Discovery of MicroRNAs from Batrachuperus yenyuanensis Using Deep Sequencing and Prediction of Their Targets
Y. Huang1,a*, J. Xiong1,b*, P. B. Brown2, and X. Sun1
1College of Animal Science and Technology, Henan University of Science and Technology, 471023 Luoyang, China2Purdue University, Department of Forestry and Natural Resources, West Lafayette, 47907 Indiana, USA
* To whom correspondence should be addressed.
Received July 14, 2018; Revised October 29, 2018; Accepted October 29, 2018
MicroRNAs (miRNAs), a family of ∼22-nucleotide non-coding single-stranded RNA molecules, are considered as key post-transcriptional regulators of gene expression that regulate various biological processes in living organism. Many miRNAs have been identified in animals; however, few have been reported in Hynobiidae species. The present study is aimed to identify a full repertoire of miRNAs in Batrachuperus yenyuanensis (Yenyuan stream salamander), which would significantly increase our knowledge of miRNAs in amphibians. A small RNA library was constructed from B. yenyuanensis and sequenced using deep sequencing. As a result, 1,717,751 clean reads were obtained, representing 356 known and 80 novel miRNAs. Additionally, expression levels of eight randomly selected miRNAs in B. yenyuanensis were confirmed using the stem-loop quantitative real-time reverse transcription PCR. In addition, 13,972 targets were predicted for these identified miRNAs, although the physiological functions of many of these targets remain unknown. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis suggested that the predicted targets are involved in a variety of physiological regulatory functions in B. yenyuanensis. These results provide useful information for further research on the miRNAs involved in the growth and development of B. yenyuanensis, as well as adaptation of this species to its high-altitude habitats.
KEY WORDS: Batrachuperus yenyuanensis, microRNA, deep sequencing, testis, stem-loop RT-qPCRDOI: 10.1134/S0006297919040059
Abbreviations: GO, gene ontology (analysis); KEGG, Kyoto Encyclopedia of Genes and Genomes (enrichment analysis); miRNA (miR), microRNA; nt, nucleotide; PC, predicted candidate; snRNA, small nuclear RNA; snoRNA, small nucleolar RNA; UTR, untranslated region.
MicroRNAs (miRNAs) are short, endogenous, non-coding RNAs of about ~22
nucleotides (nt) that act as post-transcriptional negative regulators
of mRNA stability or cause translation repression by binding to the
3´-untranslated regions (UTRs), coding regions, or 5´-UTRs
of target mRNAs [1, 2]. Mature
miRNAs are mostly transcribed from the genome non-coding regions by RNA
polymerase II into long primary transcripts that are subsequently
processed by Drosha and DGCR8 to form pre-miRNAs of approximately 70 nt
with a hairpin secondary structure [3]. Pre-miRNAs
are then exported to the cytoplasm and further processed by another
RNase III enzyme, Dicer, into the miRNA/miRNA* duplexes [4]. Finally, one strand of the RNA duplex (mature
miRNA) is predominantly incorporated in the RNA-induced silencing
complex (RISC), in which it negatively regulates gene expression by
inhibiting mRNA translation or degrading the coding target mRNAs via
perfect or imperfect complementation to them [5, 6]. According to computational analyses, a single
miRNA can bind to multiple mRNA targets, and a single target gene can
be regulated by multiple miRNAs [7, 8]. An increasing body of evidence indicates that
miRNAs play vital roles in many physiological and biochemical processes
in eukaryotes, such as cell development, proliferation,
differentiation, metabolism, homeostasis, apoptosis, immune response,
viral defense, and environmental stress [9-12].
Yenyuan stream salamander (Batrachuperus yenyuanensis, Batrachuperus, Hynobiidae, Caudata, Amphibia) is endemic to China and mainly distributed across the Hengduan Mountains and nearby mountains [13]. It is predominantly found in the south of Sichuan, including Yanyuan, Xichang, Mianning, and Puxiong counties. This salamander usually lives and spawns in mountain streams at high altitudes, where the flow is slow, and streambeds have many stones. Adults live mainly in water and feed on shrimp and aquatic insects. In traditional Tibetan and Chinese medicine, the larvae of B. yenyuanensis are used to treat injuries from falls, fractures, contusions, strains, and joint pain. Their unique medicinal value means that the market price of this species has risen continuously, leading to excessive harvesting, which has caused a sharp decline in its populations. In addition, B. yenyuanensis is threatened by habitat destruction and degeneration, in particular, those caused by infrastructure development. Currently, this species is vulnerable, endangered, and even locally extinct. Batrachuperus yenyuanensis is listed as a State Special Protected Animal (category II) under the Chinese Conservation Law and in Appendix I of CITES since 2004.
Previous studies on Hynobiidae species investigated their life habits, morphology, biodiversity, population distribution, phylogeography, and transcriptomes [14-17]. However, the information on miRNAs expressed by B. yenyuanensis is lacking. In the present study, we characterized known and novel miRNAs in B. yenyuanensis using deep sequencing. Stem-loop quantitative real-time reverse transcription PCR (qRT-PCR) was then used to validate expression levels of selected miRNAs in B. yenyuanensis. Potential miRNA targets were predicted using target prediction tools. Identification of B. yenyuanensis miRNAs will help to enrich the known repertoire of miRNAs in amphibians. Our results also provide the basis for further studying the functions of those miRNAs in physiological processes, immune response, and adaptive evolution of B. yenyuanensis.
MATERIALS AND METHODS
Animal collection. Healthy B. yenyuanensis males were collected in the Yanyuan county (Sichuan, China). Tissue samples from the whole bodies of three B. yenyuanensis specimens were pooled and frozen immediately in liquid nitrogen for further RNA extraction.
