Brain Proteome of Drosophila melanogaster Is Enriched with Nuclear Proteins
K. G. Kuznetsova1, M. V. Ivanov2, M. A. Pyatnitskiy1,3, L. I. Levitsky2, I. Y. Ilina1, A. L. Chernobrovkin4, R. A. Zubarev4,5, M. V. Gorhskov2, and S. A. Moshkovskii1,6,a*
1Institute of Biomedical Chemistry, 119121 Moscow, Russia2Institute of Energy Problems of Chemical Physics, Russian Academy of Sciences, 119334 Moscow, Russia
3Higher School of Economics, 101000 Moscow, Russia
4Karolinska Institutet, SE-171 77, Stockholm, Sweden
5Sechenov First Moscow State Medical University, 119991 Moscow, Russia
6Pirogov Russian National Research Medical University (RNRMU), 117997 Moscow, Russia
* To whom correspondence should be addressed.
Received August 30, 2018; Revised October 10, 2018; Accepted October 10, 2018
The brain proteome of Drosophila melanogaster was characterized by liquid chromatography/high-resolution mass spectrometry and compared to the earlier characterized Drosophila whole-body and head proteomes. Raw data for all the proteomes were processed in a similar manner. Approximately 4000 proteins were identified in the brain proteome that represented, as expected, the subsets of the head and body proteomes. However, after thorough data curation, we reliably identified 24 proteins unique for the brain proteome; 13 of them have never been detected before at the protein level. Fourteen of 24 identified proteins have been annotated as nuclear proteins. Comparison of three used datasets by label-free quantitation showed statistically significant enrichment of the brain proteome with nuclear proteins. Therefore, we recommend the use of isolated brain preparations in the studies of Drosophila nuclear proteins.
KEY WORDS: proteomics, mass spectrometry, Drosophila melanogaster, brain, nuclear proteinDOI: 10.1134/S0006297919010097
Abbreviations: BCA, bicinchoninic acid; LC, liquid chromatography; MS, mass spectrometry; MS/MS, tandem mass spectrometry; NSAF, normalized spectral abundance factor; TEAB, triethylammonium bicarbonate.
The fruit fly Drosophila melanogaster is a legendary model
organism in molecular biology and medicine. It is believed that
approximately three quarters of human disease-associated genes have
functional analogues in the Drosophila genome [1]. Reference proteomic maps of Drosophila have
been obtained starting from the very beginning of proteomics research
[2]. These maps can serve as a reference for
comparing proteomes, e.g., in various models used for studying
diseases, aging, or effects of environmental factors. For more than two
decades, the proteomics methods have undergone substantial
modifications; hence, the reference proteomic data should also
correspond to these modifications. An important innovation that
revolutionized the commonly used shotgun approach was introduction of
the high-resolution mass spectrometers to the filed. One of flagship
machines used for this type analysis is the Orbitrap ion trap that has
been used in biological studies since the beginning of 2000s [3]. The technical progress provided the development of
the so-called “deep” proteomic analysis, when depending on
the dynamic range of protein concentrations, two to ten thousand or
more proteins could be identified in a single sample [4].
Until recently, there has been a certain deficit of high-resolution MS proteomic data for Drosophila in the PRIDE repository [5] that contains proteomes of the entire Drosophila body obtained by one-dimensional SDS gel-electrophoresis followed by liquid chromatography-mass spectrometry (LC-MS) [6] and Drosophila head proteome obtained by analysis of protein fractions without the use of electrophoresis [7]. Only during the preparation of this article for publication, more extensive proteomic data, e.g., those obtained at different developmental stages of Drosophila flies [8], have been published. In our earlier work on the consequences of RNA editing at the proteome level, we have focused on studying the brain of Drosophila [9] because brain tissue is where most RNA editing events of interest take place. We did not describe the brain proteome itself, since we catalogued only peptide fragments containing single amino acid substitutions [9]. Meanwhile, that was the first time when Drosophila brain proteome was analyzed. In this paper, we report some properties of the brain proteome of adult D. melanogaster flies in comparison with the head and whole-body proteomes published before [6, 7].
