Effect of Leucine M196 Substitution by Histidine on Electronic Structure of the Primary Electron Donor and Electron Transfer in Reaction Centers from Rhodobacter sphaeroides
A. A. Zabelin1, T. Yu. Fufina1, A. M. Khristin1, R. A. Khatypov1, V. A. Shkuropatova1, V. A. Shuvalov1, L. G. Vasilieva1, and A. Ya. Shkuropatov1,a*
1Institute of Basic Biological Problems, Russian Academy of Sciences, Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia* To whom correspondence should be addressed.
Received November 13, 2018; Revised December 27, 2018; Accepted December 27, 2018
In our recent X-ray study, we demonstrated that substitution of the natural leucine residue M196 with histidine in the reaction center (RC) from Rhodobacter (Rba.) sphaeroides leads to formation of a close contact between the genetically introduced histidine and the primary electron donor P (bacteriochlorophylls (BChls) PA and PB dimer) creating a novel pigment–protein interaction that is not observed in native RCs. In the present work, the possible nature of this novel interaction and its effects on the electronic properties of P and the photochemical charge separation in isolated mutant RCs L(M196)H are investigated at room temperature using steady-state absorption spectroscopy, light-induced difference FTIR spectroscopy, and femtosecond transient absorption spectroscopy. The results are compared with the data obtained for the RCs from Rba. sphaeroides pseudo-wild type strain. It is shown that the L(M196)H mutation results in a decrease in intensity and broadening of the long-wavelength Qy absorption band of P at ~865 nm. Due to the mutation, there is also weakening of the electronic coupling between BChls in the radical cation P+ and increase in the positive charge localization on the PA molecule. Despite the significant perturbations of the electronic structure of P, the mutant RCs retain high electron transfer rates and quantum yield of the P+QA– state (QA is the primary quinone acceptor), which is close to the one observed in the native RCs. Comparison of our results with the literature data suggests that the imidazole group of histidine M196 forms a π-hydrogen bond with the π-electron system of the PB molecule in the P dimer. It is likely that the specific (T-shaped) spatial organization of the π-hydrogen interaction and its potential heterogeneity in relation to the bonding energy is, at least partially, the reason that this type of interaction between the protein and the pigment and quinone cofactors is not realized in the native RCs.
KEY WORDS: bacterial reaction center, amino acid replacement, primary electron donor, electronic structure, electron transfer, Rhodobacter sphaeroidesDOI: 10.1134/S0006297919050067
Abbreviations: ΔA, absorbance change; BA, monomeric bacteriochlorophyll in the active cofactors branch; BChl, bacteriochlorophyll; BPheo, bacteriopheophytin; EADS, evolution-associated decay spectra; FTIR, Fourier transform infrared spectroscopy; HA and HB, BPheo molecules in the active and inactive cofactor branches, respectively; P, primary electron donor, BChl dimer; PA and PB, BChl molecules constituting P; psWt, pseudo-wild type; QA, primary quinone acceptor; QB, secondary quinone acceptor; Rba. sphaeroides, Rhodobacter sphaeroides; RC, reaction center.
Bacterial reaction centers (RCs) are highly specialized
cofactor–protein complexes, in which the primary energy
conversion of light quanta into the electrochemical energy of separated
charges takes place via a series of fast electron transfer reactions.
The unique feature of RCs as natural photoconversion systems is their
ability to perform charge separation with extremely high quantum yield
(~100%) and high energy efficiency (tens of percent) in a molecular
donor–acceptor system involving only a few pigment molecules
(bacteriochlorins) embedded into a specifically organized protein
matrix. Obviously, the key factors for the function of this system are
both basic physicochemical properties of individual pigment molecules
and their interaction between themselves and with the local
environment. One of the approaches to investigation of the role of
intermolecular interactions in the structural and electronic
configuration of pigment cofactors and their functionality is
introduction of point amino acid substitutions to the RC protein in
order to eliminate the existing and/or create new contacts/interactions
and detailed investigation of the consequences of such mutation with
X-ray diffraction analysis in combination with the methods of optical
spectroscopy. In particular, one of the promising substitutions
affecting intermolecular interactions in the RC is the replacement of
natural amino acid residues with a histidine residue. Histidine in
proteins is capable of participating in interactions of various nature:
coordination bonds with metal cations, hydrogen bonds,
cation–π, π–hydrogen, and π–π-stacking
interactions [1].
