Western Blotting-Based Quantitative Measurement of Myosin II Regulatory Light Chain Phosphorylation in Small Amounts of Non-muscle Cells
O. A. Kazakova1, A. Y. Khapchaev1,2,a*, A. A. Ragimov3, E. L. Salimov3, and V. P. Shirinsky1,2
1National Medical Research Center for Cardiology, 121552 Moscow, Russia2Lomonosov Moscow State University, Faculty of Fundamental Medicine, 119192 Moscow, Russia
3I. M. Sechenov First Moscow State Medical University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received July 6, 2018; Revised September 10, 2018; Accepted September 10, 2018
Myosin II is the main molecular motor in the actomyosin-dependent motility in cells. Phosphorylation of the myosin regulatory light chain (RLC) at Ser19 is a prerequisite for smooth muscle/non-muscle myosin II activation and serves as a biochemical equivalent of myosin II activity. Simultaneous phosphorylation at Thr18 further promotes the myosin II ATPase activity. A number of methods have been developed to measure myosin RLC phosphorylation at Ser19 or di-phosphorylation at Thr18/Ser19. While these methods are straightforward and robust in myosin-rich muscle tissues, they demonstrate limited applicability in non-muscle cells that have low myosin II content and are usually available in lesser amounts than muscle tissue. Because of this, dynamic analysis of RLC phosphorylation in multiple samples of non-muscle cells is difficult and requires large number of cells. The use of phospho-specific antibodies increases detection sensitivity but allows estimation of only relative levels of RLC phosphorylation at specific residues, which makes it difficult to estimate the physiologic relevancy of the observed changes in RLC phosphorylation. To measure RLC phosphorylation in small amounts of non-muscle cells, we used external calibration standards of non-phosphorylated and in vitro phosphorylated RLC in standard SDS-PAGE and Western blot procedures with phospho-specific RLC antibodies. Here, we describe the method in detail and demonstrate its application for quantitative measurement of myosin RLC phosphorylation in endothelial cells in response to natural agonists (thrombin or insulin) and intact human platelets. We discuss the advantages and limitations of the proposed method vs other approaches for measuring myosin RLC phosphorylation in non-muscle cells.
KEY WORDS: myosin regulatory light chain, non-muscle cells, phosphorylation, Western blotting, quantitative measurementDOI: 10.1134/S0006297919010024
Abbreviations: CaM, calmodulin; DFP, diisopropyl fluorophosphate; DMEM, Dulbecco’s modified essential medium; DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; IPTG, isopropyl β-D-thiogalactopyranoside; MLCK, myosin light chain kinase; MOPS, 3-(N-morpholino)propanesulfonic acid; MYPT1, myosin phosphatase target subunit 1; NMII, non-muscle myosin II; PBS, phosphate buffered saline; PVDF, polyvinylidene difluoride; RLC, regulatory light chain; ROCK, Rho-associated protein kinase; SMMII, smooth muscle myosin II; TBS, Tris buffered saline.
Myosin II is a ubiquitous motor protein that interacts with actin
filaments and couples ATP hydrolysis to conformational changes in order
to generate mechanical force for a variety of intracellular processes
in eukaryotic cells. Both non-muscle myosin II (NMII) and structurally
related smooth muscle myosin II (SMMII) require activation via
phosphorylation of myosin regulatory light chains (RLC) at Ser19.
Simultaneous phosphorylation at Thr18 exerts additional stimulatory
effect on myosin II ATPase activity (see [1, 2] for reviews). There is a growing list of protein
kinases that phosphorylate these sites and activate NMII in cells (see
[3, 4] for review). However,
two protein kinases, myosin light chain kinase (MYLK1, MLCK) and
Rho-associated protein kinase (ROCK), are currently considered to be
the major myosin II activators in non-muscle cells. MLCK is a
Ca2+-calmodulin (CaM)-activated protein kinase dedicated to
activate both NMII and SMMII (reviewed recently in [5]). ROCK is regarded as a major
Ca2+-independent myosin II activator downstream of the small
GTPase RhoA [6, 7]. ROCK can
directly phosphorylate RLC in vitro and in vivo [6]; it also phosphorylates myosin phosphatase target
subunit 1 (MYPT1) and increases myosin RLC net phosphorylation due to
the inhibition of myosin phosphatase activity [8].
A variety of experimental approaches have been developed for measuring both RLC mono-phosphorylation at Ser19 (P-RLC) and di-phosphorylation at Thr18/Ser19 (PP-RLC). These methods include isoelectric focusing after myosin isolation in the presence of pyrophosphate [9], urea/glycerol polyacrylamide gel electrophoresis [10], and the Phos-tag (phosphate-binding tag) technology [11-14]. All these methods exist in various detection formats and differ in the sensitivity and ability to differentiate between specific phosphorylation sites; they generally require relatively large amounts of starting sample material and/or labor-consuming sample handling. From these standpoints, a method based on standard SDS-PAGE and Western blot would be an affordable alternative for measuring and monitoring P-RLC and/or PP-RLC dynamics in non-muscle cells. Although the use of Western blot as a quantitative method is debatable, there is a methodology to obtain reliable quantitative data from Western blots [15, 16].
In this study, we describe a method for quantitative measurement of myosin P-RLC and PP-RLC by means of standard SDS-PAGE and Western blot with phospho-specific anti-RLC antibodies and external in vitro phosphorylated RLC standards. We demonstrate the application of this method to quantify changes in RLC phosphorylation in EA.hy926 endothelial cells after their stimulation with thrombin (endothelial hyperpermeability inducer [17]) or insulin that exerts a protective effect on the endothelial barrier function [18]. Using the proposed method, we also quantified RLC phosphorylation in quiescent human platelets. Finally, we discuss the problem of choice between the proposed method and alternative techniques for evaluation of myosin RLC phosphorylation in non-muscle cells.
