsson, E., Bath, P., Modi, V., Roy-Chowdhury, S.,
Zook, J., Berntsen, P., Pandey, S., Poudyal, I., Tenboer, J., Kupitz,
C., Barty, A., Fromme, P., Koralek, J. D., Tanaka, T., Spence, J.,
Liang, M., Hunter, M. S., Boutet, S., Nango, E., Moffat, K., Groenhof,
G., Ihalainen, J., Stojkovic, E. A., Schmidt, M., and Westenhoff, S.
(2016) The room temperature crystal structure of a bacterial
phytochrome determined by serial femtosecond crystallography, Sci.
Rep., 6, 35279-35288.