MATERIALS AND METHODS
Drosophila melanogaster flies (Canton S line) were kindly donated by Dr. N. I. Romanova (Department of Genetics, Biological Faculty, Lomonosov Moscow State University). The flies were kept at 25°C in 50-ml plastic tubes (Orange Scientific, Belgium) and fed with Formula 5-24 Instant Drosophila Medium (Carolina Biological Supply Company, USA). The insects were transferred to fresh tubes after reaching the adult stage. Samples for proteome analysis contained 200 flies each with approximately the same number of males and females of different ages. The flies were sacrificed by freezing at –80°C.
Brain isolation. During the entire procedure, the flies were kept on ice in a Petri dish. The body was quickly separated with a needle, and the head was placed in 0.01 M phosphate buffered saline (PBS), pH 7.4 (Sigma-Aldrich, USA). The head capsule was torn using two forceps under a Nikon SMZ645 binocular microscope (Japan) at 10× magnification. Isolated brains were placed into PBS and centrifuged at 6000g and 4°C for 15 min (5415R centrifuge; Eppendorf, Germany). The supernatant was discarded, and the sediment was frozen at –80°C to prepare samples for proteomic analysis.
Sample preparation for proteomic analysis. The sediment (200 brains) was resuspended in 100 μl of lysis solution containing 0.1% (w/v) Protease MAX Surfactant (Promega, USA), 50 mM ammonium bicarbonate, and 10% (v/v) acetonitrile. The lysate was incubated in a shaker at 550 rpm for 60 min at room temperature and then disintegrated by sonication using a Bandelin Sonopuls HD2070 ultrasonic homogenizer (Bandelin Electronic, Germany) at 30% amplitude with short pulses (15 s) for 5 min. The homogenate was centrifuged at 15,700g for 10 min at 20°C (5415R; Eppendorf, Germany), and the supernatant was collected. Total protein in the supernatant was measured by the BCA method (BCA Kit; Sigma-Aldrich).
After that, 0.5 M dithiothreitol (DTT) solution in 50 mM triethylammonium bicarbonate (TEAB) was added to the samples up to the final DTT concentration of 10 mM, followed by incubation for 20 min at 56°C. Then, 0.5 M iodoacetamide in 50 mM TEAB was added to the final concentration of 10 mM, and the mixture was incubated in the dark at room temperature for 30 min.
Proteins in the sample were digested with trypsin (Trypsin Gold; Promega) at a 1 : 40 (w/w) enzyme to total protein ratio at 37°C overnight. The reaction was stopped by adding 5% (v/v) acetic acid.
The sample was incubated in a shaker (Eppendorf, Germany) at 500 rpm for 30 min at 45°C and then centrifuged at 15,700g at 20°C for 10 min (5415R; Eppendorf). The supernatant was added to the filter cartridge with a 10-kDa cut-off (Millipore, USA) and centrifuged at 13,400g and 20°C for 20 min. Then, 100 μl 50% (v/v) formic acid was added on the filter, and the sample was centrifuged under the same conditions. The final concentration of peptides was measured with a Peptide Assay kit (Thermo Fisher Scientific, USA) and NanoDrop spectrophotometer (Thermo Fisher Scientific). The samples were evaporated in a vacuum concentrator (Eppendorf) at 45°C in glass microtubes. Dried peptides were stored at –80°C until LC-MS/MS analysis.
Proteomic analysis by liquid chromatography followed by tandem mass spectrometry. Chromatographic separation of peptides was performed on a 25-cm C18 column of (Silica Tip 360 μm OD, 75 μm ID; New Objective, USA) attached to an UltiMate™ 3000 RSLCnano LC system (Thermo Fisher Scientific) without a pre-column. The peptides were eluted at a flow rate of 0.3 µl/min with a linear (2-26%, v/v) gradient of acetonitrile concentration in 0.1% (v/v) formic acid for 240 min. Eluted peptides were ionized with a nano-electrospray and analyzed in an Orbitrap QExactive Plus mass spectrometer (Thermo Fisher Scientific). Panoramic mass spectra were recorded with a 60,000 resolution in the 200-2000 m/z interval. The MS/MS data for 20 most intensive precursor ions, at least 2-charged, was received using high-energy collisional dissociation (HCD) with a 15,000 resolution. In order to avoid repeated analysis of the same peptides, dynamic exclusion of less than 500 precursors for 60 s was used. All procedures were performed in three technical replicates.