MATERIALS AND METHODS
Materials. All biochemical reagents were of analytical grade and purchased from Sigma-Aldrich, Calbiochem, Bio-Rad, Thermo Fisher Scientific, Merck, Enzyme Research South Bend, BD Biosciences, and Enzo Life Sciences (USA). For Western blot analysis, polyvinylidenedifluoride (PVDF) membrane (Amersham, USA) and either West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, USA) or Clarity Western ECL Substrate (BioRad, USA) were used. The primary antibodies were pan-RLC antibody (ProteinTech, USA/UK/China; #10324-1-AP), antibody against phospho-Ser19-RLC (P-RLC) (Cell Signaling, USA; #3675), antibody against phospho-Ser19/Thr18-RLC (PP-RLC) (Cell Signaling; #3674), and anti-glyceraldehyde phosphate dehydrogenase (GAPDH) antibody (Sigma-Aldrich; #G8795). The secondary antibodies were horseradish peroxidase-conjugated anti-rabbit IgG (Sigma-Aldrich; #A0545) or anti-mouse IgG (Abcam, USA; #ab97046). Thrombin from Enzyme Research; insulin from Sigma, glutathione-Sepharose from Thermo Fisher Scientific, Phos-tag from NARD Ltd. (Japan), cell culture plasticware from Corning Costar (USA) and Greiner-bio-one (Austria), Dulbecco’s modified essential medium (DMEM) from PanEco (Russia), and fetal bovine serum (FBS) from HyClone (USA) were used. The pGEX4T-1/human myosin RLC construct was a kind gift from Dr. D. M. Watterson (Northwestern University, Chicago, IL, USA). Human platelets were acquired from the Blood Center of the Sechenov First Moscow State Medical University (Moscow, Russia).
Preparation of mono- and di-phosphorylated RLC standards. Calmodulin (CaM) was isolated from calf brain as described in [19]. MLCK and myosin RLC were isolated from turkey gizzards as described in [20]. The concentrations of tissue-purified MLCK and RLC were determined by the method of Bradford [21]. Recombinant GST-fused human RLC was expressed in Escherichia coli BL21pLys cells; expression was induced with 1 mM IPTG followed by incubation of the bacterial cell culture at 37°C for 3 h. The recombinant fusion protein was isolated on glutathione-Sepharose according to the manufacturer’s instructions. The GST-tag was cleaved off with thrombin (0.77 U of thrombin per 1 mg of GST-RLC) for 30 min at 30°C in 50 mM Tris-HCl (pH 8.0) containing 10 mM reduced glutathione; the reaction was terminated by adding 1 μM diisopropyl fluorophosphate (DFP). The cleavage of GST-RLC was confirmed by SDS-PAGE using turkey gizzard RLC as a standard. The obtained RLC preparation was used in subsequent experiments without further purification.
To phosphorylate turkey gizzard RLC at Ser19, purified RLC at a final concentration of 20 μM was incubated with 6 nM MLCK for 20 min at 30°C in a buffer containing 10 mM 3-(N-morpholino)propanesulfonic acid (MOPS) (pH 7.0), 2 mM MgCl2, 100 mM KCl, 20 mM NaCl, 1 mM dithiothreitol (DTT), 0.5 mM ATP, 1 mM CaCl2, and 300 nM CaM. To obtain di-phosphorylated turkey gizzard RLC, the MLCK concentration and incubation time were increased to 1.8 μM and 100 min, respectively. To obtain recombinant human RLC phosphorylated at Ser19, the same protocol was used with minor modifications, i.e., the incubation time, temperature, MLCK, and CaM concentrations were increased to 60 min, 37°C, 0.5 μM, and 1 μM, respectively. To obtain di-phosphorylated recombinant human RLC, the reaction was let to proceed for 300 min at 37°C in a buffer containing 10 mM MOPS (pH 7.0), 10 mM MgCl2, 100 mM KCl, 20 mM NaCl, 2 mM DTT, 2 mM ATP, 5 mM CaCl2, 1.5 μM CaM, and 2 μM MLCK. The state of RLC phosphorylation was confirmed by urea/glycerol gel electrophoresis (Fig. 1a).