It was recently established based on the results of X-ray diffraction analysis in combination with the results of low-temperature absorption measurements and redox titration that the substitution of leucine by histidine at position M196 in the RCs from Rhodobacter (Rba.) sphaeroides likely resulted in formation of a new interaction between the bacteriochlorophyll (BChl) PB of the primary electron donor P and imidazole of the newly introduced histidine M196 [2]. It was suggested that such interaction could be either of π–π-stacking interaction or π–hydrogen interaction [2].
To further elucidate the nature of the discovered interaction and its effect on the electronic properties of P and primary charge separation, in this study the isolated L(M196)H RCs were investigated with the methods of steady-state absorption spectroscopy in the near- and mid-IR (Fourier transform infrared spectroscopy; FTIR) ranges and transient absorption spectroscopy with femtosecond time resolution in the visible and near-IR ranges of the spectrum. Recording of electronic absorption spectra is widely used to obtain information on the effect of mutation on electronic properties of bacterial RCs. Light-induced FTIR difference spectroscopy demonstrates extremely high sensitivity to molecular changes accompanying charge separation providing, in particular, information on the electronic structure of the primary electron donor P in its functionally important radical cation form P+ [3, 4]. Femtosecond transient absorption spectroscopy is the most appropriate method for investigation of fast electron transfer reactions occurring in bacterial RCs in the sub-picosecond time scale. The results of this work are discussed based on the analysis of key similarities and differences with the data obtained under the identical experimental conditions for the RCs from the pseudo-wild type of Rba. sphaeroides (psWt).
MATERIALS AND METHODS
Construction of a recombinant Rba. sphaeroides strain without light-harvesting antenna but containing mutant RCs with amino acid substitution of leucine in position M196 by histidine was described previously [5]. Reaction centers isolated from the puf-deficient strain transformed with a plasmid containing a wild type puf-operon were used as psWt RCs [5]. RCs were isolated using the standard procedure by treating chromatophores with lauryldimethylamine-N-oxide (LDAO) followed by purification on DEAE-cellulose [5]. The LDAO detergent was replaced with Triton X-100 on a membrane with molecular weight cut-off of 30 kDa (Millipore, USA) in an ultrafiltration cell under applied gaseous argon pressure.
Electronic absorption spectra of RCs in the range 650-1000 nm were recorded with a Shimadzu UV1800 spectrophotometer (Shimadzu, Japan).
Light-induced FTIR P+Q–/PQ difference spectra were recorded with an Equinox 55 FTIR spectrometer (Bruker, Germany) equipped with a DTGS detector and KBr beam splitter. Samples were obtained by applying a few microliters of the concentrated suspension of RCs solubilized in 20 mM Tris-HCl buffer (pH 8.0) containing 0.1% Triton X-100 onto a CaF2 plate followed by partial dehydration of the sample under a gaseous argon flow and covering it with a second CaF2 plate [6]. IR difference spectra were recorded under illumination of samples with constant light (1000 nm > λ > 620 nm). The spectral resolution was 4 cm–1. Illumination cycles were repeated hundreds of times to achieve acceptable level of signal-to-noise ratio.