Proteomic data obtained in this work were deposited to the ProteomeXchange database (http://www.proteomexchange.org/) [10] under the accession number PXD004949.
Proteomic data processing. Peptides and proteins were identified using the IdentiPy open source software [11] developed by the authors (L. L., M. I., and M. G.). This new search engine uses simplified X!Tandem algorithm [12] with improved search workflow. The search parameters were different for our data and the head and whole-body datasets [6, 7] (PRIDE accession ID PXD001712 and PXD000455, respectively). Our experiments were run on an Orbitrap QExactive mass spectrometer (Thermo Fisher Scientific), whereas the previous MS analysis used a hybrid LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). The search was performed using the Uniprot D. melanogaster Reference Proteome database (03.2018; 42,524 entries) with the following search engine parameters: enzyme – trypsin; number of missed cleavages – 1; precursor accuracy unit – ppm; precursor accuracy left – 10; precursor accuracy right – 10; precursor isotope mass error – 0; product accuracy, Da – 0.1; FDR 1.0; FDR type – psm; minimum charge – 2; maximum charge – 4; generate decoy db – yes; decoy method – reverse; decoy prefix – DECOY_; dynamic range – 100; peptide minimum length – 7: peptide maximum length – 30; peptide minimum mass – 300; peptide maximum mass – 10,000; fragments in spectra, min – 4; fragments in spectra, max – 50; product minimum m/z – 150; maximum fragments charge – 1; matched fragments, min – 1; use scoring function – RNHS (renormalized hyperscore); score threshold – 0; show unmatched spectra in results – no; report number of sequence candidates – 1; peptide mass shift – 0; deisotope – yes; deisotoping mass tolerance – 0.3. No fixed amino acid modifications were set. Methionine oxidation and cysteine alkylation by iodoacetamide were introduced as variable modifications. For the post-search validation, all parameters were used except the charge status. The same search parameters were used for analyzing head and whole-body proteomes, except product accuracy, Da – 0.3.
The output files containing lists of identified peptides and proteins assembled from these peptides were processed to compare the proteomes. In order to find peptides unique for the brain proteome, we used specially developed R script [see Supplement to this paper on the Biochemistry (Moscow) website (http://protein.bio.msu.ru/biokhimiya), File 1]. The lists of identified peptides for Drosophila brain, head, and entire body were used as input files for the script (Supplement; Files 2-4, respectively).
For further proteome comparisons and subcellular location estimates, we used the normalized spectral abundance factor (NSAF) [13] calculated with the IdentiPy sofware (Supplement, Table S1) and avaliable Gene Ontology (GO) identifiers [14].
RESULTS AND DISCUSSION
Comparison of Drosophila brain, head and whole-body proteomes. Using the same search engine, we identified 4005, 6905, and 7652 gene products in the brain proteome (own data) and the head [7] and whole-body proteomes [6], respectively. To see if these three proteomes overlap, we compared them; however, the existence of many synonyms of genes and proteins in the Uniprot database led to wrong identification of unique proteins that had to be eliminated by manual checking. Notably, the annotation of genes and proteins from non-human organism in the databases is not so thoroughly curated as for the humans. For example, a special resource (NextProt) exists for the human proteomics [15]. As a result, we decided that a more correct way to estimate the overlapping of the analyzed proteomes was their comparison at the level of tryptic peptides.
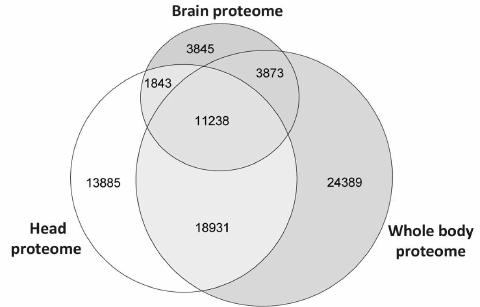
As one could see from the diagram in Fig. 1, only 18% peptides from the brain dataset were unique and did not occur in the larger datasets. The corresponding contents of unique peptides in the head and whole-body proteomes were 30 and 41%, respectively. Comparison of the proteomes at the peptide level supported the obvious conclusion that the brain proteome is a subset of the head and whole-body proteomes. However, it was interesting to identify proteins composed of those unique peptides and to characterize them in order to understand which studies could use isolated Drosophila brains instead of entire heads.
