Kinetic Measurements of Singlet Oxygen Phosphorescence in Hydrogen-Free Solvents by Time-Resolved Photon Counting
A. A. Krasnovsky1,2,a*, A. S. Benditkis1, and A. S. Kozlov1
1Federal Research Center of Biotechnology, A. N. Bach Institute of Biochemistry, Russian Academy of Sciences, 119071 Moscow, Russia2M. V. Lomonosov Moscow State University, Faculty of Biology, 119234 Moscow, Russia
* To whom correspondence should be addressed.
Received September 2, 2018; Revised October 29, 2018; Accepted October 29, 2018
Solvents lacking hydrogen atoms are very convenient models for elucidating the properties of singlet oxygen, since the lifetime of singlet oxygen in these solvents reaches tens milliseconds. Measuring intrinsic infrared (IR) phosphorescence of singlet oxygen at 1270 nm is the most reliable method of singlet oxygen detection. However, efficient application of the phosphorescence method to these models requires an equipment allowing reliable measurement of the phosphorescence kinetic parameters in the millisecond time range at low rates of singlet oxygen generation, which is a technically difficult problem. Here, we describe a highly sensitive LED (laser) spectrometer recently constructed in our laboratory for the steady-state and time-resolved measurements of the millisecond phosphorescence of singlet oxygen. In the steady-state mode, this spectrometer allows detection of singlet oxygen phosphorescence upon direct excitation of oxygen molecules in the region of dark-red absorption bands at 690 and 765 nm. For kinetic measurements, we used phenalenone as a photosensitizer, microsecond pulses of violet (405 nm) LED for excitation (irradiance intensity, ≤50 μW/cm2), a photomultiplier and a computer multichannel scaler for time-resolved photon counting. The decays of singlet oxygen in air-saturated CCl4, C6F6, and Freon 113 and quenching of singlet oxygen by phenalenone and dissolved molecules of triplet oxygen were measured. The relative values of the radiative rate constants of singlet oxygen in these media were determined. The results were compared with the absorption coefficients of oxygen measured by our group using the methods of laser photochemistry. Critical discussion of the obtained results and the data of other researchers is presented.
KEY WORDS: singlet oxygen, phosphorescence, LED (laser) spectrometer, time-resolved photon counting, lifetime, quenching by oxygen, absorption coefficients of oxygen molecules, solvents having no hydrogen atoms (perhalogenated solvents)DOI: 10.1134/S0006297919020068
Abbreviations: cw, continuous wave (waveform); DPBF, 1,3-diphenylisobenzofuran; Freon 113, 1,1,2-trifluoro-1,2,2-trichloroethane; IR, infrared; LED, light-emitting diode; TPP, tetraphenylporphin.
Photosensitized IR phosphorescence of singlet
(1Δg) oxygen (1O2)
(Fig. 1) is widely used for investigating the
properties of singlet oxygen and photodynamic photosensitizers in
connection with numerous theoretical and applied problems of
photophysics, photochemistry, and photomedicine [1,
2].
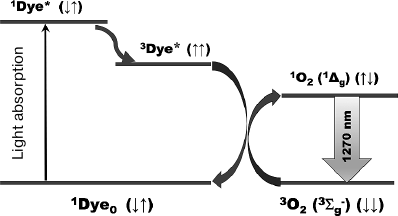
Fig. 1. Mechanism of photosensitized IR phosphorescence of singlet oxygen. 1Dye0, 1Dye* and 3Dye* are molecules of photosensitizer pigments in the ground and excited singlet and triplet states; 3O2 and 1O2 are oxygen molecules in the ground and singlet states.
The first measurements of singlet oxygen phosphorescence in hydrogen-free solvents (CCl4, Freon 113, and CS2) were performed by one of the authors of this paper in 1976-1977 ([3] and references therein). Initially, the phosphorescence had been detected using spectrometers with mechanical phosphoroscopes and photodetector systems based on cooled photomultiplier tubes with the spectral response S-1 (FEU-83). The construction and the principles of operation of these spectrometers have been described in detail in [3, 4] and in following publications ([1] and references therein). These devices allowed measurements of excitation spectra, emission spectra, and lifetimes of phosphorescence of ≥0.7 ms. It was found that the lifetime of singlet oxygen (τΔ) in these solvents is >20 ms [3], which is much greater than the value of ~1 ms previously obtained using singlet oxygen traps [5]. Detailed studies of phosphorescence in CCl4 led to the conclusion that τΔ decreases with the increase in the concentration of photosensitizer pigments due to the quenching of 1O2 by non-excited pigment molecules. The maximal τΔ ~ 30 ms was obtained in pheophytin a solution (1 μM) in CCl4 additionally purified by double distillation at the excitation light power density of 300-700 μW/cm2. Similar lifetime was observed in the commercial samples of high-purity (≥99.5%) CCl4 without additional distillation [3]. Under these conditions, no destruction of the photosensitizers was observed within the period of irradiation. Quenching of 1O2 by air oxygen was not studied in these experiments.
Therefore, it was found that the solvents, molecules of which do not have hydrogen atoms, are exceptionally convenient models for elucidating the properties and reactivity of singlet oxygen by the phosphorescence method. Using CCl4 and τΔ ~ 30 ms, the rate constants of 1O2 quenching have been measured for many biologically important compounds including porphyrins, chlorophylls, carotenoids, water, 1,4-diazobicyclo[2,2,2]octane, α-tocopherol, etc. The obtained rate constants coincided with those measured by other methods, which confirms that the above τΔ value obtained by the phosphorescence method was correct ([1, 3] and references therein).
In 1979, a group from the Institute of Physics, Minsk, constructed a spectrometer for the time-resolved phosphorescence measurements after irradiation with short pulses of lasers or flash lamps using a photomultiplier FEU-83 as a photodetector and a storage oscilloscope [6-8]. In particular, they observed biphasic nonexponential decay of oxygen phosphorescence in CCl4 upon excitation with high-power laser pulses. The authors proposed that this effect was due to the recombination of 1O2 molecules occurring at high 1O2 concentrations. The decay became exponential with the lifetime of 30 ms [6] with the decrease in the pulse power to the minimal value (7 mJ). Therefore, the authors arrived to the τΔ value similar to that obtained in our experiments. In solvents containing hydrogen atoms, the phosphorescence lifetime decreased to 10-100 μs [6-8].