Difference spectra with femtosecond time resolution were recorded using a laser spectrometer as described previously [7] with some modifications. A MaiTai titanium-sapphire laser (Spectra-Physics, USA) with a Spitfire Ace optical regenerative amplifier (Spectra-Physics) was used for generation of fundamental (800 nm) pulses with duration of 35-40 fs and energy of 3 mJ/pulse. The beam coming out of the amplifier was attenuated and split into two light beams. The first beam was used for generation of a continuum on a sapphire plate with thickness of ~2 mm. The continuum spectral components with wavelength >850 nm were separated by an RG850 light filter (Newport, USA), and the generated pulses were used for excitation of the RCs in the P absorption band. The second beam was passed over a M531.DD delay line (Physik Instrumente, Germany) and focused in a quartz cuvette with 5-mm thickness filled with water for generation of the continuum, which next was split into probing and reference beams at percent ratio 50 : 50. The spectra of probing and reference pulses were modified with spectral and spatial filtration to separate visible or near IR spectral region. Polarization of the excitation pulse was set parallel to the probing pulse. The probing and reference pulses after the cuvette with the sample were directed to the input slit of a SpectraPro 2300i spectrograph (Acton Research Corporation, USA). The spectra of pulses were recorded with a Pixis 400BR CCD camera (Princeton Instruments, USA). The RC sample (A865 ~ 0.5) was placed in a rotating quartz cuvette with optical pathlength of 1 mm (CDP Systems, Russia). Rotation speed and excitation frequency (10 Hz) were selected in a way so each excitation pulse would illuminate a new portion of RCs. Difference spectra were recorded in the visible (500-720 nm) or near IR (750-1000 nm) region in the time interval between 2 ps before excitation of the RCs and ~2 ns after it; 500 spectra were averaged for each delay. The intensity of excitation pulses was attenuated to the level that provided excitation of <20% of reaction centers in the sample.
All measurements were conducted at room temperature.
The femtosecond data were analyzed with global analysis [8] using the Glotaran program [9]. The program included mathematical description of spectral dispersion of the maximum of the instrumental function. The half width at half maximum of the instrumental function was estimated as ~35-40 fs for both investigated spectral regions.
RESULTS
The electronic absorption spectra of RCs from Rba. sphaeroides psWt and L(M196)H are presented in Fig. 1; they were recorded in the region of the Qy optical transitions (650-1000 nm) at room temperature. The spectra are normalized at the maximum of the bacteriopheophytin (BPheo) band at 759 nm based on the suggestion that the mutation does not introduce significant perturbations to the electronic structure of the BPheo molecules in the RCs and does not affect significantly their optical properties as the mutation locus is located far from the binding sites of both BPheo HA and BPheo HB.
Fig. 1. Electronic absorption spectra of Rba. sphaeroides psWt RCs (1) and L(M196)H RCs (2) recorded at room temperature. The spectra are normalized at the absorption maximum of bacteriopheophytin molecules at 759 nm.

Figure 1 shows that the absorption band of the low-energy Qy exciton transition of the primary electron donor P at ~865 nm is preserved in the mutant RCs, although it is slightly broadened and significantly lower in intensity compared to the respective band in the psWt RCs. Control experiments with the addition of reductants to the samples of mutant RCs (sodium ascorbate or sodium dithionite) revealed that the observed decrease in intensity of the P band at ~865 nm was not due to the oxidation of P (data not shown). Hence, despite the fact that the general dimeric nature of P is maintained in the mutant RCs, the electronic structure of the dimer is likely subjected to substantial modification. The minor short wavelength shifts of the absorption bands of the monomeric BChl molecules at ~805 nm and of the BPheo molecules at ~760 nm are also characteristic for the mutant (Fig. 1). The decrease in intensity of the Qy-band of P and its spectral broadening in the L(M196)H RCs in comparison with the psWt RCs was observed previously in the low-temperature (90 K) absorption spectrum [2]. It is interesting that in this case maximum of the Qy absorption band of P in the L(M196)H RC spectrum demonstrated larger long-wavelength shift than in the spectrum of the psWt RCs (894 versus 888 nm). It is well known that the spectral position of the long wavelength Qy-band of P depends on a number of factors including, in particular, formation of hydrogen bonds [10] and change of the energy of the charge-transfer states between the molecules PA and PB in the P dimer [11].