Fig. 1. Overlap of Drosophila brain proteome with previously published whole-body [6] and head [7] proteomes represented as Venn’s diagram. Since names and accession numbers of Drosophila proteins are redundant in the databases, the data are presented for unique tryptic peptides identified using the same search parameters.
Proteins identified in Drosophila brain only. Since automatic analysis and comparison of Drosophila proteomes using protein identifiers was complicated due to the database redundancy, the best way to identify brain-specific proteins using our data was to assemble them de novo from the unique peptides. Protein identification a using a single unique peptide is attractive but, according to the commonly accepted notion, unreliable at the same time. Proteomic search engines often cannot distinguish between two peptides with different structure due to the full match between the molecular weights of unchanged amino acid residue and some other residue that was chemically modified [16]. Thus, deamidation of asparagine and glutamine side chains, which often occurs in vivo and in vitro, is indistinguishable from the genetically encoded substitution, and this not a unique case [17]. Therefore, to identify brain-specific proteins, we used a heuristic rule of identification of at least two unique peptides for each protein (see Supplement, Table S2, for the list of all brain-specific unique peptides). Using this principle, we found 24 brain-specific proteins, six of which were annotated in the Uniprot database as “Proteins predicted”, i.e., never found before at the transcript or protein levels (table). Analysis of proteins