In 1987, Schmidt and Brauer constructed a setup with a mercury lamp for phosphorescence excitation and a germanium photodiode for phosphorescence detection. The exciting light was passed through a mechanical chopper to obtain light pulses with 10 ms duration and 2.5 Hz repetition rate. The time constant of the photodiode and its amplifying system was 5.1 ms. To eliminate distortion of the kinetic curves by the instrument hardware function, the signal was processed with a computer program to account for a slow response of the photodetector system. Hence, the resulting curves depended on the efficiency of the applied software. Using this setup, the following values of τΔ were obtained: 87 ms in CCl4, 99 ms in Freon 113, and 24 ms in hexafluorobenzene [9, 10].
In parallel, Losev et al. reported the results of time-resolved and steady-state measurements of τΔ in CCl4 and CS2 using photodiodes as photodetectors [11, 12]. The authors found that at the minimal power of excitation light and the minimal photosensitizer (Pd-mesoporphyrin) concentration, τΔ was ~30 ms at room temperature in both solvents. At lower temperature (193K), a 30% increase in the lifetime was observed. The discrepancy with the data of the Schmidt’s group was explained by a higher degree of solvent purification in Schmidt’s experiments. It is of interest that some authors [6, 9-12] raised the question of the involvement of air oxygen in the quenching of singlet oxygen phosphorescence. However, they all came to the conclusion that this process is not efficient.
In 1991, Schmidt’s group improved the procedure of data processing and established that after removal of air, the phosphorescence lifetime increased by 1.5-2 times. Increasing the concentration of oxygen to a partial pressure of 1 bar decreased τΔ to 10-20 ms [13]. It was concluded that air oxygen reduces the lifetime of singlet oxygen in aerated solutions. In aerated CCl4, the lifetime of singlet oxygen was 59 ± 5 ms, which is 1.5 times less than the value reported by this group earlier (87 ms) [9, 10]. Smaller τΔ values were also obtained in Freon 113 (72 ms) and hexafluorobenzene (21 ms) [13]. The same values were reported in the review paper of Schweitzer and Schmidt [14] with no clear explanation of the reasons for the discrepancy with the earlier data [9, 10]. Until now, these experiments have not been repeated by anyone, although it is obvious that their verification is of fundamental importance.
More recently, kinetic parameters of singlet oxygen phosphorescence in CCl4 were investigated by several groups. Bagrov et al. used a flash lamp, a germanium photodetector, and fullerenes C70 and C60 as photosensitizers. At room temperature and excitation power density of 10 mJ/cm2, τΔ varied between 22 and 55 ms [15, 16]. In agreement with Salokhiddinov et al. [6] and Losev et al. [11, 12], the authors observed that the phosphorescence lifetime in CCl4 decreased with the increase in the excitation power and solution temperature.
Wang et al. reproduced these experiments using a Hamamatsu IR photomultiplier. At the lowest power of laser pulses (10 mJ), the lifetime in C60 (200 µM) solution in CCl4 was 16-17 ms [17]. Simultaneously, they observed oxygenation of C60 and quenching of 1O2 by the oxygenation products. Hasebe et al. [18] used Pt(II)-meso-tetra(pentafluorophenyl)porphin (7.4 μM) as a sensitizer and pulses of Nd:YAG laser (8 ns, 10 Hz) for excitation. The obtained τΔ value in CCl4 was 5.04 ms. Extrapolation of the data to the photosensitizer zero concentration leads to τΔ ~ 14 ms (no information about the pulse energy was provided).
The discrepancies in the determined values of the singlet oxygen lifetime in hydrogen-free solvents have caused doubts in the reliability of the used model systems. As a result, some researchers question or even refuse to use them. To obtain reliable information, one needs instrumentation that could measure the stationary intensity and temporal parameters of phosphorescence under a very low excitation power, at which the nonlinear and destructive effects described above are excluded. The first spectrometers developed in our laboratory [3] have solved this problem in general. Although they provided the measurements of the spectral parameters and average lifetime of phosphorescence, these devices did not allow recording the phosphorescence decay traces.
By the present time, the technique for detection of singlet oxygen phosphorescence has been significantly improved. The instruments with time-resolved photon counting are the most effective for kinetic measurements. The first setup of this type designed for measurement of singlet oxygen phosphorescence with microsecond resolution was constructed in our laboratory [19, 20]. Later, its nanosecond modification operating in the regime of time-correlated single photon counting was constructed [21]. At present, such devices are widely used by many researchers ([1, 2] and references therein). However, as mentioned above, all of them have been designed for studying rapidly decaying delayed light emissions.
A new setup recently constructed by our group and briefly described in our preliminary publications [22, 23] has been specially designed for investigation of slowly decaying delayed light emissions using the time-resolved photon counting technique. Here, we used this setup for kinetic measurements of photosensitized phosphorescence of singlet oxygen in solvents weakly deactivating 1O2 upon excitation by LED pulses, whose energy is 3-4 orders lower than in all the above-mentioned papers employing the pulsed technique. We also demonstrated that this device can be used to record phosphorescence of singlet oxygen in pigment-free solvents occurring upon direct excitation of oxygen by dark-red light that corresponds to the oxygen absorption bands at 690 and 765 nm.