The light-induced FTIR difference spectra P+Q–/PQ (Q is a quinone acceptor) for reaction centers from psWt and L(M196)H are presented in Figs. 2 and 3. The positive and negative peaks in the spectra reflect an appearance of the state P+Q– and a disappearance of the state PQ, respectively. The FTIR spectra of psWt and L(M196)H (Figs. 2 and 3) were normalized to the amplitude of the differential signal (+)1750/(–)1740 cm–1 attributed to the shift of stretching vibrations of the 133-ester groups of the BChl [6, 12]. In agreement with the data published previously [6, 12], the FTIR spectrum P+Q–/PQ for the psWt RCs (Fig. 2, curve 1; Fig. 3, curve 1) demonstrates a broad band of the low-energy electronic transition at ~2700 cm–1 related to the transfer of the positive charge (hole) within the dimeric P+ radical cation and three phase-phonon IR bands at ~1550, 1477, and ~1290 cm–1. The band at ~2700 cm–1 is not observed for the monomeric BChl+ radical cation [12], which makes it a unique characteristic of the dimeric structure of the oxidized primary electron donor in the RCs of purple bacteria [4]. Vibrational modes of phase-phonon type are formally symmetry-forbidden in the monomeric BChl; however, they are amplified due to coupling with the hole-transfer transition, which make them also markers of the dimeric structure of P+ [13]. The intensity of all indicated bands depends significantly on the degree of electronic coupling between the PA and PB molecules in the P dimer [4, 13].
Fig. 2. Light-induced difference (light-minus-dark) FTIR spectra P+Q–/PQ for Rba. sphaeroides psWt RCs (1) and L(M196)H RCs (2) recorded at room temperature in the range 4800-1200 cm–1. The spectra are normalized at the amplitude of the differential signal (+)1750/(–)1740 cm–1. Region ~3700-3100 cm–1 is saturated due to the strong absorption of the sample and water and is excluded from the figure.
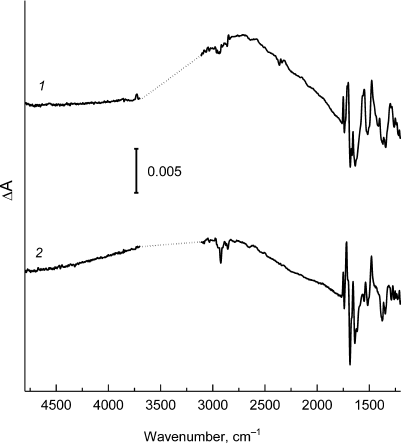
Fig. 3. Low-frequency region (1800-1200 cm–1) of the FTIR spectra P+Q–/PQ for Rba. sphaeroides psWt RCs (1) and L(M196)H RCs (2), taken from Fig. 2.
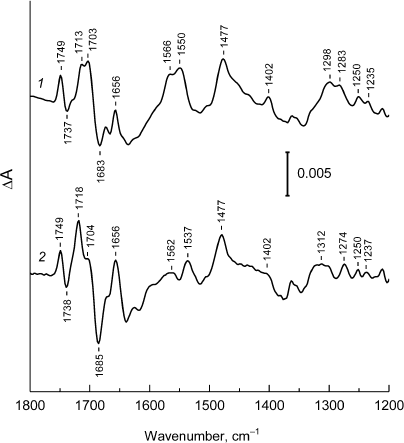
The FTIR P+Q–/PQ spectrum for the L(M196)H RCs also contains the hole-transfer band in the region of ~2700 cm–1 (Fig. 2, curve 2) and phase-phonon bands at 1537, 1477, and ~1300 cm–1 (Fig. 3, curve 2), which is in agreement with preservation of the dimeric structure of the electron donor P in the mutant [2] (Fig. 1). However, the pronounced decrease in intensity of the hole-transfer band and large changes in the intensity and frequencies of the phase-phonon bands in the spectrum of the mutant RCs indicate reduced electronic coupling and enhanced asymmetry in distribution of the positive charge between the molecules of BChl in the P+ radical cation [13].