presented in this table leads to another observation: more than a half of them (13 out of 24) have been annotated as localized in the cell nucleus. Among them, there are DNA binding factors and other chromatin components. The enrichment with nuclear proteins of a sample including 24 gene products only cannot be proven statistically. However, we tried to solve the problem using proteome-wide analysis.
Proteins unique for D. melanogaster brain proteome identified by
comparison with previously published proteomes of the insect head [7] and entire body [6]. The
proteins were selected based on presence of two peptides unique for the
brain dataset. Gene and protein names, intercellular localization, and
database status are listed according to the UniProt database [18]
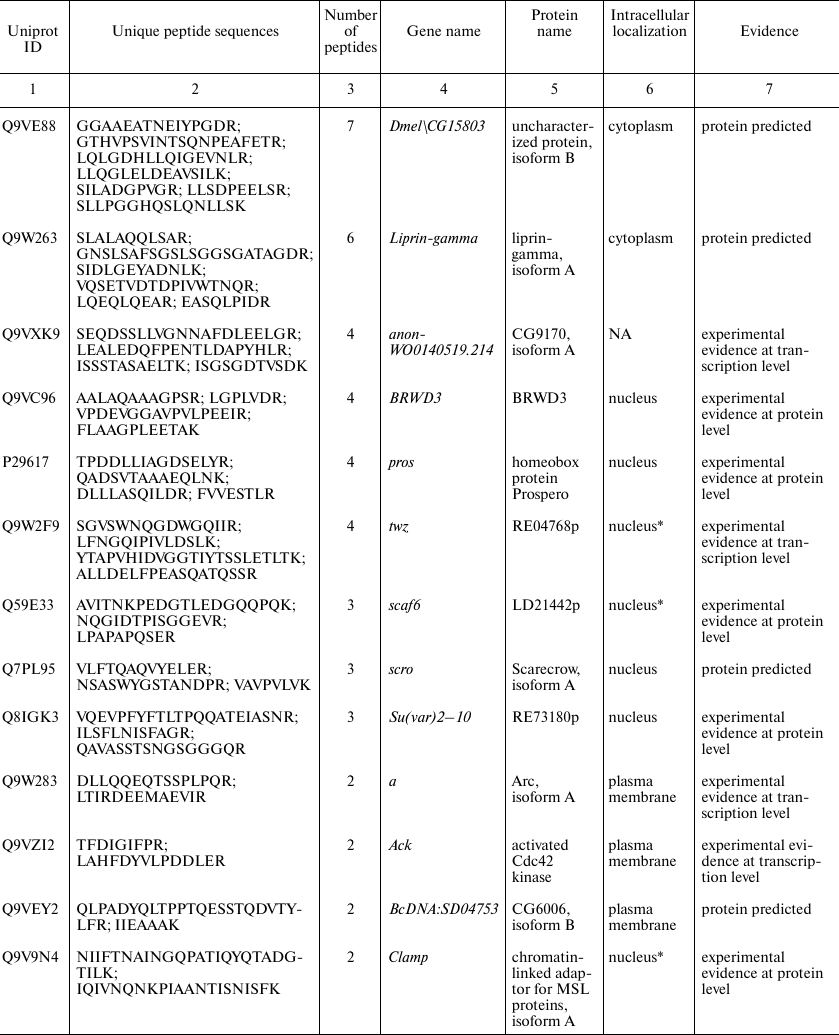
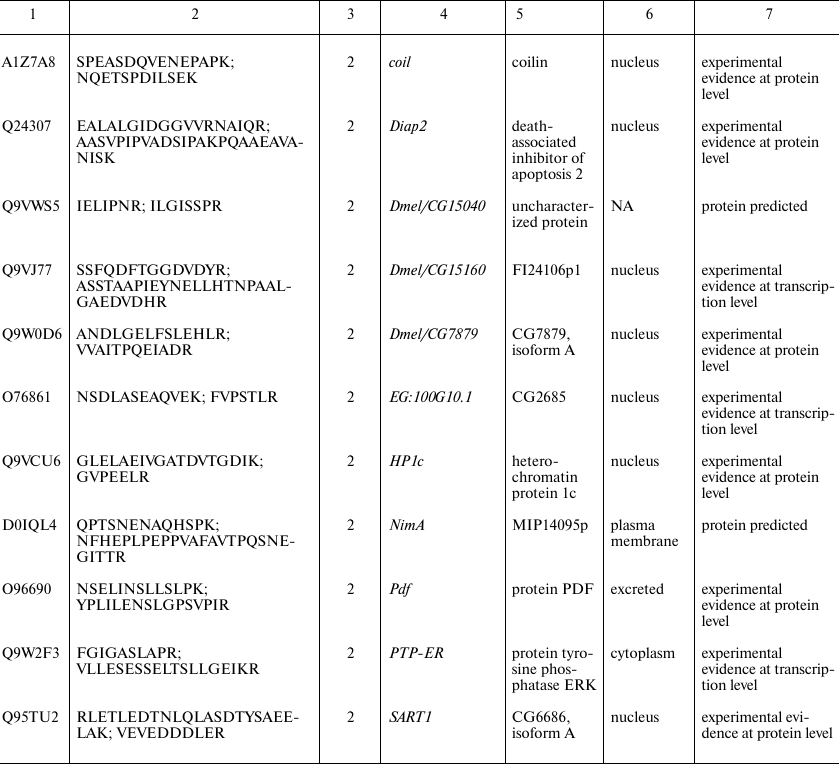
* Intercellular localization is not specified in UniProt and predicted
based on the analysis of the database and literature data; NA, data not
available.
Quantitative enrichment of Drosophila brain proteome by nuclear proteins. With few proteins specific for the brain proteome, enrichment of their list with nuclear proteins remains speculative. Analysis of data of label-free quantitation of all three studied proteomes (Supplement, Table S1) led to another relevant observation. When the proteins were sorted by their relative abundance, histones occupied much higher position in the brain proteome list than in the other two datasets. This observation also suggested that the Drosophila brain proteome is enriched with nuclear proteins. Since this is an indirect evidence, how can it be proven?