MATERIALS AND METHODS
The functional layout of the new spectrometer is shown in Fig. 2. For the steady-state luminescence measurements, a light-emitting diode (LED) was used with the emission maximum at 399 nm and a half-width of 14 nm (Polironik, Russia). The photodiode radiation was focused into a 5-mm spot on the surface of a quartz cell containing the analyzed solution. The intensity of the exciting light was monitored by a ThorLabs PM-100D power meter with a S120VC sensor head (Thorlabs, USA). In separate experiments, diode lasers with the emission maxima of 765 nm (half-width, 2 nm; power, up to 1 W) (LAMI Helios, Russia) and 690 nm (half-width, 3 nm; power, up to 5 W) (Milon-Lakhta, Russia) were used. The laser radiation power was measured using an Ophir ORION-TH instrument with a 20C-SH sensor head (Ophir, Israel). The phosphorescence of singlet oxygen was measured at a 90° angle with a cooled photomultiplier FEU-112 (S1 spectral response) (Ekran, Russia) through IR filters transmitting IR light at λ ≥ 950 nm and one of three interchangeable interference light filters with the transmission peaks at 1230, 1270, and 1310 nm and a half-width of 10 nm. The steady-state phosphorescence intensity was measured with a digital high-resistance millivoltmeter (Econix-Expert, Russia); the obtained data were processed with a computer.
Fig. 2. Block diagram of laser (LED) spectrometers for stationary and time-resolved measurements of singlet oxygen IR phosphorescence (1270 nm) in the solvents that weakly deactivate singlet oxygen.
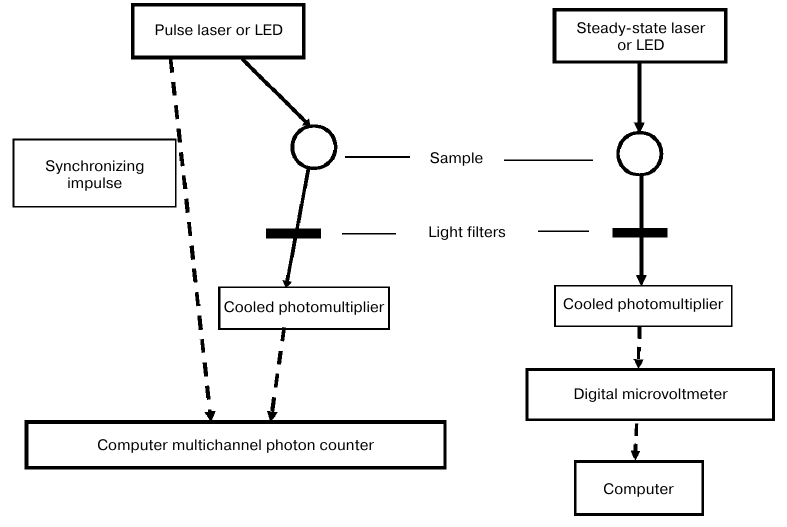
For kinetic measurements, a pulse LED (Polironik) with an operating unit (Alcom Medica, Russia) was used for sample irradiation with 405-nm monochromatic light (band half-width, 14 nm) with a pulse duration of 10 μs and pulse repetition rate of 5 or 10 Hz. The average power of the exciting light was 15-30 μW; the energy of one pulse was 3 μJ. The photomultiplier signal was sent through a preamplifier to a computer board that worked in two modes – time-resolved multichannel photon counting or time-resolved single photon counting (Parsek, Russia). The time constant of the recording system was ~10 ns. The counting board was started by an additional pulse of the operating unit synchronized with the LED pulse. The counting board divided the time interval between the pulses into 1024 channels; the kinetic curves were obtained by accumulating the photopulses in each channel. In this work, we did not use the method of time-correlated single photon counting.
The analyzed solution (1.5 ml) was placed in a 1-cm quartz cell. Phenalenone (perinaphthenone, 1H-phenalen-1-one) or tetraphenylporphyrin (Aldrich, EU) were used as photosensitizers. The concentration of phenalenone was calculated using the molar absorption coefficient at the absorbance maximum (9700 M–1·cm–1) [24]. In some experiments, the singlet oxygen trap 1,3-diphenylisobenzofuran (DPBF) was used (Acros Organics, Belgium). CCl4 (Labtech, Russia), hexafluorobenzene (Piminvest, Russia), and Freon 113 (1,1,2-trifluoro-1,2,2-trichloroethane) (Ruskhimprom, Russia) with ≥99.5-99.8% purity were used as solvents. The absorption spectra were recorded with an SF-56 spectrophotometer (LOMO Spektr, Russia).
RESULTS AND DISCUSSION
Steady-state measurements. Measurements of the steady-state photomultiplier signal in the direct photocurrent registration mode showed that illumination of air-saturated solutions of phenalenone or tetraphenylporphyrin in CCl4, hexafluorobenzene, or Freon 113 with continuous or pulsed LED radiation at ~400 nm resulted in 1O2 phosphorescence with the spectral maximum at 1270 nm (Fig. 3). Addition of 50% acetone led to a sharp decrease in the 1O2 lifetime and weakened the photosensitized phosphorescence ~500 times (Fig. 3). The IR phosphorescence in the air-saturated solvents was also observed in the absence of photosensitizers upon irradiation with LEDs at 400 nm [16, 25, 26]. However, the quantum efficiency of this phosphorescence was several orders of magnitude lower than in the photosensitizer-containing solutions. Therefore, it did not affect the overall intensity of photosensitized phosphorescence.
Fig. 3. Intensity and spectrum of 1O2 phosphorescence upon photosensitized (a, d) and direct (b-d) excitation of oxygen in air-saturated CCl4. a) Solution of phenalenone excited with cw LED 399 nm (4 mW) in the absence of acetone (upper curve) and after addition of 50% acetone (lower curve); b) in the absence of phenalenone upon oxygen excitation with a 765-nm diode laser (900 mW); c) in the absence of phenalenone upon oxygen excitation with a 690-nm diode laser (1-3 W); d) phosphorescence spectra corresponding to the curves (a)-(c) after subtraction of luminescence observed in the presence of acetone (from left to right).