The signals predominately related to the shifts of 131-keto-C=O-groups in the PA and PB molecules upon P photooxidation are observed in the region 1710-1650 cm–1 of the IR spectrum of the psWt RCs (Fig. 3, curve 1) [12]. These groups are conjugated with the π-electron system of the macrocycle and are sensitive to changes in the electronic structure of the P+ dimer. The relative increase in intensity of the stretching vibrations of the 131-keto-group of PA+ at 1718 cm–1 in comparison with the similar band of the 131-keto-group of PB+ at 1704 cm–1 in the spectrum of the mutant L(M196)H RCs (Fig. 3, curve 2 ) can be interpreted as an increase in localization of the positive charge on the PA molecule [14]. Redistribution of the positive charge towards PA+ due to the wakening of electronic coupling between molecules PA and PB is likely due to the interaction of the introduced histidine M196 with the π-electron system of P.
The results of investigation of the dynamics and mechanism of charge separation in the mutant RCs provided by femtosecond time-resolved difference absorption spectroscopy are presented in Figs. 4-6. The data were analyzed using global analysis [8].
Fig. 4. a) Normalized decay kinetics of the stimulated emission from the P* excited state at 936 nm for the Rba. sphaeroides psWt RCs (1) and at 930 nm for the L(M196)H RCs (2). Monoexponential approximation of the kinetics is presented by solid lines. b) Normalized kinetics of absorption change at 836 nm for the psWt RCs (1) and at 830 nm for the L(M196)H RCs (2).
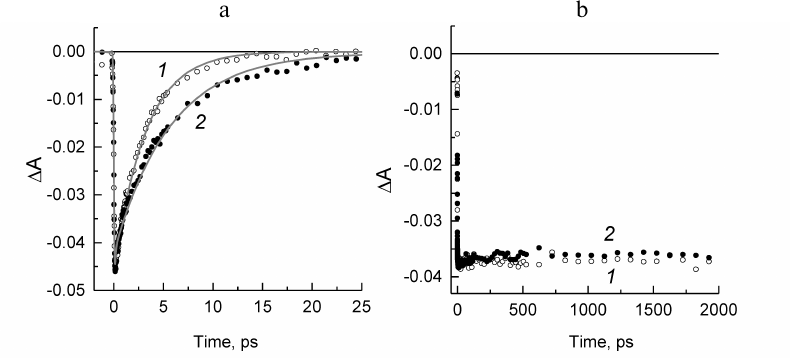
Fig. 5. Difference spectra associated with evolution of P* (1), P+HA (2), and P+QA– (3) states obtained from global analysis of the spectral–temporal data in the Qx (a) and Qy (b) regions for the Rba. sphaeroides psWt RCs. Lifetimes of the states P* and P+HA are 3.2 and 190 ps, respectively. The state P+QA– does not decay within the investigated time scale (2 ns).
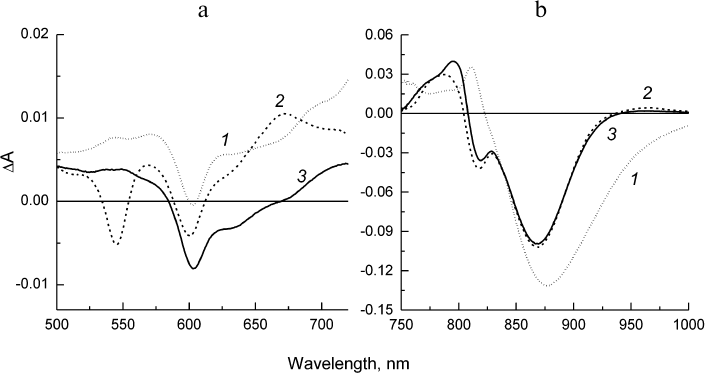
Fig. 6. Difference spectra associated with evolution of P* (1), P+HA (2), and P+QA– (3) states obtained from global analysis of the spectral–temporal data in the Qx (a) and Qy (b) regions for the Rba. sphaeroides L(M196)H RCs. Lifetimes of the states P* and P+HA are 5.7 and 219 ps, respectively. The state P+QA– does not decay within the investigated time scale (2 ns).