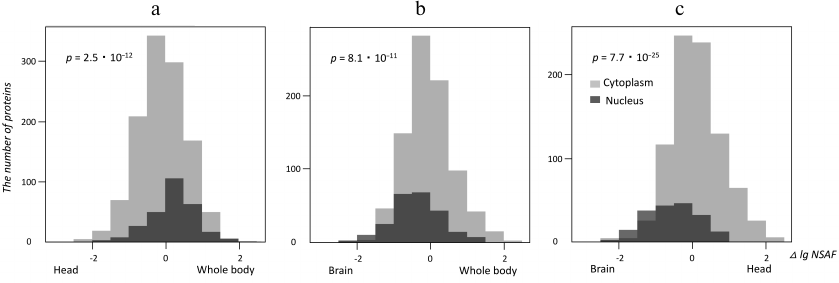
Using the Gene Ontology [14], we selected proteins annotated as localized in the nucleus or the cytoplasm in all three studies datasets (brain, head and whole body of Drosophila) in order to elucidate if relative abundances of nuclear and cytoplasmic proteins determined using the mass-spectrometric signal transformed by the NSAF method [13] were different in these datasets. To answer this question, logarithmically transformed NSAF values for the same cytoplasmic and nuclear proteins in different proteomes were subtracted pair-wise and presented as histograms in Fig. 2. The shifts between the “nuclear” and “cytoplasmic” histograms visualize the differences between the corresponding protein abundances, whose significance was estimated using the Kruskal–Wallis test [19]. As one could see from Fig. 2, nuclear proteins were more abundant than cytoplasmic proteins in the whole-body proteome as compared with the head proteome. As expected, the brain proteome had the highest relative content of nuclear proteins as compared to both reference proteomes. The lower level of nuclear proteins in the head proteome might be explained by a high content of eye tissue in this sample, which contained many large ommatidia cells rich with the cytoplasm.
Fig. 2. Quantitative enrichment of Drosophila brain proteome with nuclear proteins localization. Histograms illustrate label-free quantitative data for proteins annotated with “cytoplasm” or “nucleus” Gene Ontology terms [14]. Horizontal axis depicts differences of decimal logariphms of the NSAF quantitative parameter [13] for the same proteins in different proteomes (Δ log NSAF). The shifts between the “nuclear” and “cytoplasmic” data reflect quantitative enrichment of proteins with these annotations in the studied proteomes; the p-value of significant difference between the abundances of nuclear vs. cytoplasmic proteins is calculated using the Kruskal–Wallis test. a) Head and whole-body proteomes compared; b) brain and whole-body proteomes compared; c) brain and head proteomes compared.
In conclusion, we analyzed for the first time the Drosophila brain proteome and found several gene products that had not been identified before at the protein level. As expected, the brain proteome was a subset of previously published whole-body and head proteomes. Importantly, the brain proteome was found to be quantitatively enriched with nuclear proteins. Therefore, we believe it is reasonable to use isolated brain preparations instead of Drosophila heads in the studies of nuclear brain proteins (e.g., transcription factors) by proteomic analysis.
Funding
The work was supported by the Russian Science Foundation (project no. 17-15-01229).
Acknowledgements
The authors are thankful to Dr. N. I. Romanova (Department of Genetics, Biological Faculty, Lomonosov Moscow State University) for providing D. melanogaster flies.
Conflict of Interest
The authors declare no conflict of interest in financial or any other sphere.
Ethical Approval
All procedures with insects were conducted in accordance with the legal requirements adopted by the Russian Federation and international organizations (EU Directive 2010/63/EU and Appendix A of Convention ETS).
REFERENCES
1.Pandey, U. B., and Nichols, C. D. (2011) Human
disease models in Drosophila melanogaster and the role of the fly in
therapeutic drug discovery, Pharmacol. Rev., 63,
411-436.
2.Vierstraete, E., Cerstiaens, A., Baggerman, G., Van
den Bergh, G., De Loof, A., and Schoofs, L. (2003) Proteomics in
Drosophila melanogaster: first 2D database of larval hemolymph
proteins, Biochem. Biophys. Res. Commun., 304,
831-838.
3.Scigelova, M., and Makarov, A. (2006) Orbitrap mass
analyzer – overview and applications in proteomics,
Proteomics, 6 (Suppl. 2), 16-21.
4.Tyanova, S., Albrechtsen, R., Kronqvist, P., Cox,
J., Mann, M., and Geiger, T. (2016) Proteomic maps of breast cancer
subtypes, Nat. Commun., 7, 10259.