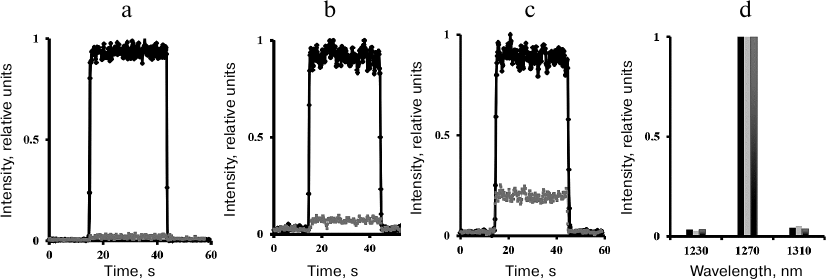
In the stationary mode, the spectrometer allows the recording of the oxygen phosphorescence upon irradiation of pigment-free solutions with 765- and 690-nm laser diodes (1-3 W), i.e., at the wavelengths corresponding to the oxygen absorption bands. The ability of a 765-nm laser to excite oxygen phosphorescence has already been described by Ogilby’s group [27] and in our previous publications [22, 23]. In this work, we report the excitation of oxygen by irradiation at 690 nm (1-3 W) (Fig. 3) (see [23] for preliminary data).
Acetone quenched the phosphorescence (Fig. 3): the phosphorescence intensity in the presence of 50% acetone corresponded to the level of the intrinsic luminescence of the quartz cell. It should be noted that Fig. 3 shows the phosphorescence intensity curves normalized to 1. In reality, the quantum efficiency of photosensitized phosphorescence is about five orders of magnitude higher than the quantum efficiency of phosphorescence caused by the dark-red light in the absence of the photosensitizer. Detailed analysis of the emission caused by dark-red lasers is beyond the scope of this work and will be presented in a separate publication.
In all solvents under cw excitation at 399 nm, the intensity of phosphorescence photosensitized by phenalenone linearly depended on the intensity of the exciting light (Iex) in the range of 0.05-4 mW (data not shown). At higher intensities, some deviation from linearity was observed, which was relatively weak in hexafluorobenzene but distinctly noticeable in CCl4 and Freon 113. For example, at the maximum photodiode power (200 mW), the phosphorescence intensity in CCl4 was twice as low as it would be expected at the linear dependence on the exciting light intensity observed at Iex ≤ 4 mW.
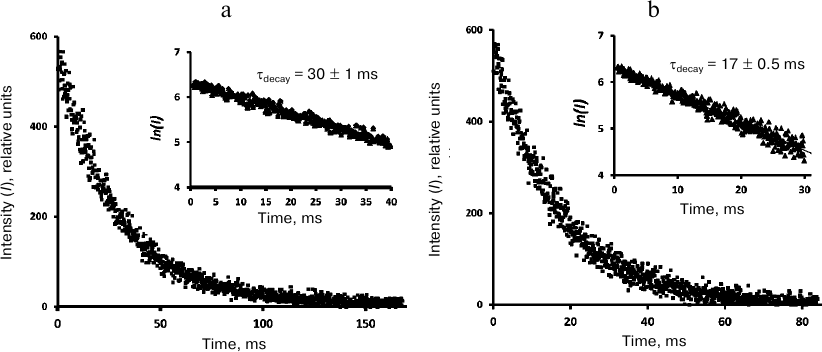
Kinetic measurements. The phosphorescence kinetic curves recorded at 1270 nm using the time-resolved photon counting setup after irradiation of phenalenone solutions with 10-μs LED pulses (405 nm) are shown in Figs. 4-6. The decay times were determined from the slopes of the kinetic curves on a semi-logarithmic scale. To obtain this parameter, the averaged luminescence traces from a quartz cell with a pure solvent were subtracted from the phosphorescence kinetic curves of phenalenone solutions obtained under the same irradiation conditions. The first 10 μs of the resulting kinetic curve were omitted due to possible contribution of 1O2 accumulation by the triplet molecules of the photosensitizer. The final part of the curves was discarded because of significant distortions due to the signal noise. Figure 4 demonstrates that the phosphorescence decays are exponential. In air-saturated CCl4 containing ~30 μM phenalenone, the phosphorescence decay time was 30 ± 1 ms (Fig. 4). Therefore, it coincides with the value reported in our earlier papers [3] and publications of the Minsk group [6-8, 11, 12], being half as much as reported by the Schmidt’s group (59 ms) [13, 14].
Fig. 4. Decays of 1O2 phosphorescence in phenalenone (35 μM) solutions in CCl4 after irradiation by 10 μs pulses of LED (405 nm) in the presence of air (a) and after saturation with oxygen (b). The curves were obtained at a pulse repetition rate of 5 Hz as a result of 30-min signal accumulation; duration of one channel, 164 μs; average excitation power, 15 μW. Insets, decay curves in the semi-log scale.
In the air-saturated hexafluorobenzene (Fig. 5), the lifetime was 16 ms. This value coincides with the result of our previous measurements performed with a setup with a phosphoroscope [3, 28] and is close enough to the latest data from the Schmidt’s group (21 ms) [13, 14]. In aerated Freon 113, the lifetime was ~37 ms (Fig. 6), which is 2 times smaller than in Schmidt’s work (72 ± 5 ms) [13, 14].
Fig. 5. Decays of 1O2 phosphorescence in phenalenone (35 μM) solutions in hexafluorobenzene after irradiation with 10-μs LED pulses (405 nm) in the presence of air (a) and after saturation with oxygen (b). The curves were recorded at a pulse repetition rate of 10 Hz as a result of 20-min signal accumulation; duration of one channel, 82 μs; average excitation power, 30 μW. Insets, decay curve in the semi-log scale.
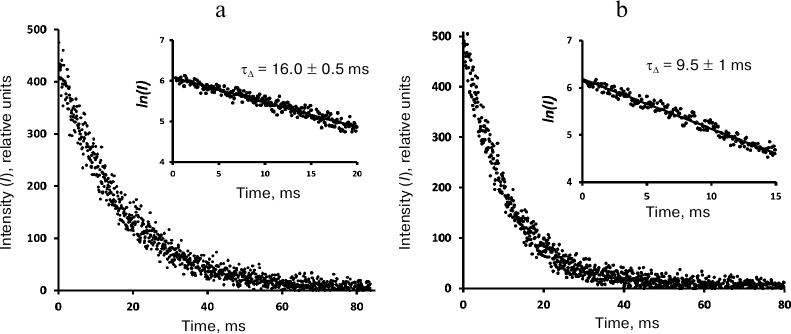
Fig. 6. Decays of 1O2 phosphorescence in phenalenone (35 μM) solutions in Freon 113 after irradiation by 10-μs LED pulses (405 nm) in the presence of air (a) and after saturation with oxygen (b). The curves were recorded at the pulse repetition rates of 5 Hz as a result of 30-min signal accumulation; duration of one channel, 164 μs; average excitation power, 15 μW. Insets, decay curves in the semi-log scale.