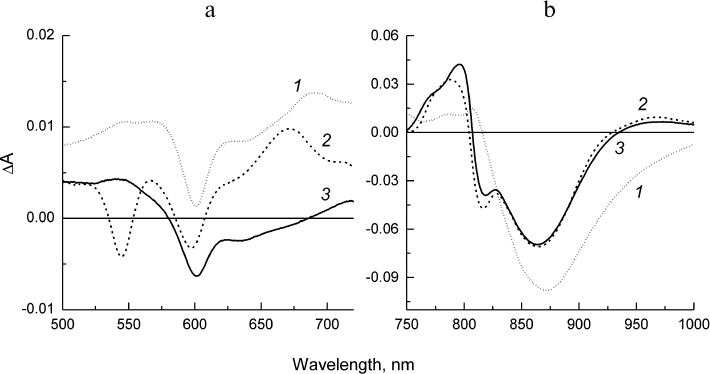
The kinetics of the stimulated emission decay from the excited state of the primary donor P* reflecting the electron transfer from P* to BPheo HA in the psWt RCs (curve 1) and mutant RCs (curve 2) are compared in Fig. 4a. The monoexponential analysis of the kinetic traces revealed that the lifetime of P* (that is the time constant of the reaction P* → P+HA–) in the psWt RCs is 3.2 ps, which is in good agreement with the literature data for the wild type RCs. The lifetime of P* in the L(M196)H RCs increases to 5.7 ps. Investigation of the bleaching kinetics on the short-wavelength side of the P absorption band at ~830-840 nm (region, where contribution of the stimulated emission is minimal) provides information on the quantum yield of the charge separation in the RCs. It can be seen in Fig. 4b (curve 1) that the amplitude of bleaching in the psWt RCs remains practically unchanged in the time interval of 2 ns. This implies that the charge separation is not accompanied by the return of P* and P+ to the ground state, and that the quantum yield of the P+QA– state is close to 100%. Comparison of the kinetics 1 and 2 in Fig. 4b reveals that within the accuracy of our measurements a similar conclusion can be made for the L(M196)H RCs.
The difference spectra associated with the evolution of the states involved in the electron transfer from P* to QA (EADS, evolution-associated decay spectra) [8] in the psWt and L(M196)H RCs are presented in Figs. 5 and 6, respectively. The EADS were obtained from the global analysis of the complete set of the experimental absorption difference spectra recorded at different time delays between the pump and probe laser pulses using the sequential kinetic model with increasing lifetimes of the states (1 → 2 → 3 →… ). It follows from Figs. 5 and 6 that the EADS spectral profiles and corresponding lifetimes are close for the two types of RCs and are in agreement with the data published for the wild type RCs (see, for example, Sun et al. [15]). The EADS associated with lifetimes 3.2 and 5.7 ps show spectral features typical for the excited state P* (Figs. 5 and 6, curves 1). A broad negative band is observed in the Qy-region of both spectra that include contributions from the bleaching P absorption band at ~865 nm and from stimulated emission from P* in the region of ~920 nm, as well as the characteristic positive peak at 810 nm. The negative band at 600 nm is present in the Qx spectral regions on the background of the weakly structured absorption of P* in the range 500-720 nm. The P* state evolves into the charge-separated state P+HA– with EADS demonstrating the preserved negative band at 600 nm, bleaching of the Qx-absorption band of the BPheo HA at 545 nm, and emerging of the absorption bands of the radical anion HA– at ~670 and 960 nm. Bleaching of the P absorption band at ~865 nm is observed in the Qy-region as well as electrochromic shift in the region of ~800 nm (Figs. 5 and 6, curves 2). The amplitude of the bleaching at ~865 nm in the EADS of the mutant RCs is reduced relative to the amplitude in the EADS of the psWt RCs in accordance with the differences observed in the RC absorption spectra (Fig. 1). In the course of several hundreds of picoseconds (τ ~ 200 ps), the P+HA– state in both types of RCs is evolved to the terminal state P+QA–, which does not decay on the time scale of our measurements (Figs. 5 and 6, curves 3).