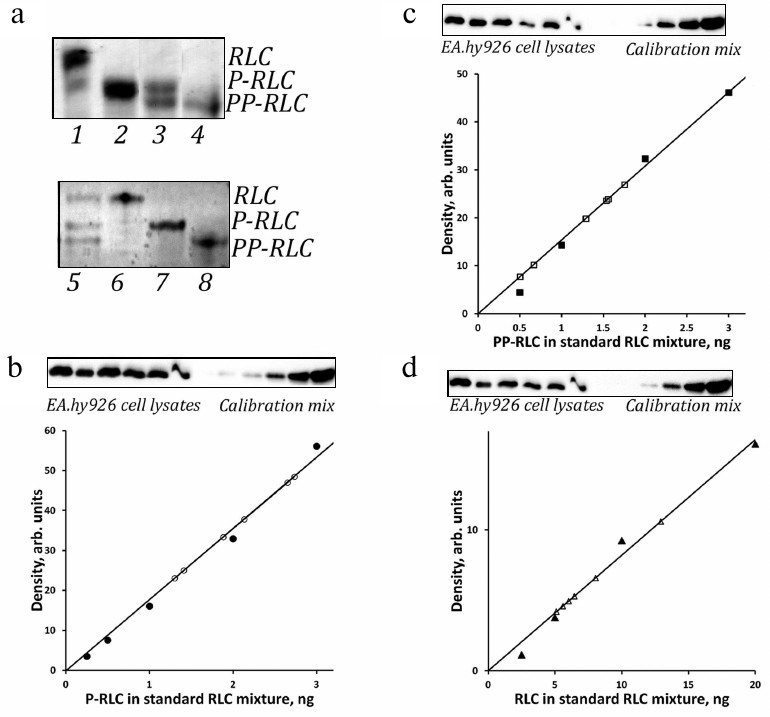
Fig. 1. Quantitation of P-RLC, PP-RLC, and total RLC in lysates of endothelial cells using a single blot with RLC standards. a) Top panel, representative Coomassie R-250-stained urea/glycerol gel with turkey gizzard smooth muscle myosin RLC (lane 1), P-RLC (lane 2), an equimolar mixture of P-RLC and PP-RLC (lane 3), and PP-RLC (lane 4). Bottom panel: representative Coomassie R-250-stained urea/glycerol gel with an equimolar mixture of recombinant human RLC, P-RLC, and PP-RLC (lane 5), RLC (lane 6), P-RLC (lane 7), and PP-RLC (lane 8). Positions of RLC species are shown on the right. b-d) Representative calibration curves for P-RLC (b), PP-RLC (c), and total RLC (d) derived by incrementally loading RLC standard mixtures (closed circles); open circles, signals obtained from endothelial cell samples. The corresponding Western blots demonstrating empirically adjusted endothelial cell samples and RLC calibration standards are shown above the plots. Calibration plots represent linear parts of the full range calibration curves derived from the same set of RLC calibration standards that were exposed for different time intervals in order to align with the ECL signals from experimental RLC species.
Electrophoresis and Western blotting. For standard SDS-PAGE [22], purified proteins and cell lysates were resolved in 12% gels. For Phos-tag gel electrophoresis, the same Laemmli samples were resolved in 12% gels containing the Phos-tag reagent as described in [11]. Electrophoresis in urea/glycerol polyacrylamide gels was carried out as described in [10]. Briefly, the samples were dissolved in a buffer containing 20 mM Tris-HCl (pH 6.8), 9 M urea, 10 mM DTT, and 0.05% bromophenol blue; electrophoresis (MiniProtean III; BioRad) was carried out in a buffer containing 4 mM Tris/4.6 mM glycine, pH 8.6, 14 mM 2-mercaptoethanol at 330 V for 50 min and at 400 V for the next 240 min. The gels were stained with Coomassie R-250; alternatively, proteins were electrotransferred onto PVDF membrane by the method of Towbin et al. [23] in 25 mM Tris/192 mM glycine, pH 8.6, 20% ethanol at 300 mA for 1 h. After the transfer, the membranes were incubated in 0.25% glutaraldehyde solution in H2O for 20 min at room temperature to covalently link the proteins to the membrane. For Western blotting, the membranes were blocked in TBS (20 mM Tris-HCl, pH 7.6, 165 mM NaCl) containing 0.05% Tween-20 and 5% fat-free milk for 1 h at room temperature. The membranes were washed with TBS to remove residual non-bound milk proteins and then incubated with anti-pan RLC, phosphospecific anti-RLC and then secondary peroxidase-conjugated antibodies overnight at 4°C in TBS supplemented with 0.1% bovine serum albumin and 0.1% NaN3. Primary antibodies were used in the following working dilutions: pan-RLC antibody, 1 : 1000; antibody against P-RLC(Ser19), 1 : 2000; antibody against PP-RLC(Ser19/Thr18), 1 : 2000; anti-GAPDH antibody, 1 : 10,000. For multiple re-probing, bound antibodies were stripped by the method of Yeung and Stanley [24]. Briefly, after exposure, PVDF membranes were washed 3 times for 5 min in TBS at room temperature and then stripped by two 5-min incubations in 20 mM Tris-HCl (pH 8.0) containing 6 M guanidine, 0.2% NP-40, and freshly added 0.1 M 2-mercaptoethanol. Then, the membranes were washed 4 times for 5 min in TBS containing 0.1% Tween 20 at room temperature, rinsed in distilled water, dried out on air, and stored at room temperature until re-probing. Chemiluminescent signals were acquired with a Fusion SL3500W detection system (Vilber Lourmat, France) in the video mode. To ensure the linearity of the chemiluminescence signal, images obtained at varying exposure times were quantitatively processed using the ImageJ v1.48 software (NIH, USA).
Cells and treatments. EA.hy926 cells (ATCC, USA) were cultured in gelatin-precoated 35-mm dishes (100-mm dishes were used when the cells were analyzed by urea/glycerol electrophoresis) at 37°C in 5% CO2 atmosphere in DMEM supplemented with 10% FBS and antibiotics. The medium was changed every other day. In all experiments, the cells were incubated in serum-free DMEM for 1 h before the challenges. α-Thrombin and insulin were dissolved in DMEM and 0.1% BSA in PBS, respectively, and used at a final concentration of 100 nM. Freshly prepared thrombin and insulin working solutions were used. The effect of thrombin or insulin on RLC phosphorylation in EA.hy926 cells was analyzed after 12- and 15-min incubation with thrombin and insulin, respectively.
Platelets from 50 ml of platelet-rich human plasma were washed off from the serum with Hanks’ solution (30 ml) by three consecutive centrifugation/resuspension steps (800g, 10 min, 4°C) within 2 h after isolation.