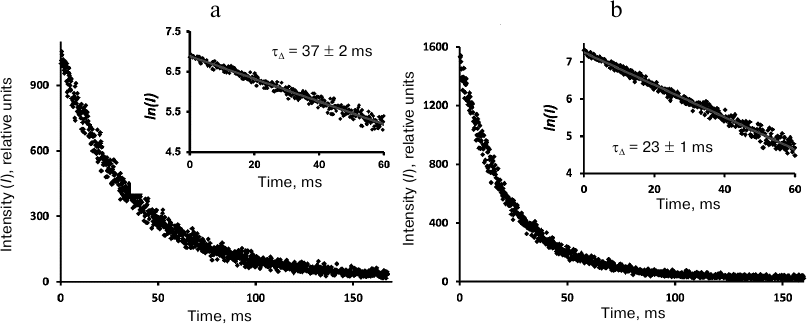
We also studied the influence of the concentrations of the photosensitizer and oxygen on the phosphorescence decay times. Figures 4-6 show that saturation of the solutions with pure oxygen (bubbling oxygen for 20 min increases its concentration in the solution by 4.8 times) caused the 1.5-1.7-fold increase in the phosphorescence decay rates. Also, the decay rates increased with the phenalenone concentration increase. These facts indicate that 1O2 is physically quenched by phenalenone (Ph) and triplet oxygen. The quenching efficiency can be described by the following version of the Stern–Volmer equation:
kobs = k0 + kox[O2] + kph[Ph], (1)
where kobs is the observed phosphorescence decay rate (kobs = 1/τdecay) in solutions containing phenalenone and oxygen; k0 is the decay rate in pure solvent; kox and kph are the rate constants for 1O2 quenching by triplet oxygen and phenalenone, respectively. It follows from the Eq. (1) that:
(kobs)oxygen – (kobs)air = kox([O2]oxygen – [O2]air). (2)
Table 1 shows the kox values calculated using Eq. (2), the decay times indicated in Figs. 4-6, and the literature data on the oxygen concentration in air-saturated solutions [29, 30]. The kox values obtained in CCl4 and hexafluorobenzene reasonably correlate with the data of Schmidt’s and other research groups [13, 14, 31]. A somewhat smaller constant was obtained in Freon 113. However, it should be noted that to our knowledge, there is no reliable information on the solubility of oxygen in this solvent. Therefore, to estimate kox, we calculated [O2]air using the data of the present work on the phosphorescence quantum efficiency and oxygen absorption coefficients. The results of these calculation are presented in Table 2. The obtained kox values appear to be reasonable. It should be noted that the boiling point of Freon 113 is 47°C; therefore, at room temperature, the volatility of this solvent is higher compared to other solvents. Hence, Freon 113 saturation by oxygen after oxygen purging might be less complete.
Table 1. Rates constants for
1O2 quenching by triplet oxygen
(kox) and phenalenone (kph) and the
decay rate constants (kobs) and lifetimes
(τΔ) of singlet oxygen phosphorescence in
solutions saturated with air and oxygen
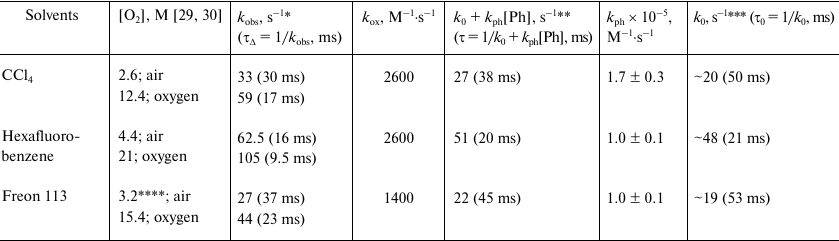
* Measurements in the presence of air and 35 μM phenalenone.
** Calculated decay rate constants and lifetimes of
1O2 in solvents in the absence of oxygen.
*** Calculated decay rate constants and the lifetimes of
1O2 in solvents in the absence of oxygen and
phenalenone.
**** Oxygen concentration was taken from the data of Table 2 (see below).
Table 2. Relative values of
kr for IR 1O2 emission band and
relative values of molar absorption coefficients in the oxygen
absorption maximum at about 1270 nm (ε1270)
calculated using relative kr values. Comparison with
the absolute values of A1270 and ε1270
obtained by the method of laser photochemistry [22, 34, 35]
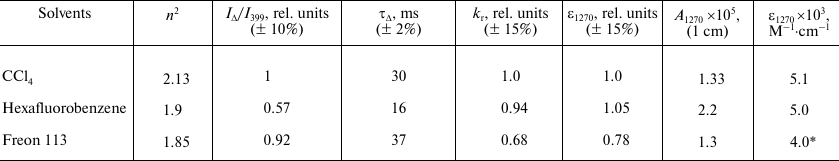
* This value was obtained by multiplying the relative value of
ε1270 in Freon 113 (0.78) by the absolute value of
ε1270 in
M–1·cm–1 in CCl4
(5.1).
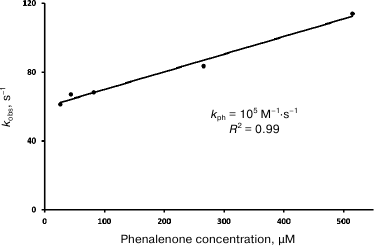
The dependences of the decay rates of 1O2 phosphorescence on phenalenone concentration in hexafluorobenzene and Freon 113 are linear in the Stern–Volmer coordinates (Fig. 7). Similar linear dependence was found in CCl4, although in this solvent, the data were more scattered (not shown). The kph values obtained in our experiments are shown in Table 1. These values are somewhat lower than the values measured in our laboratory earlier (3·105 M–1·s–1 in CCl4) [28], and slightly greater than the values reported by the Schmidt’s group (3.5·104 M–1·s–1) [13, 14].