Small RNA library construction and deep sequencing. Total RNA was extracted from the pooled tissue samples of three B. yenyuanensis specimens using the RNAiso reagent (Takara, China) according to the manufacturer’s instructions. The quantity and purity of total RNA were determined with a NanoDrop ND-1000 spectrophotometer (NanoDrop, USA) from the A260/A280 ratio (A260/A280 ratio in the RNA samples was >2.0). The integrity of the total RNA was analyzed with an Agilent 2100 Bioanalyzer system (Agilent Technologies, USA) at the RNA integrity value (RIN) >8.0. In brief, a fraction of small RNAs (15-50 nt) was isolated from the total testis RNA by electrophoresis in 15% Tris-borate/EDTA/urea polyacrylamide gel. Then, 5′- and 3′-adapters (Illumina, USA) were sequentially ligated to the isolated small RNAs. After reverse transcription, the obtained cDNAs were used for cluster generation with an Illumina cluster station and then sequenced with an Illumina GAIIx instrument following manufacturer’s instructions. The raw data were deposited in the NCBI database under the accession number SRP153555.
Sequencing data analysis and miRNA identification. Low-quality reads including contaminated reads, sequences containing adapters, sequences without insert tags, and reads containing polyA, polyT, polyG, or polyC were removed from the sequencing data. Sequences with the length of ≥18 and ≤26 nt were selected for further analysis. The remaining clean reads were searched against the NCBI, Rfam, and Repbase databases to remove known classes of RNAs (mRNA, rRNA, tRNA, snRNA, snoRNA, and repeats), so that every unique small RNA mapped to only one annotation. The remaining reads were searched against the miRBase 21.0 (ftp://mirbase.org) to identify known miRNAs. The Mireap software (http://sourceforge.net/projects/mireap/) was used to predict novel miRNAs. The criteria for the secondary structure prediction were: (i) number of nucleotides in one bulge in the stem, ≤12; (ii) number of base pairs in the stem region of the predicted hairpin, ≥16; (iii) free energy cutoff, ≤−15 kcal/mol; (iv) hairpin length (upper and lower stems + terminal loop), ≥50; (v) hairpin loop length, ≤20; (vi) number of nucleotides in one bulge in the mature region, ≤8; (vii) number of biased errors in one bulge in the mature region, ≤4; (viii) number of biased bulges in the mature region, ≤2; (ix) number of errors in the mature region, ≤7; (x) number of base pairs in the mature region of the predicted hairpin, ≥12); and (xi) percent of nucleotides in the stem of mature miRNA, ≥80%.
Stem-loop qRT-PCR validation of miRNA expression. To validate the presence and expression of the identified miRNAs, eight miRNAs were randomly selected for qRT-PCR validation using a stem-loop reverse transcription primer. The primers were designed using previously described methods [18, 19]. The qPCR reaction regime included one cycle at 95°C for 15 min, followed by 40 cycles at 94°C for 15 s, 55°C for 30 s, and 70°C for 30 s. All reactions were performed in triplicate. The fold change in gene expression was estimated in terms of the threshold cycles using the 2−ΔΔCt method. 16S rRNA was used as the internal reference. All primers used for miRNA validation and expression analysis are listed in Table S1 [see Supplement to this paper at the web site of Biochemistry (Moscow) (http://protein.bio.msu.ru/biokhimiya) and Springer site (Link.springer.com)].
Target gene prediction and pathway analysis. Based on the sequences of known and novel miRNAs, target gene candidates were predicted using the miRanda (v.3.3a) and Targetscan (v.7.0) software. The parameters were set as: (i) perfect miRNA seed complementarity (positions 2 to 8) with transcripts; (ii) score >50 and free energy <−20 kcal/mol. The overlapping results from the predictions using both software packages were considered as miRNA target genes. The biological functions of the target genes were assessed using the Gene Ontology (GO) enrichment analysis, and the gene numbers of each GO term were calculated. To identify biological pathways in which the target genes are involved, the predicted targets were classified into functional pathways using the Kyoto Encyclopedia of Genes and Genomes (KEGG).
RESULTS AND DISCUSSION
Deep sequencing of small RNAs. Using the Illumina Hiseq 3000 deep sequencing platform, a total of 11,514,774 raw reads were obtained from the small RNA library. After removing the reads identified with the 3ADT&length filter, junk reads, Rfam (rRNA, tRNA, snRNA, snoRNA, and other Rfam RNAs) and Repbase sequences, a total of 1,717,751 unique reads representing 869,818 valid reads of 18-26 nt were obtained (Table 1). The length distribution of the valid reads is shown in Fig. 1. The length of the majority of small RNAs was between 19 and 24 nt, which is typical of Dicer-processed small RNAs in animals [20, 21]. The most abundant size class was 22 nt, which accounted for 33.69% of the library, followed by 21, 20, and 23 nt (21.17, 13.17 and 11.88%, respectively). Small RNAs of more than 25 nt may be rasiRNAs (repeat-associated small interfering RNAs) that mediate the silencing of genomic repeats and transposons [22].
Table 1. Analysis of small RNA sequences
from B. yenyuanensis
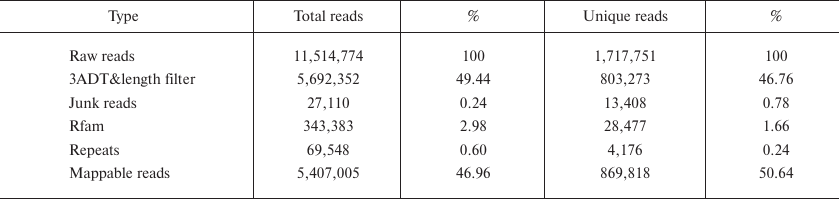
Fig. 1. Length distribution of sequenced B. yenyuanensis small RNAs.