Hence, all the results of femtosecond measurements indicate that the sequence P* → P+HA– → P+QA– and dynamics of the electron transfer involved in the charge separation as well as the total yield of the process that are observed for psWt RCs are practically fully maintained in the L(M196)H RCs.
Note that formation of the P+BA– intermediate state [16-18], which decays faster than forms in the native RCs causing a low level of its population and difficulties in its experimental detection [19], has not been considered in this work during the global analysis. However, preliminary analysis of the difference spectra of the L(M196)H RCs with the reduced QA revealed a weak absorption band at ~1025 nm, which could be assigned to the BA– radical anion [16, 18] (data not shown). It is likely that the two-step mechanism of the initial electron transfer P* → P+BA– → P+HA– typical for the native RCs (see, for example, Khatypov et al. [20]) is conserved also in the mutant preparations.
DISCUSSION
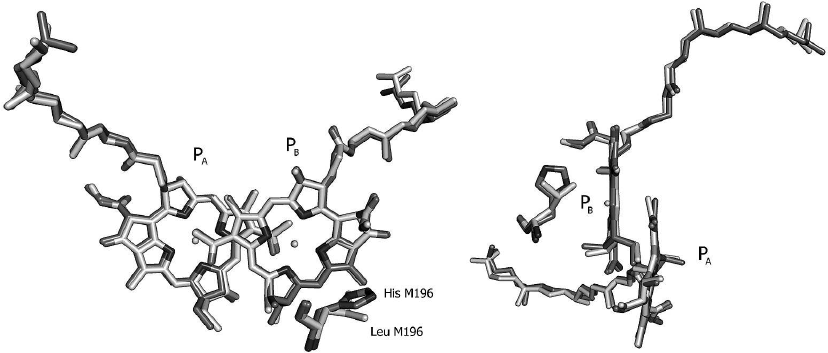
According to the X-ray diffraction data for the mutant L(M196)H RCs [2], the angle between the imidazole group of histidine M196 and the plane of the PB macrocycle is estimated as 103 ± 10°, which corresponds to the T-shaped geometry of the mutual arrangement of the histidine residue and BChl. The distance between the histidine nitrogen atom ND1 and ring C in the PB molecule is 3.8 ± 0.15 Å. Hence, the introduced histidine and the PB molecule are located quite close to each other, ensuring their interaction.
It can be seen from the crystal structure of the L(M196)H RCs (Fig. 7) that the mutation does not cause global changes in the geometry of the P dimer. The observed minor deviations between the RC structures of the mutant and psWt are likely beyond the limit of resolution. Hence, it can be concluded that the experimentally observed changes in the electronic structures of P and P+ (Figs. 1-3) are caused by the interaction between histidine M196 and the π-electron system of the PB molecule. According to the literature data [1], the T-shaped orientation of the imidazole ring relative to the plane of the aromatic molecule is characteristic for π-hydrogen interaction. Formation of the π-hydrogen interaction in the mutant RCs would be in agreement with the fact that the L(M196)H mutation increases the redox potential of the P+/P pair by ~60 mV in comparison with the psWt RCs [2]. Apparently, formation of the hydrogen bond between the histidine and the conjugated system of P destabilizes the P+ state in comparison with the neutral state, thus hindering P oxidation and shifting the redox potential of the P+/P pair towards the positive region as has been observed in the mutant RCs during formation of hydrogen bonds between the carbonyl groups of P and histidines [23, 24]. In this connection, it is worth mentioning that the long wavelength shift of the P band in the low temperature spectrum of the L(M196)H RCs [2] is in agreement with the theoretically estimated effect of formation of the hydrogen bond on the energy of the Qy-transition in P [10].