Fig. 7. Quenching of singlet oxygen phosphorescence by phenalenone in air-saturated hexafluorobenzene; kobs is the rate constant of deactivation of singlet oxygen phosphorescence; R2 is the coefficient of determination.
Radiative constants and absorption coefficients. It is known that the lifetime of singlet oxygen (τΔ) is mainly characterized by nonradiative deactivation of 1O2, whereas the intensity of 1O2 phosphorescence is determined by the radiative rate constant (kr); its phosphorescence quantum yield (Φr) can be calculated as:
Φr = kr·τΔ. (3)
The radiative rate constant characterizes the electronic structure and the state of oxygen molecule and, according to the generally known Einstein relation, is directly proportional to the molar absorption coefficient:
ε1270 ~ kr/n2. (4)
Our spectrometer allowed us to compare the radiative rate constants of 1O2 deactivation in the studied solvents. Phenalenone solutions with the same optical density of 0.12 ± 0.005 at 399 nm were used for the measurements. The kr constants were calculated using the following equation for the steady-state intensity (IΔ) of photosensitized phosphorescence:
IΔ = (K/n2)ΦΔ I399 α (1 – 10–Aph)kr·τΔ, (5)
where K is a proportionality coefficient accounting for the device design; IΔ and I399 are the intensities (in photons per second) of photosensitized phosphorescence and the exciting light, respectively; n is the solvent refractive index; ΦΔ is the quantum yield of singlet oxygen generation by phenalenone (equal to ~1) [24, 32]; Aph is the phenalenone absorbance in the excitation region (399 nm); α is a coefficient showing the extent of overlapping of the LED emission band with the absorption band of phenalenone. Therefore,
kr = (n2 IΔ/I399)/τΔ KΦΔ α(1 – 10–Aph), (6)
where KΦΔ α(1 – 10–Aph) is the same for all solutions, therefore it was not taken into account in calculating the relative values of kr. The relative values of the molar absorption coefficient for the same band were calculated from the relative values of kr using the Einstein equation (4). The obtained values are summarized in Table 2. The IΔ/I399 ratio and the values of k0 and ε1270 in CCl4 were set to be 1. It follows from Table 2 that in all solvents studied in this work, the constants kr and ε1270 are rather similar within the indicated confidence interval. There is a tendency for some decrease in kr and ε1270 in Freon 113, which has been mentioned earlier [33].
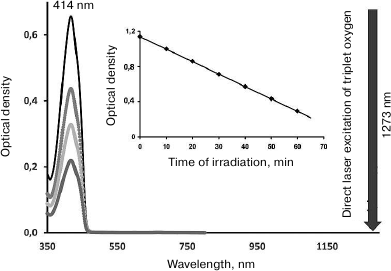
Over the past 15 years, our laboratory has developed a photochemical method for measuring absorbance (A1270 and A765) and molar absorption coefficients (ε1270 and ε765) for the absorption maxima of oxygen dissolved in many organic solvents and water. This method is based on the kinetic analyses of the rates of chemical trapping of singlet oxygen upon direct laser excitation of oxygen molecules (Fig. 8) [22, 33-35]. The most accurate results were obtained from comparative analysis of the chemical trapping rates upon direct (Vdir, M/s) and photosensitized (Vpsr, M/s) oxygen excitation [22, 33-35]. In this work, the absorbance of oxygen A1270 (A765 was not considered in the present paper) was calculated using the following equation:
A1270 = (Vdir/I1270)/(Vpsr/I511) ΦΔ(1 – 10–Aps)/(αlas 2.3), (7)
where I1270 and I511 are the fluence rates (in photons/second) for direct and photosensitized [tetraphenylporphine (TPP) was used as a photosensitizer in most cases mainly] excitation of oxygen, respectively; ΦΔ is the quantum yield of singlet oxygen generation by porphyrin (ΦΔ = 0.73); Aps is the optical density of TPP at the excitation wavelength (511 nm); αlas is the extent of overlapping of the diode laser emission band and the oxygen absorption spectrum [22, 34, 35]. The obtained values of A1270 are shown in Table 2. Given that
A1270 = ε1270 [O2] l, (8)
where l = 1 cm, one can easily obtain the values of ε1270 using known oxygen concentrations (see Table 2).
Fig. 8. Principles of singlet oxygen detection using the laser photochemistry method. The absorption spectrum of the singlet oxygen trap 1,3-diphenylisobenzofuran (maximum, 414 nm). The trap is bleached by the laser radiation at 1273 nm corresponding to the absorption maximum of dissolved oxygen. The absorbance of oxygen at 1270 nm is ~10–5 [22, 33-35], therefore, on the scale of this figure, this maximum is not seen.
To our knowledge, the exact value of oxygen solubility in Freon 113 has not been described yet, and only the estimates are available [13, 31]. This value can be determined using our data from Table 2. For this purpose, we used the ε1270 value in CCl4 obtained by the laser photochemistry methods [22, 34, 35]. The ε1270 value in Freon 113 was calculated by multiplying ε1270 in CCl4 by the relative value of ε1270, which has been obtained from the photosensitized phosphorescence of oxygen. Using A1270 in Freon 113 measured by the method of laser photochemistry and Eq. (8), we arrived to [O2] = 3.2 mM. In Table 2, this value was used to calculate the rate constant for 1O2 quenching by triplet oxygen. Interestingly, a similar procedure, after some improvement, can be applied to estimating the oxygen solubility in any medium that allows spectral measurements.