Identification and characterization of known miRNAs. To identify known miRNAs in B. yenyuanensis, we analyzed the valid reads in comparison with known animal miRNAs. All small RNA sequences were subjected to BLASTN search against the known animal miRNAs in the miRBase 21.0. A total of 356 known mature miRNAs (5p and 3p) were identified that belonged to 98 families (Table S2, see Supplement). Deep sequencing technology is a powerful tool to estimate the expression levels of miRNAs and has the ability to generate millions of small RNA reads. Moreover, the results of sequencing generally reflect the relative abundance of various miRNAs and can be used to assess the expression levels of these miRNAs [23]. Intriguingly, we found that the number of reads of identified known miRNAs varied drastically. For example, the top ten most abundant miRNAs (dre-miR-202-5p_R-1, aca-miR-99b-5p_R-1, dre-miR-122_R-1, hsa-miR-30c-5p_R-2, hsa-let-7a-5p, aca-miR-21-5p, aca-miR-451-5p_R-1, hhi-miR-26_R-1, aca-miR-126-3p_R-1, and aca-miR-16b-5p) had more than 8000 reads (Table 2), while many miRNAs were sequenced only once (e.g., ccr-miR-27c-3p_R-1, ipu-miR-30d_R-2_1ss9TC, aca-miR-202-3p.1_R+2_1ss10CT, ola-miR-192-3p_R-1_1ss8GA and hsa-miR-500a-3p_L+1R-2_1ss18AG). This suggested that the highly abundant miRNAs might play important roles in the development or physiology of B. yenyuanensis, whereas low-abundant miRNAs might function in a tissue-specific manner, at certain developmental stages, or under particular physiological conditions [24, 25]. miRNAs with a high number of reads have been reported to play important regulatory roles. For example, previous studies have shown that miR-202-5p is abundantly expressed in mouse, Xenopus, chicken, and Atlantic halibut testes [26-28]. Similarly, dre-miR-202-5p_R-1 exhibited high expression levels (with 206,216 reads in B. yenyuanensis). miR-99b-5p has been reported to display abnormal expression in various malignant tumors; this miRNA promotes apoptosis by targeting mTOR in human esophageal squamous cell carcinoma and regulates DNA damage response [29, 30]. miR-26a is involved in the control of cell cycle and differentiation [31, 32]; miR-21 was found to regulate the self-renewal of spermatogonial stem cells and spermatogenesis [33, 34]. In addition, miR-21 functions as an anti-apoptotic factor and an oncogene related to cell growth [35, 36]. Let-7 and its family members are highly conserved across different species in terms of sequence and function. miRNA let-7 was found to play a major role in developmental timing in Caenorhabditis elegans [37].
Table 2. Sequences and abundance of top 10
known miRNAs in B. yenyuanensis

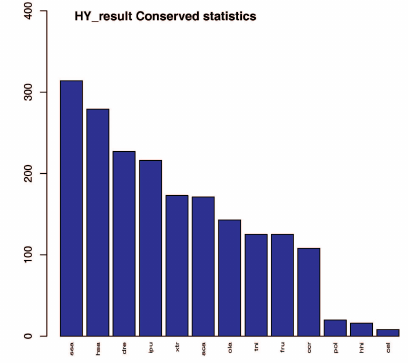
Conserved miRNAs are found in many animal species and have important functions in animal development and physiological processes [38, 39]. We searched our dataset of identified known miRNAs for conserved miRNAs using BLAST and miRBase 21.0. A total of 314 miRNAs were found to be conserved in B. yenyuanensis that matched miRNAs of other species. The top six species were Salmo salar, Homo sapiens, Danio rerio, Ictalurus punctatus, Xenopus tropicalis, and Anolis carolinensis with 314, 279, 227, 216, 173, and 171 conserved miRNAs similar to those in B. yenyuanensis, respectively. The details of the sequenced miRNA tags and statistical detection rates are presented in Fig. 2 and Table S3 (see Supplement). Some identified conserved miRNAs have been found in a large variety of fish species, indicating their highly conserved structure or role in evolution.
Fig. 2. Conservation of the identified B. yenyuanensis miRNAs in 13 other species (according to miRbase); x-axis, queried species; y-axis, number of conserved miRNAs. A total of 314 miRNAs were found to be conserved in B. yenyuanensis (the first bar).
Discovery of novel miRNAs. One of the greatest advantages of deep sequencing is the ability to identify novel miRNAs with low expression levels in a small RNA library [40]. To found possible novel miRNAs, we used the Mireap software that identifies potential stem-loop structures in the sequences flanking unannotated reads [41]. No B. yenyuanensis genome data are currently available; therefore, in this study, unannotated small RNAs that could be mapped to B. yenyuanensis transcripts (unpublished data) were used to predict novel miRNAs. Finally, 80 putative novel miRNAs were predicted to be derived from their miRNA precursors, of which 36 miRNAs were derived from the 3´ arm and 44 from the 5´ arm (Table S4, see Supplement), indicating they might be species-specific miRNAs in the gene regulatory network. These novel miRNAs were named predicted candidates (PCs). The length of the precursors for the putative novel miRNAs varied between 50 and 160 nt, and the length of the mature miRNAs ranged from 18 to 26 nt, which was similar to those found in Andrias davidianus and X. tropicalis [42, 43]. The minimum folding free energies (MFEs) of the novel miRNA precursors were −15.4 to −82.6 kcal/mol, with an average of −37.61 kcal/mol. The G + C content of all 80 miRNA precursors ranged from 24.7 to 69.8%, with an average of 46.7%. The length variations have been mainly attributed to enzymatic modifications, such as RNA editing, 3´-editing, and exonuclease activities [44]. In this study, we also observed that these PC miRNAs had 10-310 reads, which reflects extremely low expression levels compared to most of known miRNAs in B. yenyuanensis. This phenomenon was in accordance with the low expression observed for novel miRNAs and for those involved in the organizational or specific developmental periods [45, 46].