5.Vizcaino, J. A., Cote, R. G., Csordas, A., Dianes,
J. A., Fabregat, A., Foster, J. M., Griss, J., Alpi, E., Birim, M.,
Contell, J., O’Kelly, G., Schoenegger, A., Ovelleiro, D.,
Perez-Riverol, Y., Reisinger, F., Rios, D., Wang, R., and Hermjakob, H.
(2013) The PRoteomics IDEntifications (PRIDE) database and associated
tools: status in 2013, Nucleic Acids Res., 41,
D1063-D1069.
6.Xing, X., Zhang, C., Li, N., Zhai, L., Zhu, Y.,
Yang, X., and Xu, P. (2014) Qualitative and quantitative analysis of
the adult Drosophila melanogaster proteome, Proteomics,
14, 286-290.
7.Aradska, J., Bulat, T., Sialana, F. J.,
Birner-Gruenberger, R., Erich, B., and Lubec, G. (2015) Gel-free mass
spectrometry analysis of Drosophila melanogaster heads,
Proteomics, 15, 3356-3360.
8.Casas-Vila, N., Bluhm, A., Sayols, S., Dinges, N.,
Dejung, M., Altenhein, T., Kappei, D., Altenhein, B., Roignant, J.-Y.,
and Butter, F. (2017) The developmental proteome of Drosophila
melanogaster, Genome Res., 27, 1273-1285.
9.Kuznetsova, K. G., Kliuchnikova, A. A., Ilina, I.
U., Chernobrovkin, A. L., Novikova, S. E., Farafonova, T. E., Karpov,
D. S., Ivanov, M. V., Goncharov, A. O., Ilgisonis, E. V., Voronko, O.
E., Nasaev, S. S., Zgoda, V. G., Zubarev, R. A., Gorshkov, M. V., and
Moshkovskii, S. A. (2018) Proteogenomics of adenosine-to-inosine RNA
editing in fruit fly, J. Proteome Res., 17,
3889-3903.
10.Vizcaino, J. A., Deutsch, E. W., Wang, R.,
Csordas, A., Reisinger, F., Rios, D., Dianes, J. A., Sun, Z., Farrah,
T., Bandeira, N., Binz, P.-A., Xenarios, I., Eisenacher, M., Mayer, G.,
Gatto, L., Campos, A., Chalkley, R. J., Kraus, H.-J., Albar, J. P.,
Martinez-Bartolome, S., Apweiler, R., Omenn, G. S., Martens, L., Jones,
A. R., and Hermjakob, H. (2014) ProteomeXchange provides globally
coordinated proteomics data submission and dissemination, Nat.
Biotech., 32, 223-226.
11.Levitsky, L. I., Ivanov, M. V., Lobas, A. A.,
Bubis, J. A., Tarasova, I. A., Solovyeva, E. M., Pridatchenko, M. L.,
and Gorshkov, M. V. (2018) IdentiPy: an extensible search engine for
protein identification in shotgun proteomics, J. Proteome Res.,
17, 2249-2255.
12.Craig, R., and Beavis, R. C. (2003) A method for
reducing the time required to match protein sequences with tandem mass
spectra, Rapid Commun. Mass Spectrom., 17, 2310-2316.
13.Paoletti, A. C., Parmely, T. J., Tomomori-Sato,
C., Sato, S., Zhu, D., Conaway, R. C., Conaway, J. W., Florens, L., and
Washburn, M. P. (2006) Quantitative proteomic analysis of distinct
mammalian Mediator complexes using normalized spectral abundance
factors, Proc. Natl. Acad. Sci. USA, 103,
18928-18933.
14.Blake, J. A., Dolan, M., Drabkin, H., Hill, D.