Quantitation of RLC phosphorylation in cells. EA.hy926 cells were cultured in 35-mm dishes to confluence. On the day of the experiment, the cells were deprived of serum and challenged with 100 nM α-thrombin or insulin. At defined time points, the cells were washed with ice-cold PBS and lysed in 100 μl of 2× Laemmli buffer. For analysis of RLC phosphorylation in intact platelets, equal volumes of washed platelet suspension from each donor were placed in two 1.5-ml tubes (final volume, 0.5-0.7 ml). Platelets were pelleted by centrifugation at 5000g for 5 min at 4°C and lysed in 200 μl of 3× Laemmli buffer. The standard RLC mixture was prepared by combining non-phosphorylated RLC, P-RLC, and PP-RLC at a 60 : 20 : 20 molar ratio. Increasing amounts of the standard RLC mixture and empirically established volumes of cell lysates were transferred onto the same PVDF membrane and sequentially stained with RLC antibodies. Between the staining procedures, the membranes were stripped from previous antibodies as described above. Complete removal of the antibodies from the membranes was confirmed in separate experiments by developing the washed membranes after preincubation with secondary antibodies. No detectable ECL signal was observed in the region corresponding to RLC. The amounts of P-RLC and PP-RLC in the lysates were determined from the calibration curves using P-RLC and PP-RLC antibodies, respectively (Fig. 1, b and c). The total RLC load was determined using the pan-RLC antibody (Fig. 1d). Anti-GAPDH antibody was used as an additional protein loading control in parallel with normalization using the pan-RLC antibody. The fraction of phosphorylated RLC was calculated as a ratio between the amount of phosphorylated RLC and total RLC amount.
Statistical analysis of data. The data from at least three independent experiments were pooled and presented as mean ± standard deviation (SD). Differences between the groups were considered significant at p < 0.05 (Student’s t-test).
RESULTS
Validation of phospho-RLC standards and anti-RLC antibodies. Firstly, we prepared and validated RLC standards for analysis of RLC phosphorylation in cells. MLCK is known to readily phosphorylate RLC at Ser19, while subsequent phosphorylation at Thr18 requires prolonged incubation times and significantly higher amounts of the enzyme [25]. Purified turkey gizzard myosin RLC and human myosin RLC were phosphorylated with purified MLCK in vitro to obtain P-RLC(Ser19) and PP-RLC(Thr18/Ser19) as described in “Materials and Methods”. The completeness of RLC mono- and di-phosphorylation was confirmed by the electrophoretic shift of phosphorylated RLC forms in the urea/glycerol polyacrylamide gel (Fig. 1a). As shown in Fig. 1a, tissue-purified non-phosphorylated RLC preparation (top panel, lane 1) contained 3% of mono-phosphorylated RLC (based on the densitometry of the Coomassie-stained gel). After incubation of 20 μM RLC with 6 nM purified MLCK for 20 min, RLC appeared in the mono-phosphorylated form (Fig. 1a, top panel, lane 2). Prolonged RLC incubation with high MLCK concentrations resulted in apparently complete RLC di-phosphorylation (Fig. 1a, top panel, lane 4). Figure 1a, bottom panel, demonstrates recombinant human RLC standards.
Next, to validate the specificity of phospho-RLC antibodies, we resolved EA.hy926 cell lysates in either urea/glycerol or Phos-tag gels and confirmed that the chosen P-RLC(Ser19) and PP-RLC(Ser19/Thr18) antibodies did not cross-react with other RLC phospho-species that could potentially be generated in thrombin/insulin-stimulated EA.hy926 cells (data not shown). As anticipated, these antibodies recognized the corresponding phospho-RLC standards (but not other RLC phospho-species). These experiments validated the applicability of the chosen antibodies for monitoring phosphorylation levels of endothelial myosin RLC in EA.hy926 cells after stimulation with thrombin or insulin.
A general limitation of the ELC-based quantification of Western blots is a narrow linear range of ECL signal hardly exceeding one order of magnitude. Due to intrinsic differences in the reactivity of various RLC antibodies, we could not use the same linear range for the detection of phospho-RLC and total RLC. At the optimal exposure for P-RLC and PP-RLC antibodies, the signal from total RLC antibodies was below the linear range, whereas bringing the total RLC calibration signal to the linear range resulted in the overexposure of phospho-RLC signals and loss of linearity. To circumvent this problem, we developed RLC calibration standards for different periods of time in order to achieve the linear dependence of the ECL signal on the RLC load that would also cover the optimal range for quantification of the phospho-RLC or total RLC. We also loaded on the gel a range of incremental volumes of endothelial cell samples to make sure that at least some ECL signals from these samples will fall within the linear calibration range for particular RLC forms. By using this approach, we were able to correctly measure the absolute amounts of P-RLC, PP-RLC, and total RLC in endothelial cell lysates.
Further, our experiments demonstrated that running Western blots for each of the analyzed RLC species may not be necessary as all the information could be obtained from a single Western blot subjected to multiple re-probing with appropriate RLC antibodies. Once the PVDF membrane is treated with glutaraldehyde after electrotransfer of the RLC standards and cell proteins, it can be subjected to at least three rounds of re-probing with different RLC antibodies without losing the ECL signal intensity (Fig. 2, a and b). This approach allows to save valuable experimental samples, as well as to avoid variations in the sample load.