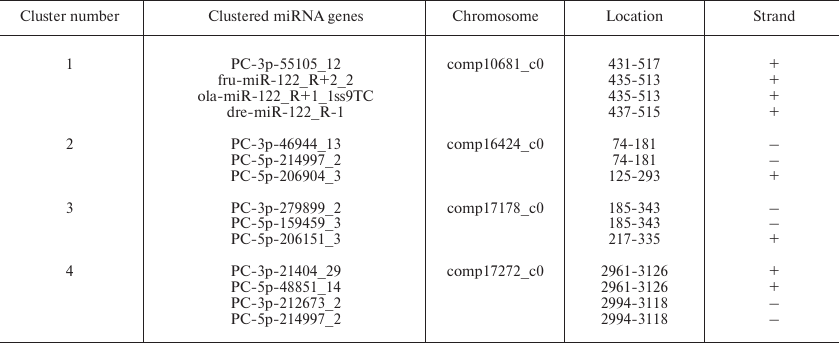
miRNA gene clusters in B. yenyuanensis. Frequently, miRNA genes form clusters in the genome. Clusters of miRNA genes have been reported in animal species, for example, S. salar and Gadus morhua [47, 48]. Expression of miRNAs from a specific cluster may reflect developmental stage-specific transcription or differential pre-miRNA post-transcriptional processing [49, 50]. Based on the definition given in the miRBase, a miRNA cluster should include two or more miRNA genes that are less than 10 kb. By applying this definition, we classified 14 miRNAs (3 conserved and 11 PC miRNAs) into four clusters (Table 3). Each cluster contained at least two miRNAs. One of the novel miRNA genes (PC-3p-55105_12) was attributed to the gene cluster containing three conserved miRNAs genes (fru-miR-122_R+2_2, ola-miR-122_R+1_1ss9TC, and dre-miR-122_R-1), suggesting that they might be functionally related and act cooperatively in regulating multiple biological processes [51]. It is known that certain miRNAs in gene clusters share the same set of control sequences and are found on the same transcript. It was earlier demonstrated that a miRNA gene can produce two different miRNAs through duplex transcription and thus control different target genes [45]. In our study, we speculated that only four clusters might involve this phenomenon; however, it could not be confirmed because of the lack of genome sequence data and the limited the ability to discover gene clusters in B. yenyuanensis.
Table 3. Clusters of identified miRNA
genes
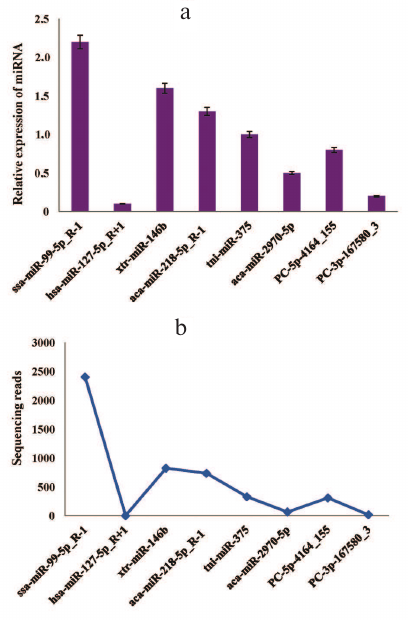
Stem-loop qRT-PCR validation of known and novel miRNAs. Generally, the timing and location of miRNA expression are tightly regulated, which might be useful for understanding the functions of this miRNA [52]. To verify the presence of the predicted miRNAs and to determine their relative expression, we randomly selected six known miRNAs and two novel miRNAs from the deep sequencing data for further verification by stem-loop qRT-PCR. The same samples used for deep sequencing were used for experimental validation. All miRNAs (ssa-miR-99-5p_R-1, hsa-miR-127-5p_R+1, xtr-miR-146b, aca-miR-218-5p_R-1, tni-miR-375, aca-miR-2970-5p, PC-5p-4164_155, and PC-3p-167580_3) showed varying expression levels (as normalized to the amounts of 16S RNA) (Fig. 3a). The expression patterns of these miRNAs corresponded to the original miRNA sequencing results (Fig. 3b), although there were slight differences.
Fig. 3. Validation of the selected known and novel miRNAs by the stem loop RT-qPCR: a) relative expression of eight (six known and two novel) miRNAs identified by RT-qPCR; b) sequencing results for the same miRNAs show similar expression pattern. The transcript levels of both known and novel miRNAs were normalized to the amounts of 16S RNA. The RT-qPCR data represent mean ± SD from three independent experiments.