Thus, the spectrometers developed in our lab provide reliable measurements of the kinetic parameters of singlet oxygen phosphorescence in solvents, in which the lifetime of 1O2 reaches tens of milliseconds. From the technical point of view, the designed spectrometers are more advanced and informative than all the instruments used previously to study the kinetic parameters of oxygen phosphorescence in such solvents. As indicated, the kinetic curves were measured upon the irradiation with a very low-intensity exciting light (15-30 μW), which is orders of magnitude lower than used previously. This excludes the contribution of nonlinear effects observed earlier [6]. The temporal resolution of the photodetector system (nanoseconds) could not introduce distortions in the decay kinetics. The kinetic measurements performed with these instruments led to the value of the singlet oxygen lifetime in CCl4 (30 ms) similar to those obtained in our first papers [3] and subsequent works of the Minsk researchers [6-8, 11, 12]. However, this value is twice as little as the value obtained by the Schmidt’s group [13, 14]. In Freon 113, the measured lifetime was 37 ms, which is also a half of the value obtained by the Schmidt’s group [13, 14]. In hexafluorobenzene, our data and the data of the Schmidt’s group [13, 14] were rather similar (Table 1 and Figs. 5 and 6).
More recently, other groups have reported the values of τΔ in CCl4, which were within 14-55 ms [15-18]. Unfortunately, the information on the conditions for their measurements is insufficient for detailed analysis of the results. The authors mostly assumed that the higher τΔ values obtained by the Schmidt’s group were due to a higher purity of the solvents used in the experiments. Indeed, Schmidt and colleagues indicated that “all solvents were purified by repeated chromatography on columns with Al2O3”. In our experiments and in the experiments of other groups, highly purified commercial solvents were used without further purification or after additional distillation. On the other hand, it is obvious that the phosphorescence setup developed by the Schmidt’s group is not perfect (see the introductory section). So, it cannot be ruled out that the delay of the most slowly decaying curves was due to technical problems. Nevertheless, our measurements confirm the observation of the Schmidt’s group that 1O2 is quenched by triplet air oxygen and phenalenone. The quenching rate constants obtained in our experiments are in a reasonable agreement with the Schmidt’s data.
By comparing the phosphorescence quantum yields in phenalenone solutions, the relative rates of 1O2 radiative deactivation were determined. It was established that the relative values of radiative constants and relative absorption coefficients obtained using the laser photochemistry methods are virtually similar. The rationale for this idea in regard to a wider range of solvents has been given in our previous publications [1, 22, 33-35]. In addition, as mentioned above, the designed devices open up new possibilities for studying 1O2 generation and quenching. Also, as Fig. 3 indicates, they open new prospects in studying the generation of singlet oxygen by dark-red light in pigment-free aerated systems.
Funding
This work was supported in part by the Program of the Presidium of the Russian Academy of Sciences “Basic Science – for Medicine” and by the Russian Foundation for Basic Research (projects nos. 15-04-05500, 19-04-00331).
Acknowledgements
The authors are grateful to the Innovative Surgical Technologies Ltd, Moscow, and Milon-Lakhta, Ltd, St. Petersburg, for the diode laser and technical support.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
1.Krasnovsky, A. A., Jr. (2015) Singlet oxygen and
primary mechanisms of photodynamic and laser medicine, in Basic
Science for Medicine: Biophysical and Medical Technology
(Grigoriev, A. I., and Vladimirov, Yu. A., eds.) [in Russian], Maks
Press Ltd., Moscow, pp. 173-217.
2.Nonell, S., and Flors, C. (2016) Singlet Oxygen:
Applications in Biosciences and Nanosciences (Comprehensive
Series in Photochemistry and Photobiology, No. 13), The
Royal Society of Chemistry, European Society for Photobiology, UK.
3.Krasnovsky, A. A., Jr. (1979) Photoluminescence of
singlet oxygen in pigment solutions, Photochem. Photobiol.,
29, 29-36.
4.Krasnovsky, A. A., Jr. (1993) Detection of
photosensitized singlet oxygen luminescence in systems of biomedical
importance. Steady-state and time-resolved spectral measurements based
on application of S-1 photomultiplier tubes, SPIE Proc.,
1887, 177-186.
5.Merkel, P. B., and Kearns, D. R. (1972)
Radiationless decay of singlet molecular oxygen in solution. An
experimental and theoretical study of electronic-to-vibrational energy
transfer, J. Amer. Chem. Soc., 94, 7244-7253.
6.Salokhiddinov, K. I., Byteva, I. M., and Dzhagarov,
B. M. (1979) Duration of luminescence of singlet oxygen in solutions
under pulsed laser excitation, Russ. Opt. Spektr., 47,
881-886.
7.Byteva, I. M., and Gurinovich, G. P. (1979)
Sensitized luminescence of oxygen in solutions, J. Luminescence,
21, 17-20.
8.Byteva, I. M. (1979) Investigation of luminescence
of oxygen in solutions by the method of pulsed spectroscopy, Zh.
Prikl. Spektr. (J. Appl. Spektr., Minsk), 31,
333-335.
9.Schmidt, R., and Brauer, H.-D. (1987) Radiationless
deactivation of singlet oxygen (1Δg) by
solvent molecules, J. Am. Chem. Soc., 109, 6976-6981.
10.Schmidt, R. (1989) Influence of heavy
atoms on the deactivation of singlet oxygen
(1Δg) in solution, J. Am. Chem.
Soc., 111, 6983-6987.
11.Losev, A. P., Byteva, I. M., and Gurinovich, G.
P. (1988) Singlet oxygen luminescence yield in organic solvents and
water, Chem. Phys. Lett., 143, 127-129.
12.Losev, A. P., Byteva, I. M., and Gurinovich, G.
P. (1989) Deactivation of singlet oxygen in CCl4 and
CS2, Russ. Chem. Phys., 8, 732-739.
13.Afshari, E., and Schmidt, R. (1991)
Isotope-dependent quenching of singlet molecular oxygen
(1Δg) by ground-state oxygen in several
perhalogenated solvents, Chem. Phys. Lett., 184,
128-132.
14.Schweitzer, C., and Schmidt, R. (2003) Physical
mechanisms of generation and deactivation of singlet oxygen, Chem.
Rev., 103, 1685-1757.