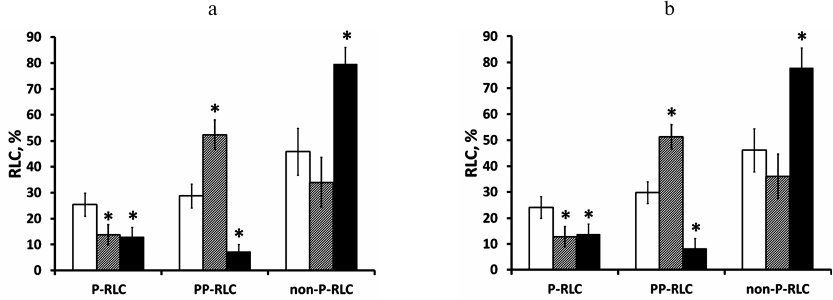
Fig. 2. P-RLC and PP-RLC measurement in EA.hy926 endothelial cells is reproducible after Western blot reprobing and does not depend on the order of RLC antibody application. Confluent EA.hy926 cells were stimulated with either 100 nM thrombin for 12 min or with 100 nM insulin for 15 min. Cell lysates were electrotransferred to PVDF membranes along with RLC standards. a) Western blots were probed with the primary antibodies/secondary antibodies in the following order: P-RLC, PP-RLC, pan-RLC, and P-RLC (the membrane was stripped between each antibody staining). ECL signals were acquired using a digital camera after each staining. ECL signals from after staining with the first and the last antibody pairs were compared and found similar. Obtained results were averaged and shown as absolute values of endothelial myosin P-RLC or PP-RLC levels in unstimulated cells (empty bars) and cells stimulated with thrombin (grey bars) or insulin (closed bars). The fractions of non-phosphorylated RLC (non-P-RLC) were calculated as 100% minus the sum of P-RLC and PP-RLC fractions (%). b) The same cell lysates were analyzed by Western blot using the same algorithm as above but with altered primary antibody order: PP-RLC, P-RLC, pan-RLC, and PP-RLC. Again, ECL signals from the first and the last antibody pairs were comparable. Data are presented as mean ± SD for three independent experiments; * p < 0.05 vs. basal phosphorylation level.
Lastly, the cost-effectiveness of the proposed method can be increased by using the same working solutions of primary antibodies that can be saved after staining and stored at 4°C for up to 3 months. The working solutions of primary antibodies can be used for up to three subsequent re-probing of PVDF membranes without losing the ECL signal reproducibility (data not shown).
Measurement of P-RLC and PP-RLC levels in EA.hy926 human endothelial cells stimulated with thrombin and insulin. To demonstrate the practical application of the proposed method, we measured the levels of P-RLC and PP-RLC in EA.hy926 endothelial cells stimulated with natural agonists such as thrombin and insulin. As shown in Fig. 2a, the basal levels of P-RLC and PP-RLC in quiescent EA.hy926 cells were 25.4 ± 4.4 and 28.8 ± 4.6%, correspondingly (Fig. 2, open bars). After 12 min of treatment with 100 nM thrombin, the level of P-RLC decreased to 13.7 ± 3.9%, whereas the PP-RLC level increased to 52.3 ± 5.7% (Fig. 2, grey bars). Compared to the untreated control, a 15-min challenge of endothelial cells with 100 nM insulin induced a 2-fold decrease in P-RLC (13.0 ± 3.6%) and 3-fold decrease in PP-RLC (7.3 ± 2.8%) (Fig. 2, closed bars). As mentioned above, changing the order of antibody application while re-probing the same blot resulted in similar values for the P-RLC and PP-RLC levels (Fig. 2b) indicating the absence of RLC antigen leakage from the PVDF filter or irreversible binding of RLC antibodies.
Comparable P-RLC/PP-RLC levels were observed in EA.hy926 cells when human standard RLC mixture was used instead of turkey gizzard standard RLC mixture (data not shown).
Additionally, we assessed the levels of RLC phosphorylation in quiescent human platelets from two donors and found no PP-RLC and less than 10% of P-RLC, which is in a good agreement with the earlier measurements using radioactive phosphate labelled platelets [26].
DISCUSSION
Myosin RLC phosphorylation at Ser19 (P-RLC) is sufficient to fully activate SMMII and NMII (reviewed in [27]), however frequently observed RLC di-phosphorylation at Thr18/Ser19 (PP-RLC) (reviewed in [28]) may be responsible for prolonged myosin activation because of significantly slower RLC dephosphorylation [29]. Based on these findings, mono-phosphorylation at Ser19 and/or di-phosphorylation at Thr18/Ser19 are widely used to biochemically characterize myosin II motor activity and its involvement in (patho)physiological responses.
A variety of experimental approaches has been developed for analysis of P-RLC and PP-RLC. One of these approaches is electrophoretic separation of non-phosphorylated RLC, P-RLC, and PP-RLC followed by staining with a protein dye and densitometric analysis of the stained protein bands. RLC phospho-species can be separated by isoelectric focusing in the presence of pyrophosphate [9], urea/glycerol polyacrylamide electrophoresis [10], or recently introduced Phos-tag technology [11-14]. Originally, these methods required myosin isolation prior to quantitative measurements and, therefore, demanded relatively large amounts of starting sample material. Hence, they cannot be used for analysis of myosin II phosphorylation in non-muscle cells that are usually available in much smaller amounts. To address this problem, the sensitivity of detection could be increased by measuring myosin phosphorylation by radioactive phosphate incorporation in the RLC followed by either one-dimensional SDS-PAGE or two-dimensional phosphopeptide mapping [30-32]. However, this approach does not allow estimating the phosphorylation levels of individual residues. In order to overcome this limitation, phosphorylated protein can be analyzed by mass-spectrometry [12], but again, this requires relatively large sample amounts.