Prediction of potential miRNA target genes and GO and KEGG enrichment analysis. Identification of miRNA targets is an important step in understanding their roles. A total of 118,668 target genes were predicted from B. yenyuanensis transcriptome data (unpublished data) using the TargetScan and miRanda software (Table S5, see Supplement). Previous studies demonstrated that a single miRNA can target mRNAs from one to hundreds of genes, and occasionally, numerous different miRNAs can regulate a single mRNA [53]. The number of potential targets for each miRNA ranged from 64 (xtr-miR-449b-3p_R-2_1ss10AC) to 444 (aca-let-7c-1-3p_1ss9CT), with an average of 272 targets. Unsurprisingly, the regulation network of miRNA target genes is extremely complicated. In our study, these predicted targets are involved in transcription, immunity, signaling, metabolism, transport, growth, development, and other processes. Among the targets, most were genes encoding transcription factor (e.g., zinc finger proteins), RNA-binding proteins, protease-activated receptors, translation initiation factors, GTP-binding nuclear proteins, Ras-related proteins, Ran-specific GTPase-activating proteins, TATA box-binding proteins, and nuclear receptors, i.e., genes playing critical regulatory roles in B. yenyuanensis. miRNAs frequently target transcription factor genes involved in the life activities of animals, indicating that miRNAs act as regulators [54, 55]. However, further biological experimental evidence is needed to confirm the relationship between miRNAs and their target genes in B. yenyuanensis.
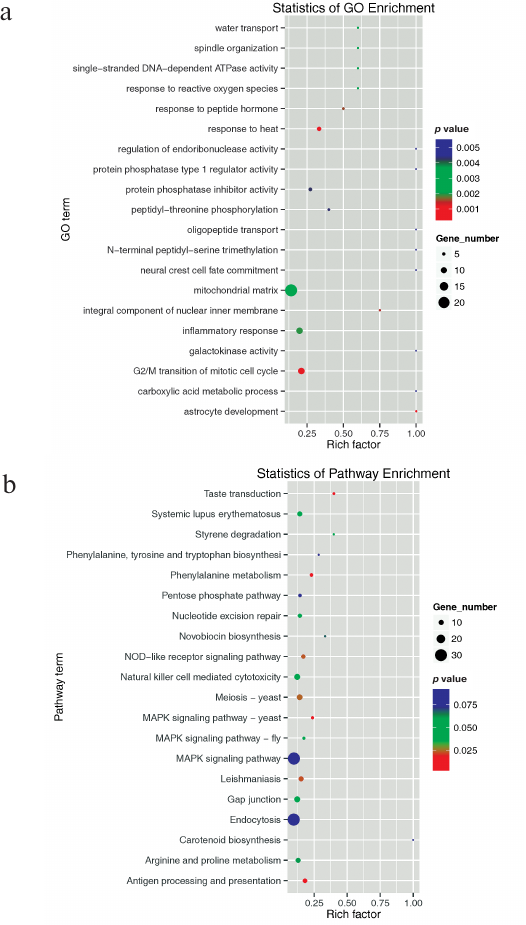
Although a large number of miRNAs have been identified in animals, miRNAs in salamanders have rarely been studied, except for those in A. davidianus (Chinese giant salamander) [56]. To further investigate physiological processes and pathways regulated by the identified miRNAs, their predicted target genes were subjected to GO and KEGG pathway analysis. Predicted target genes were classified into three major GO terms − biological process, cellular component, and molecular function. A total of 18,741 target genes were predicted to take part in 3636 biological processes; 15,343 target genes were involved in 1898 different molecular functions; and 14,480 target genes participated in 735 cellular component functions (Fig. S1 and Table S6, see Supplement). The top 20 enriched terms were mainly involved in biological processes (vesicle-mediated transport, tricarboxylic acid cycle, RNA splicing, protein transport, protein K48-linked ubiquitination, protein folding, mRNA transport, mRNA processing, mitotic nuclear division, glycogen metabolic process, G2/M transition of mitotic cell cycle, and cell division), cellular components (nucleolus, mitochondrion, mitochondrial matrix, melanosome, endoplasmic reticulum membrane, centriole), and molecular functions (RNA binding and GTPase activity) (Fig. 4a).
All of these results implied that many target genes function differentially and interdependently. Batrachuperus yenyuanensis is an endangered species; therefore, targets involved in immunity are particularly important [for example, viral myocarditis (GO05416), intestinal immune network for IgA production (GO04672), and autoimmune thyroid disease (GO05320)]. Using the KEGG pathway analysis, 6082 predicted target genes were grouped into 250 pathways. Among them, 39 pathways were significantly enriched (Table S7, see Supplement). The top 20 enriched pathway terms are shown in Fig. 4b. Oocyte meiosis was the most significantly enriched pathway (KO04144), followed by pyrimidine metabolism (KO00240) and meiosis (KO04113). The results also highlighted major biological pathways related to gonad cell proliferation and differentiation, as well as tissue metabolism and development, suggesting that these targets are likely to be involved in high-altitude adaptation processes in B. yenyuanensis.
Fig. 4. The top 20 enriched GO terms (a) and KEGG pathways (b). Gene number, the number of target genes in each term or pathway. Rich factor, the ratio of target genes to all genes in each term or pathway.
Despite many miRNAs being discovered in a wide variety of organisms, miRNA research in B. yenyuanensis lags behind. In the present study, we successfully characterized 356 known and 80 potential novel miRNAs from B. yenyuanensis. The expression levels of eight selected miRNAs were validated using the stem-loop qRT-PCR. A total of 13,972 target genes were predicted for the known and novel miRNAs. GO and KEGG enrichment analyses of the putative target genes indicated that identified miRNAs regulate genes involved in diverse physiological processes. The results of our study supplement current public miRNA databases with miRNA sequences from a non-model animal whose genome is still not sequenced. These data provide a strong foundation for studying miRNAs and may contribute to our understanding of the role of miRNAs in the regulation of their targets involved in immune response, adaptation to the environment, and reproduction in B. yenyuanensis.