15.Bagrov, I. V., Belousova, I. M., Danilov, O. B.,
Kiselev, V. M., Murav’eva, T. D., and Sosnov, E. N. (2007)
Photoinduced quenching of luminescence of singlet oxygen in solutions
of fullerenes, Opt. Spectrosc., 102, 52-59.
16.Bagrov, I. V., Kiselev, V. M., Kislyakov, I. M.,
and Sosnov, E. N. (2014) Direct optical excitation of singlet oxygen in
organic solvents, Opt. Spectrosc., 116, 567-574.
17.Wang, J., Leng, J., Yang, H., Sha, G., and Zhang,
C. (2014) Luminescence properties and kinetic analysis of singlet
oxygen from fullerene solutions, J. Luminescence, 149,
267-271.
18.Hasebe, N., Suzuki, K., Horiuchi, H., Suzuki, H.,
Yoshihara, T., Okutsu, T., and Tobita, S. (2015) Absolute
phosphorescence quantum yields of singlet molecular oxygen in solution
determined using an integrating sphere instrument, Anal. Chem.,
87, 2360-2366.
19.Egorov, S. Yu., and Krasnovsky, A. A., Jr. (1983)
Photosensitized luminescence of singlet oxygen under pulse laser
excitation. Decay kinetics in aqueous solutions, Biophysics,
28, 497-498.
20.Krasnovsky, A. A., Jr., Egorov, S. Yu., Nasarova,
O. V., Yartsev, E. I., and Ponomarev, G. V. (1988) Photosensitized
formation of singlet molecular oxygen in solutions of water-soluble
porphyrins. Direct luminescence measurements, Stud. Biophys.,
124, 123-142.
21.Egorov, S. Yu., Kamalov, V. F., Koroteev, N. I.,
Krasnovsky, A. A., Jr., Toleutaev, B. N., and Zinukov, S. V. (1989)
Rise and decay kinetics of photosensitized singlet oxygen luminescence
in water. Measurements with nanosecond time-correlated photon counting
technique, Chem. Phys. Lett., 163, 421-424.
22.Krasnovsky, A. A., Jr., and Kozlov, A. S. (2017)
Laser photochemistry of oxygen. Application to studies of the
absorption spectra of dissolved oxygen molecules, J. Biomed. Photon.
Eng., 3, 1-10.
23.Benditkis, A. S., Kozlov, A. S., Goncharov, S.
E., and Krasnovsky, A. A., Jr. (2018) Absorption of dark red laser
light by oxygen molecules in organic media. Results of photochemical
and luminescence measurements, Proc. Int. Conf, on Laser Optics
(ICLO 2018), IEEE Xplor Digital Library, p. 598.
24.Oliveros, E., Suardi-Murasecco, P.,
Aminian-Saghafi, T., and Braun, A. M. (1991) 1H-phenalen-1-one:
photophysical properties and singlet oxygen production, Helv. Chim.
Acta, 74, 79-90.
25.Krasnovsky, A. A., Jr., Sukhorukov, V. L.,
Egorov, S. Yu., and Potapenko, A. Ya. (1986) Generation and quenching
of singlet molecular oxygen by furocoumarins. Direct luminescence
measurements, Stud. Biophys., 114, 149-158.
26.Scurlock, R. D., and Ogilby, P. R. (1989) Singlet
molecular oxygen (1Δ2O2)
formation upon irradiation of an oxygen
(3Σg–O2)
organic molecule charge-transfer absorption band absorption band, J.
Phys. Chem., 93, 5493-5500.
27.Bregnhoj, M., Krǣgpoth, M. V., Serensen, R.
J., Westberg, M., and Ogilby, P. R. (2016) Solvent and heavy-atom
effects on the
O2(X3Σg–)
→ O2(b1Σg+)
absorption transition, J. Phys. Chem. A, 120,
8285-8296.
28.Krasnovsky, A. A., Jr., and Neverov, K. V. (2010)
On the mechanism of photosensitized luminescence of singlet oxygen
dimols in air-saturated pigment solutions, Biophysics,
55, 349-352.
29.Batino, R., Rettich, T. R., and Tominaga, T.
(1983) The solubility of oxygen and ozone in liquids, J. Phys. Chem.
Ref. Data, 12, 163-178.
30.Murov, S. L., Charmichael, I., and Hug, G. L.
(1993) Handbook of Photochemistry, Marcel Dekker Inc., New
York-Basel-Hong Kong.
31.Matheson, I. B. C., Lee, J., Yamanashi, B. S.,
and Wolbarsht, M. L. (1974) Measurement of the absolute rate constants
for singlet molecular oxygen (1Δg) reaction
with 1,3-diphenylisobenzofuran and physical quenching by ground state
molecular oxygen, J. Am. Chem. Soc., 96, 3343-3358.
32.Schmidt, R., Tanelian, C., Dunsbach, R., and
Wolff, C. J. (1994) Phenalenone, a universal reference compound for the
determination of yields of singlet oxygen
O2(1Δg) sensitization, J.
Photochem. Photobiol. A: Chem., 79, 11-17.
33.Krasnovsky, A. A., Jr., Roumbal, Ya. V., Ivanov,
A. V., and Ambartzumian, R. V. (2006) Solvent dependence of the
steady-state rate of 1O2 generation upon
excitation of dissolved oxygen by cw 1267 nm laser radiation in
air-saturated solutions. Estimates of the absorbance and molar
absorption coefficients of oxygen at the excitation wavelength,
Chem. Phys. Lett., 430, 260-264.
34.Krasnovsky, A. A., Jr., and Kozlov, A. S. (2014)
New approach to measurement of IR absorption spectra of dissolved
oxygen molecules based on photochemical activity of oxygen upon direct
laser excitation, Biofizika, 59, 199-205.
35.Krasnovsky, A. A., Jr., and Kozlov, A. S. (2016)
Photonics of dissolved oxygen molecules. Comparison of the rates of
direct and photosensitized excitation of oxygen and reevaluation of the
oxygen absorption coefficients, J. Photochem. Photobiol., A:
Chemistry, 329, 167-174.