P., Ni, L., Sitnikov, D., Bridges, S., Burgess, S., Buza, T., McCarthy,
F., Peddinti, D., Pillai, L., Carbon, S., Dietze, H., Ireland, A.,
Lewis, S. E., Mungall, C. J., Gaudet, P., Chisholm, R. L., Fey, P.,
Kibbe, W. A., Basu, S., Siegele, D. A., McIntosh, B. K., Renfro, D. P.,
Zweifel, A. E., Hu, J. C., Brown, N. H., Tweedie, S., Alam-Faruque, Y.,
Apweiler, R., Auchinchloss, A., Axelsen, K., Bely, B., Blatter, M.-C.,
Bonilla, C., Bougueleret, L., Boutet, E., Breuza, L., Bridge, A., Chan,
W. M., Chavali, G., Coudert, E., Dimmer, E., Estreicher, A.,
Famiglietti, L., Feuermann, M., Gos, A., Gruaz-Gumowski, N., Hieta, R.,
Hinz, U., Hulo, C., Huntley, R., James, J., Jungo, F., Keller, G.,
Laiho, K., Legge, D., Lemercier, P., Lieberherr, D., Magrane, M.,
Martin, M. J., Masson, P., Mutowo-Muellenet, P., O’Donovan, C.,
Pedruzzi, I., Pichler, K., Poggioli, D., Porras Millan, P., Poux, S.,
Rivoire, C., Roechert, B., Sawford, T., Schneider, M., Stutz, A.,
Sundaram, S., Tognolli, M., Xenarios, I., Foulger, R., Lomax, J.,
Roncaglia, P., Khodiyar, V. K., Lovering, R. C., Talmud, P. J.,
Chibucos, M., Gwinn Giglio, M., Chang, H.-Y., Hunter, S., McAnulla, C.,
Mitchell, A., Sangrador, A., Stephan, R., Harris, M. A., Oliver, S. G.,
Rutherford, K., Wood, V., Bahler, J., Lock, A., Kersey, P. J.,
McDowall, M. D., Staines, D. M., Dwinell, M., Shimoyama, M.,
Laulederkind, S., Hayman, T., Wang, S.-J., Petri, V., Lowry, T.,
D’Eustachio, P., Matthews, L., Balakrishnan, R., Binkley, G.,
Cherry, J. M., Costanzo, M. C., Dwight, S. S., Engel, S. R., Fisk, D.
G., Hitz, B. C., Hong, E. L., Karra, K., Miyasato, S. R., Nash, R. S.,
Park, J., Skrzypek, M. S., Weng, S., Wong, E. D., Berardini, T. Z., Li,
D., Huala, E., Mi, H., Thomas, P. D., Chan, J., Kishore, R., Sternberg,
P., Van Auken, K., Howe, D., and Westerfield, M. (2013) Gene ontology
annotations and resources, Nucleic Acids Res., 41,
D530-D535.
15.Gaudet, P., Michel, P.-A., Zahn-Zabal, M.,
Britan, A., Cusin, I., Domagalski, M., Duek, P. D., Gateau, A.,
Gleizes, A., Hinard, V., Rech de Laval, V., Lin, J., Nikitin, F.,
Schaeffer, M., Teixeira, D., Lane, L., and Bairoch, A. (2017) The
neXtProt knowledgebase on human proteins: 2017 update, Nucleic Acids
Res., 45, D177-D182.
16.Moshkovskii, S. A., Ivanov, M. V., Kuznetsova, K.
G., and Gorshkov, M. V. (2018) Identification of single amino acid
substitutions in proteogenomics, Biochemistry (Moscow),
83, 250-258.
17.Chernobrovkin, A. L., Kopylov, A. T., Zgoda, V.
G., Moysa, A. A., Pyatnitskiy, M. A., Kuznetsova, K. G., Ilina, I. Y.,
Karpova, M. A., Karpov, D. S., Veselovsky, A. V., Ivanov, M. V.,
Gorshkov, M. V., Archakov, A. I., and Moshkovskii, S. A. (2015)
Methionine to isothreonine conversion as a source of false discovery
identifications of genetically encoded variants in proteogenomics,
J. Proteomics, 120, 169-178.
18.UniProt Consortium (2015) UniProt: a hub for
protein information, Nucleic Acids Res., 43,
D204-D212.
19.Kruskal, W., and Wallis, W. A. (1952) Use of
ranks in one-criterion variance analysis, J. Am. Stat. Assoc.,
47, 583-621.