Funding
This work was supported by the Natural Science Foundation of China (projects 1471971 and 31402263), Natural Science Foundation of Henan Province of China (project 182300410087), and the Foundation of Henan Educational Committee of China (project 2015GGJS-054).
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Statement
This study was approved by the Institutional Animal Welfare Committee of the College of Animal Science and Technology, Henan University of Science and Technology, Henan, China.
REFERENCES
1.Alwin Prem Anand, A., Huber, C., Asnet Mary, J.,
Gallus, N., Leucht, C., Klafke, R., Hirt, B., and Wizenmann, A. (2018)
Expression and function of microRNA-9 in the mid-hindbrain area of
embryonic chick, BMC Dev. Biol., 18, 3.
2.Sun, K., and Lai, E. C. (2013) Adult-specific
functions of animal microRNAs, Nat. Rev. Genet., 14,
535-548.
3.Warf, M. B., Johnson, W. E., and Bass, B. L. (2011)
Improved annotation of C. elegans microRNAs by deep sequencing
reveals structures associated with processing by Drosha and Dicer,
RNA, 17, 563-577.
4.Seitz, H., Tushir, J. S., and Zamore, P. D. (2011)
A 5′-uridine amplifies miRNA/miRNA* asymmetry in
Drosophila by promoting RNA-induced silencing complex formation,
Silence, 2, 4.
5.Cambronne, X. A., Shen, R., Auer, P. L., and
Goodman, R. H. (2012) Capturing microRNA targets using an RNA-induced
silencing complex (RISC)-trap approach, Proc. Natl. Acad. Sci.
USA, 109, 20473-20478.
6.Ota, H., Sakurai, M., Gupta, R., Valente, L.,
Wulff, B. E., Ariyoshi, K., Iizasa, H., Davuluri, R. V., and Nishikura,
K. (2013) ADAR1 forms a complex with Dicer to promote microRNA
processing and RNA-induced gene silencing, Cell, 153,
575-589.
7.Luo, J., Wang, Y., Yuan, J., Zhao, Z., and Lu, J.
(2018) MicroRNA duplication accelerates the recruitment of new targets
during vertebrate evolution, RNA, 24, 787-802.
8.Krek, A., Grun, D., Poy, M. N., Wolf, R.,
Rosenberg, L., Epstein, E. J., MacMenamin, P., da Piedade, I.,
Gunsalus, K. C., Stoffel, M., and Rajewsky, N. (2005) Combinatorial
microRNA target predictions, Nat. Genet., 37,
495-500.
9.Ameres, S. L., and Zamore, P. D. (2013)
Diversifying microRNA sequence and function, Nat. Rev. Mol. Cell
Biol., 14, 475-488.
10.Maltby, S., Plank, M., Tay, H. L., Collison, A.,
and Foster, P. S. (2016) Targeting microRNA function in respiratory
diseases: mini-review, Front. Physiol., 7, 21.
11.Clark, E. A., Kalomoiris, S., Nolta, J. A., and
Fierro, F. A. (2014) Concise review: microRNA function in multipotent
mesenchymal stromal cells, Stem Cells, 32, 1074-1082.
12.Huang, Y., Shen, X. J., Zou, Q., Wang, S. P.,
Tang, S. M., and Zhang, G. Z. (2011) Biological functions of microRNAs:
a review, J. Physiol. Biochem., 67, 129-139.
13.Fu, J., and Zeng, X. (2008) How many species are
in the genus Batrachuperus? A phylogeographical analysis of the
stream salamanders (family Hynobiidae) from southwestern China, Mol.
Ecol., 17, 1469-1488.
14.Lu, B., Zheng, Y., Murphy, R. W., and Zeng, X.
(2012) Coalescence patterns of endemic Tibetan species of stream
salamanders (Hynobiidae: Batrachuperus), Mol. Ecol.,
21, 3308-3324.
15.Huang, Z. S., Yu, F. L., Gong, H. S., Song, Y.
L., Zeng, Z. G., and Zhang, Q. (2017) Phylogeographical structure and
demographic expansion in the endemic alpine stream salamander
(Hynobiidae: Batrachuperus) of the Qinling Mountains, Sci.
Rep., 7, 1871.
16.Jiang, J. P., Jia, J., Zhang, M., and Gao, K. Q.
(2018) Osteology of Batrachuperus londongensis (Urodela,
Hynobiidae): study of bony anatomy of a facultatively neotenic
salamander from Mount Emei, Sichuan Province, China, Peer J.,
6, e4517.
17.Che, R., Sun, Y., Wang, R., and Xu, T. (2014)
Transcriptomic analysis of endangered Chinese salamander:
identification of immune, sex and reproduction-related genes and
genetic markers, PLoS One, 9, e87940.
18.Huang, Y., Gong, W., Ren, H., Xiong, J., Gao, X.,
and Sun, X. (2017) Identification of the conserved and novel microRNAs
by deep sequencing and prediction of their targets in Topmouth culter,
Gene, 626, 298-304.
19.Sui, W., Liu, F., Chen, J., Ou, M., and Dai, Y.
(2014) Evaluating a particular circulating microRNA species from an SLE
patient using stem-loop qRT-PCR, Methods Mol. Biol.,
1134, 201-209.
20.Ju, Z., Jiang, Q., Liu, G., Wang, X., Luo, G.,
Zhang, Y., Zhang, J., Zhong, J., and Huang, J. (2018) Solexa sequencing
and custom microRNA chip reveal repertoire of microRNAs in mammary
gland of bovine suffering from natural infectious mastitis, Anim.