For several decades, urea/glycerol gel electrophoresis was the method of choice for separating myosin RLC, P-RLC, and PP-RLC in cell and tissue samples. Subsequent visualization of RLC phospho-species with pan-RLC antibody after Western blotting allows calculation of relative amounts of P-RLC and PP-RLC [33, 34]; however, it is impossible to claim without doubt that the phosphorylated residues are Ser19 and/or Thr18, since phosphate group could be potentially incorporated at other site(s). Despite that RLC phosphorylation at Ser19 and/or di-phosphorylation at Thr18/Ser19 are considered major contributors to the net RLC phosphorylation and actomyosin contraction, mono-phosphorylation at Thr18 was observed upon sustained smooth muscle relaxation [12]. Moreover, human non-sarcomeric myosin RLC2 contains about a dozen residues that can be phosphorylated (see http://www.phosphosite.org resource [35] for more details), indicating a potential to generate multiple combinatorial RLC phospho-species. Indeed, simultaneous presence of multiple RLC phospho-species has been demonstrated in cultured uterine myocytes [13]. These findings imply that analysis of RLC phosphorylation should not leave out sites other than Ser19 and Thr18. Therefore, the site-specificity of RLC phosphorylation has to be established prior to considering its physiological significance.
RLC immunodetection after protein transfer from the urea/glycerol gels is significantly less sensitive than after the standard SDS-PAGE. This discrepancy has not been investigated in detail but might be due to inefficient solubilization of RLC in the presence of urea as compared to SDS; regardless, Western blotting after standard SDS-PAGE is a preferable method for detection of myosin II RLC in small amounts of non-muscle cells and muscle tissue [14].
Recently introduced Phos-tag separation technology combines the high sensitivity of Western blotting after SDS-PAGE with the resolving capacity of urea/glycerol gel electrophoresis. Phos-tag technology provides identification of RLC phospho-species phosphorylated at different sites. On the other hand, the electrophoretic mobility of P-RLC depends on the position of phosphorylated residues in the RLC sequence [13]. Phos-tag generates an impressive pattern of RLC bands that could be unique for certain agonists/stimuli in various cell/tissue types. To quantify RLC phospho-species by the Phos-tag technique using pan-RLC antibodies, it is necessary to assign RLC protein bands to RLC forms phosphorylated by Ser19 and/or Thr18 using phospho-specific RLC antibodies.
It should be noted that several commercial pan-RLC antibodies have been raised against synthetic peptides or recombinant antigens that included Ser19 and Thr18 residues. Hence, these pan-RLC antibodies could potentially be sensitive to phosphorylation and may contribute to the misinterpretation of phosphorylation data, in particular, overestimation of phosphate incorporation in RLC. Similarly, recent reports have shown that individual anti-RLC antibodies claimed to be strictly phospho- or site-specific, in fact, may cross-react with another RLC phospho-species [11-14]. Hence, validation of RLC antibodies prior to quantitation of Ser19 and Thr18 phosphorylation is critical for reliable measurements. In preliminary experiments, we established that the pan-RLC antibody used in this study (ProteinTech; #10324-1-AP) demonstrates no phospho-sensitivity; moreover, using both urea/glycerol gel and Phos-tag electrophoreses, we confirmed that the chosen phosphospecific antibodies do not cross-react with other RLC phospho-species that could be generated in thrombin/insulin-stimulated EA.hy926 cells.
Once reliable phosphospecific RLC antibodies are selected, direct dynamics of RLC mono-phosphorylation at Ser19 or di-phosphorylation at Thr18/Ser19 could be studied by standard SDS-PAGE and Western blotting. In its widely used format, this method does not allow determining the stoichiometry of RLC phosphorylation [36]. However, quantitative data could be obtained from the measurements relatively easily using external phospho-RLC standards (Fig. 1). We found that P-RLC and PP-RLC could be reproducibly quantified in samples containing ~105 non-muscle cells. As seen from Fig. 1, b-d, our method allows detecting 0.5 to 1 ng of P(P)-RLC and ~3-5 ng of total RLC in the experimental samples. It is possible that the sensitivity of detection can be increased further by using tertiary antibody, as it has been demonstrated in [14].
Ideally, species-specific RLC standards should be used to achieve the most precise quantitation of RLC phospho-species. However, in practice, we observed no differences in the results of phospho-RLC quantification when using turkey gizzard myosin or human myosin RLC standards. Indeed, the amino acid sequence homology between these RLCs is 98% or higher (2-3 differing amino acid residues depending on the RLC isoform), and the use of polyclonal pan-RLC antibodies for RLC detection brings possible variations below the error limit. Still, for other RLC sources outside avian and mammalian species, the method might require some modification based on the RLC sequence homology and relative reactivity of RLC antibodies.
Using the outlined approach, we measured the constitutive levels of P-RLC and PP-RLC in nonstimulated EA.hy926 endothelial cells and analyzed the dynamics of P-RLC and PP-RLC changes in these cells in response to thrombin or insulin. Thrombin induced reciprocal changes in the P-RLC and PP-RLC levels (Fig. 2); however, there was a net increase in myosin RLC phosphorylation after 12-min cell treatment with thrombin (see the decrease in the level of non-phosphorylated RLC vs nonstimulated cells). These findings are consistent with the published reports that demonstrated thrombin-induced myosin phosphorylation in endothelial cells and subsequent cell rounding, contraction, and hyperpermeability development [33, 37]. On the other hand, insulin decreased 2 to 3 times the levels of both P-RLC and PP-RLC in the endothelial cells (Fig. 2). The decrease in the RLC net phosphorylation is consistent with insulin action as a barrier-protecting agent [18]. Therefore, the results of measurements of myosin II activation adequately reflected changes in the endothelial cell physiology. It should be emphasized that the described method of quantification by immunoblotting allowed discrimination between myosin activation from 30 to 60% vs changes from 3 to 6%. Although physiological significance of these changes might be essentially different, assessment of phospho-RLC levels by standard Western blotting would demonstrate 2-fold myosin activation in both cases, which might lead to misinterpretation of experimental data.