Genet., 49, 3-18.
21.Meng, Y., Tian, H., Hu, Q., Liang, H., Zeng, L.,
and Xiao, H. (2018) MicroRNA repertoire and comparative analysis of
Andrias davidianus infected with ranavirus using deep
sequencing, Dev. Comp. Immunol., 85, 108-114.
22.Feng, X., Zhou, X., Zhou, S., Wang, J., and Hu,
W. (2018) Analysis of microRNA profile of Anopheles sinensis by
deep sequencing and bioinformatic approaches, Parasit. Vectors,
11, 172.
23.Chen, X., Li, Q., Wang, J., Guo, X., Jiang, X.,
Ren, Z., Weng, C., Sun, G., Wang, X., Liu, Y., Ma, L., Chen, J. Y.,
Zen, K., Zhang, J., and Zhang, C. Y. (2009) Identification and
characterization of novel amphioxus microRNAs by Solexa sequencing,
Genome Biol., 10, R78.
24.Cowled, C., Stewart, C. R., Likic, V. A.,
Friedlander, M. R., Tachedjian, M., Jenkins, K. A., Tizard, M. L.,
Cottee, P., Marsh, G. A., Zhou, P., Baker, M. L., Bean, A. G., and
Wang, L. F. (2014) Characterization of novel microRNAs in the Black
flying fox (Pteropus alecto) by deep sequencing, BMC
Genomics, 15, 682.
25.Gao, Z. H., Wei, J. H., Yang, Y., Zhang, Z.,
Xiong, H. Y., and Zhao, W. T. (2012) Identification of conserved and
novel microRNAs in Aquilaria sinensis based on small RNA
sequencing and transcriptome sequence data, Gene, 505,
167-175.
26.Ro, S., Song, R., Park, C., Zheng, H., Sanders,
K. M., and Yan, W. (2007) Cloning and expression profiling of small
RNAs expressed in the mouse ovary, RNA, 13,
2366-2380.
27.Michalak, P., and Malone, J. H. (2008)
Testis-derived microRNA profiles of African clawed frogs
(Xenopus) and their sterile hybrids, Genomics, 91,
158-164.
28.Bannister, S. C., Tizard, M. L., Doran, T. J.,
Sinclair, A. H., and Smith, C. A. (2009) Sexually dimorphic microRNA
expression during chicken embryonic gonadal development, Biol.
Reprod., 81, 165-176.
29.Mueller, A. C., Sun, D., and Dutta, A. (2013) The
miR-99 family regulates the DNA damage response through its target
SNF2H, Oncogene, 32, 1164-1172.
30.Sun, J., Chen, Z., Tan, X., Zhou, F., Tan, F.,
Gao, Y., Sun, N., Xu, X., Shao, K., and He, J. (2013) MicroRNA-99a/100
promotes apoptosis by targeting mTOR in human esophageal squamous cell
carcinoma, Med. Oncol., 30, 411.
31.Dey, B. K., Gagan, J., Yan, Z., and Dutta, A.
(2012) miR-26a is required for skeletal muscle differentiation and
regeneration in mice, Genes Dev., 26, 2180-2191.
32.Zhang, Y. F., Zhang, A. R., Zhang, B. C., Rao, Z.
G., Gao, J. F., Lv, M. H., Wu, Y. Y., Wang, S. M., Wang, R. Q., and
Fang, D. C. (2013) MiR-26a regulates cell cycle and anoikis of human
esophageal adenocarcinoma cells through Rb1-E2F1 signaling pathway,
Mol. Biol. Rep., 40, 1711-1720.
33.Bouhallier, F., Allioli, N., Lavial, F., Chalmel,
F., Perrard, M. H., Durand, P., Samarut, J., Pain, B., and Rouault, J.
P. (2010) Role of miR-34c microRNA in the late steps of
spermatogenesis, RNA, 16, 720-731.
34.Niu, Z., Goodyear, S. M., Rao, S., Wu, X.,
Tobias, J. W., Avarbock, M. R., and Brinster, R. L. (2011) MicroRNA-21
regulates the self-renewal of mouse spermatogonial stem cells, Proc.
Natl. Acad. Sci. USA, 108, 12740-12745.
35.Si, M. L., Zhu, S., Wu, H., Lu, Z., Wu, F., and
Mo, Y. Y. (2007) miR-21-mediated tumor growth, Oncogene,
26, 2799-2803.
36.Liu, H., Cheng, L., Cao, D., and Zhang, H. (2018)
Suppression of miR-21 expression inhibits cell proliferation and
migration of liver cancer cells by targeting phosphatase and tensin
homolog (PTEN), Med. Sci. Monit., 24, 3571-3577.
37.Reinhart, B. J., Slack, F. J., Basson, M.,
Pasquinelli, A. E., Bettinger, J. C., Rougvie, A. E., Horvitz, H. R.,
and Ruvkun, G. (2000) The 21-nucleotide let-7 RNA regulates
developmental timing in Caenorhabditis elegans,
Nature, 403, 901-906.
38.Tan, T. T., Chen, M., Harikrishna, J. A.,
Khairuddin, N., Mohd Shamsudin, M. I., Zhang, G., and Bhassu, S. (2013)
Deep parallel sequencing reveals conserved and novel miRNAs in gill and
hepatopancreas of giant freshwater prawn, Fish. Shellfish.
Immunol., 35, 1061-1069.
39.Yakovlev, I. A., and Fossdal, C. G. (2017) In
silico analysis of small RNAs suggest roles for novel and conserved
miRNAs in the formation of epigenetic memory in somatic embryos of
Norway spruce, Front. Physiol., 8, 674.
40.Juan, L., Tong, H. L., Zhang, P., Guo, G., Wang,
Z., Wen, X., Dong, Z., and Tian, Y. P. (2014) Identification and
characterization of novel serum microRNA candidates from deep
sequencing in cervical cancer patients, Sci. Rep., 4,
6277.
41.Jiang, P., Wu, H., Wang, W., Ma, W., Sun, X., and
Lu, Z. (2007) MiPred: classification of real and pseudo microRNA
precursors using random forest prediction model with combined features,
Nucleic Acids Res., 35, W339-344.
42.Huang, Y., Ren, H. T., Xiong, J. L., Gao, X. C.,
and Sun, X. H. (2017) Identification and characterization of known and
novel microRNAs in three tissues of Chinese giant salamander base on
deep sequencing approach, Genomics, 109, 258-264.
43.Harding, J. L., Horswell, S., Heliot, C.,
Armisen, J., Zimmerman, L. B., Luscombe, N. M., Miska, E. A., and Hill,
C. S. (2014) Small RNA profiling of Xenopus embryos reveals
novel miRNAs and a new class of small RNAs derived from intronic
transposable elements, Genome Res., 24, 96-106.
44.Islam, M. T., Ferdous, A. S., Najnin, R. A.,
Sarker, S. K., and Khan, H. (2015) High-throughput sequencing reveals
diverse sets of conserved, nonconserved, and species-specific miRNAs in
jute, Int. J. Genomics, 2015, 125048.
45.Zhang, J., Liu, Y., Zhang, X., Pan, J., Nie, Z.,
Zhang, W., Yu, W., Chen, J., Liu, L., Li, J., Zhang, Y., Guo, J., Wu,
W., Zhu, H., and Lv, Z. (2013) The identification of microRNAs in the
whitespotted bamboo shark (Chiloscyllium plagiosum) liver by
Illumina sequencing, Gene, 527, 259-265.
46.Huang, S., Cao, X., Tian, X., and Wang, W. (2016)
High-throughput sequencing identifies microRNAs from posterior
intestine of loach (Misgurnus anguillicaudatus) and their
response to intestinal air-breathing inhibition, PLoS One,
11, e0149123.
47.Andreassen, R., Worren, M. M., and Hoyheim, B.
(2013) Discovery and characterization of miRNA genes in Atlantic salmon
(Salmo salar) by use of a deep sequencing approach, BMC
Genomics, 14, 482.
48.Andreassen, R., Rangnes, F., Sivertsen, M.,
Chiang, M., Tran, M., and Worren, M. M. (2016) Discovery of miRNAs and
their corresponding miRNA genes in Atlantic cod (Gadus morhua):
use of stable miRNAs as reference genes reveals subgroups of miRNAs
that are highly expressed in particular organs, PLoS One,
11, e0153324.
49.Bartel, D. P. (2004) MicroRNAs: genomics,
biogenesis, mechanism, and function, Cell, 116,
281-297.
50.Hoss, A. G., Kartha, V. K., Dong, X., Latourelle,
J. C., Dumitriu, A., Hadzi, T. C., Macdonald, M. E., Gusella, J. F.,
Akbarian, S., Chen, J. F., Weng, Z., and Myers, R. H. (2014) MicroRNAs
located in the Hox gene clusters are implicated in
Huntington’s disease pathogenesis, PLoS Genet., 10,
e1004188.
51.Castellano, L., Rizzi, E., Krell, J., Di
Cristina, M., Galizi, R., Mori, A., Tam, J., De Bellis, G., Stebbing,
J., Crisanti, A., and Nolan, T. (2015) The germline of the malaria
mosquito produces abundant miRNAs, endo-siRNAs, piRNAs and 29-nt small
RNAs, BMC Genomics, 16, 100.
52.Xia, J. H., He, X. P., Bai, Z. Y., and Yue, G. H.
(2011) Identification and characterization of 63 microRNAs in the Asian
seabass Lates calcarifer, PLoS One, 6, e17537.
53.Samad, A. F. A., Nazaruddin, N., Murad, A. M. A.,
Jani, J., Zainal, Z., and Ismail, I. (2018) Deep sequencing and in
silico analysis of small RNA library reveals novel miRNA from leaf
Persicaria minor transcriptome, 3 Biotech.,
8, 136.
54.Alizadeh, E., Akbarzadeh, A., Eslaminejad, M. B.,
Barzegar, A., Hashemzadeh, S., Nejati-Koshki, K., and Zarghami, N.
(2015) Up-regulation of liver-enriched transcription factors HNF4a and
HNF6 and liver-specific microRNA (miR-122) by inhibition of let-7b in
mesenchymal stem cells, Chem. Biol. Drug Des., 85,
268-279.
55.Ow, M. C., Martinez, N. J., Olsen, P. H.,
Silverman, H. S., Barrasa, M. I., Conradt, B., Walhout, A. J., and
Ambros, V. (2008) The FLYWCH transcription factors FLH-1, FLH-2, and
FLH-3 repress embryonic expression of microRNA genes in C.
elegans, Genes Dev., 22, 2520-2534.
56.Su, S., Wang, Y., Wang, H., Huang, W., Chen, J.,
Xing, J., Xu, P., Yuan, X., Huang, C., and Zhou, Y. (2018) Comparative
expression analysis identifies the respiratory transition-related
miRNAs and their target genes in tissues of metamorphosing Chinese
giant salamander (Andrias davidianus), BMC Genomics,
19, 406.
Supplementary Materials (ZIP): Figure S1, Tables S1-S7.