Altogether, our results demonstrate that the proposed method of quantitative analysis of myosin RLC phosphorylation in non-muscle cells can be successively used in research. In order to assist in choosing a method to study myosin II activation in cells, we provide a table that summarizes advantages, limitations, and practical aspects of various methods of measuring Ser19 and/or Thr18/Ser19 phosphorylation in non-muscle cells. As indicated in the table, the method described in this report is highly sensitive and time/resource-saving; however, its major disadvantage is the requirement for phosphorylated RLC standards, which, to the best of our knowledge, are currently commercially unavailable.
Characteristics of analytical methods for assessment of myosin II RLC
phosphorylation at Ser19 and Thr18/Ser19 in non-muscle cells
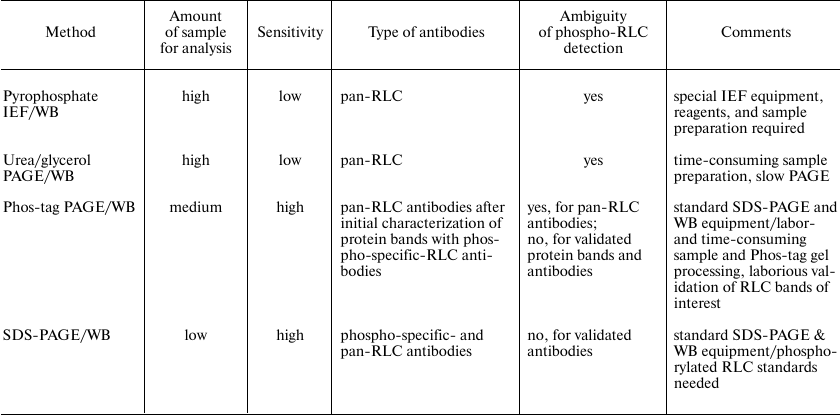
Notes: IEF, isoelectric focusing; PAGE, polyacrylamide gel
electrophoresis; RLC, myosin regulatory light chain; SDS, sodium
dodecyl sulfate; WB, Western blot.
In conclusion, we describe an easy-to-use Western blotting-based method for quantification of myosin P-RLC and PP-RLC in biological samples using external calibration standards. The method provides reliable assessment of myosin II activation in small samples of non-muscle cells and allows processing of many samples at once, thereby reducing the use of costly reagents commonly used for cell treatment and analysis. Quantitative information regarding myosin RLC phosphorylation in cells vs relative changes in P-RLC and PP-RLC levels proves the link between myosin activation and cellular responses.
Funding
This work was supported by the Russian Foundation for Basic Research (project no. 14-04-32039 to OAK) and the Russian Science Foundation (project no. 14-35-00026).
Conflict of Interests
The authors declare no conflict of interests in financial or any other sphere.
Compliance with Ethical Standards
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants involved in the study.
REFERENCES
1.Betapudi, V. (2014) Life without double-headed
non-muscle myosin II motor proteins, Front. Chem.,
2, 45.
2.Heissler, S. M., and Sellers, J. R. (2016) Various
themes of myosin regulation, J. Mol. Biol., 428,
1927-1946.
3.Heissler, S. M., and Manstein, D. J. (2013)
Nonmuscle myosin-2: mix and match, Cell. Mol. Life Sci.,
70, 1-21.
4.Matsumura, F., Yamakita, Y., and Yamashiro, S.
(2011) Myosin light chain kinases and phosphatase in mitosis and
cytokinesis, Arch. Biochem. Biophys., 510,
76-82.
5.Khapchaev, A. Y., and Shirinsky, V. P. (2016)
Myosin light chain kinase MYLK1: anatomy, interactions, functions, and
regulation, Biochemistry (Moscow), 81,
1676-1697.
6.Amano, M., Ito, M., Kimura, K., Fukata, Y.,
Chihara, K., Nakano, T., Matsuura, Y., and Kaibuchi, K. (1996)
Phosphorylation and activation of myosin by Rho-associated kinase
(Rho-kinase), J. Biol. Chem., 271,
20246-20249.
7.Amano, M., Nakayama, M., and Kaibuchi, K. (2010)
Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity,
Cytoskeleton, 67, 545-554.
8.Kimura, K., Ito, M., Amano, M., Chihara, K.,
Fukata, Y., Nakafuku, M., Yamamori, B., Feng, J. H., Nakano, T., Okawa,
K., Iwamatsu, A., and Kaibuchi, K. (1996) Regulation of myosin
phosphatase by Rho and Rho-associated kinase (Rho-kinase),
Science, 273, 245-248.
9.Silver, P. J., and Stull, J. T. (1982) Quantitation
of myosin light chain phosphorylation in small tissue samples, J.
Biol. Chem., 257, 6137-6144.
10.Persechini, A., Kamm, K. E., and Stull, J. T.
(1986) Different phosphorylated forms of myosin in contracting tracheal
smooth-muscle, J. Biol. Chem., 261, 6293-6299.
11.Hirano, M., and Hirano, K. (2016) Myosin
di-phosphorylation and peripheral actin bundle formation as initial
events during endothelial barrier disruption, Sci. Rep.,
6, 20989.
12.Puetz, S., Schroeter, M. M., Piechura, H.,
Reimann, L., Hunger, M. S., Lubomirov, L. T., Metzler, D., Warscheid,
B., and Pfitzer, G. (2012) New insights into myosin phosphorylation
during cyclic nucleotide-mediated smooth muscle relaxation, J.
Muscle Res. Cell Motil., 33, 471-483.
13.Aguilar, H. N., Tracey, C. N., Tsang, S. C. F.,
McGinnis, J. M., and Mitchell, B. F. (2011) Phos-tag-based analysis of
myosin regulatory light chain phosphorylation in human uterine
myocytes, Plos One, 6, e20903.
14.Takeya, K., Loutzenhiser, K., Shiraishi, M.,
Loutzenhiser, R., and Walsh, M. P. (2008) A highly sensitive technique
to measure myosin regulatory light chain phosphorylation: the first
quantification in renal arterioles, Am. J. Physiol. Renal.,
294, F1487-F1492.
15.Taylor, S. C., Berkelman, T., Yadav, G., and
Hammond, M. (2013) A defined methodology for reliable quantification of
Western blot data, Mol. Biotechnol., 55,
217-226.
16.Taylor, S. C., and Posch, A. (2014) The design of
a quantitative Western blot experiment, Biomed. Res. Int.,
2014, 361590.
17.Siller-Matula, J. M., Schwameis, M., Blann, A.,
Mannhalter, C., and Jilma, B. (2011) Thrombin as a multi-functional
enzyme focus on in vitro and in vivo effects, Thromb.
Haemostasis, 106, 1020-1033.
18.Liu, Y., Chen, X. L., Wang, L., and
Martins-Green, M. (2017) Insulin antagonizes thrombin-induced
microvessel leakage, J. Vasc. Res., 54,
143-155.
19.Gopalakrishna, R., and Anderson, W. B. (1982)
Ca2+-induced hydrophobic site on calmodulin: application for
purification of calmodulin by phenyl-Sepharose affinity chromatography,
Biochem. Biophys. Res. Commun., 104, 830-836.
20.Adelstein, R. S., and Klee, C. B. (1981)
Purification and characterization of smooth-muscle myosin light chain
kinase, J. Biol. Chem., 256, 7501-7509.
21.Bradford, M. M. (1976) Rapid and sensitive method
for quantitation of microgram quantities of protein utilizing principle
of protein–dye binding, Anal. Biochem., 72,
248-254.
22.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
23.Towbin, H., Staehelin, T., and Gordon, J. (1979)
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: procedure and some applications, Proc. Natl.
Acad. Sci. USA, 76, 4350-4354.
24.Yeung, Y. G., and Stanley, E. R. (2009) A
solution for stripping antibodies from polyvinylidene fluoride
immunoblots for multiple reprobing, Anal. Biochem.,
389, 89-91.
25.Ikebe, M., Hartshorne, D. J., and Elzinga, M.
(1986) Identification, phosphorylation, and dephosphorylation of a 2nd
site for myosin light chain kinase on the 20,000-dalton light chain of
smooth-muscle myosin, J. Biol. Chem., 261,
36-39.
26.Daniel, J. L., Molish, I. R., and Holmsen, H.
(1981) Myosin phosphorylation in intact platelets, J. Biol.
Chem., 256, 7510-7514.
27.Somlyo, A. P., and Somlyo, A. V. (2003)
Ca2+ sensitivity of smooth muscle and nonmuscle myosin II:
modulated by G proteins, kinases, and myosin phosphatase, Physiol.
Rev., 83, 1325-1358.
28.Walsh, M. P. (2011) Vascular smooth muscle myosin
light chain diphosphorylation: mechanism, function, and pathological
implications, IUBMB Life, 63, 987-1000.
29.Takeya, K., Wang, X., Sutherland, C., Kathol, I.,
Loutzenhiser, K., Loutzenhiser, R. D., and Walsh, M. P. (2014) The
involvement of myosin regulatory light chain diphosphorylation in
sustained vasoconstriction under pathophysiological conditions, J.
Smooth Muscle Res., 50, 18-28.
30.Sandquist, J. C., Swenson, K. I., DeMali, K. A.,
Burridge, K., and Means, A. R. (2006) Rho kinase differentially
regulates phosphorylation of nonmuscle myosin II isoforms A and B
during cell rounding and migration, J. Biol. Chem.,
281, 35873-35883.
31.Colburn, J. C., Michnoff, C. H., Hsu, L. C.,
Slaughter, C. A., Kamm, K. E., and Stull, J. T. (1988) Sites
phosphorylated in myosin light chain in contracting smooth-muscle,
J. Biol. Chem., 263, 19166-19173.
32.Kamm, K. E., Hsu, L. C., Kubota, Y., and Stull,
J. T. (1989) Phosphorylation of smooth-muscle myosin heavy and
light-chains. Effects of phorbol dibutyrate and agonists, J. Biol.
Chem., 264, 21223-21229.
33.Garcia, J. G. N., Davis, H. W., and Patterson, C.
E. (1995) Regulation of endothelial-cell gap formation and barrier
dysfunction: role of myosin light-chain phosphorylation, J. Cell.
Physiol., 163, 510-522.
34.Garcia, J. G. N., Verin, A. D., Schaphorst, K.,
Siddiqui, R., Patterson, C. E., Csortos, C., and Natarajan, V. (1999)
Regulation of endothelial cell myosin light chain kinase by Rho,
cortactin, and p60(src), Am. J. Physiol. Lung Cell. Mol.
Physiol., 276, L989-L998.
35.Hornbeck, P. V., Zhang, B., Murray, B.,
Kornhauser, J. M., Latham, V., and Skrzypek, E. (2015) PhosphoSitePlus,
2014: mutations, PTMs and recalibrations, Nucleic Acids Res.,
43, D512-D520.
36.Aguilar, H. N., Zielnik, B., Tracey, C. N., and
Mitchell, B. F. (2010) Quantification of rapid myosin regulatory light
chain phosphorylation using high-throughput in-cell Western assays:
comparison to Western immunoblots, Plos One, 5,
e9965.
37.Vouret-Craviari, V., Boquet, P., Pouyssegur, J.,
and Van Obberghen-Schilling, E. (1998) Regulation of the actin
cytoskeleton by thrombin in human endothelial cells: role of Rho
proteins in endothelial barrier function, Mol. Biol. Cell,
9, 2639-2653.