Fig. 7. Overlay of crystal structures of the Rba. sphaeroides psWt RCs (PDB: 3V3Y [21]) and of the L(M196)H mutant [2] in the region of the primary electron donor P dimer in two projections. The respective amino acid residues at position M196 are marked. Crystal structure resolution for the psWt and L(M196)H RCs was 2.8 and 2.4 Å, respectively. The PyMOL program was used for generating this figure [22].
It is interesting in this context that a similar effect on the low-temperature absorption spectrum of RCs, redox potential of P, P+ electronic structure, and primary photochemistry was observed previously for the Rba. sphaeroides L(M160)H mutant RCs, where a common hydrogen bond was introduced between the histidine M160 and 131-keto-carbonyl group of the BChl PB molecule [12, 25]. The shift of the absorption band of P towards longer wavelengths and decrease of its intensity was observed in the low-temperature (20 K) absorption spectrum of the L(M160)H RC mutant [25]. The redox potential of the P+/P pair in the mutant RCs increased by 55 mV in comparison with the wild type RCs [25]. The hole-transfer band and one of the phase-phonon bands in the P+QA–/PQA FTIR spectrum of the L(M160)H mutant were shifted towards the high-frequency region to ~2800 and ~1300 cm–1, respectively, and the intensity of both bands decreased [12]. It is significant that the decrease in intensity of the IR bands in the spectrum of L(M160)H [12] was pronounced to a considerably lower degree than in the L(M196)H RCs (Figs. 2 and 3, curves 2), not affecting, in particular, the phase-phonon band at 1550 cm–1. These data can be explained by the fact that during formation of the common hydrogen bond in the L(M160)H RCs the hydrogen atom of the N-H-group in imidazole interacts with the electron density of oxygen in the 131-keto-carbonyl group of PB, while in the case of π-hydrogen bond the hydrogen atom interacts directly with the π-electron cloud of the PB BChl macrocycle, causing greater perturbation of the P+ electronic structure in the L(M196)H RCs. The time constants of electron transfer from P* to HA and from HA– to QA in the mutant L(M160)H RCs (5.7 and 210 ps, respectively) [25] are practically identical to the ones observed for the L(M196)H RCs (5.7 and 219 ps, respectively; this study).
It was shown previously using quantum-chemical calculations and EPR data that the semiquinone form of the quinone acceptor A1 could be stabilized in complexes of photosystem 1 (PS1) reconstructed with modified quinones with short side chains via re-orientation of the quinone molecule followed by formation of a π-hydrogen bond with tryptophan [26]. It was also suggested based on the quantum-chemical calculations that the CH–π-interactions between β-carotene and chlorophyll together with the π–π-stacking played an important role in the binding of a β-carotene molecule in the PS1 from Synechococcus elongatus [27]. The mutant Rba. sphaeroides L(M196)H RCs are likely to be the first example of formation of a π–hydrogen bond between the protein and one of the electron transfer cofactors in bacterial RCs. To the best of our knowledge, cofactor–protein bonds of this type have not been detected in the native photosynthetic RCs, while the formation of common hydrogen bonds between the protein and redox-active pigment and quinone cofactors plays an important role in optimization of the function (and, likely, maintenance of the structure) of the native RC complexes [28-30]. According to the literature data, the most favorable orientation of the π–hydrogen bond is the perpendicular arrangement of the X–H bond of the donor (X is atom N, O, or C) with respect to the plane of π–acceptor [1, 31, 32]. It was also shown that unlike for the common hydrogen bond, a “softer” geometry is characteristic for the π–hydrogen interaction, which makes formation of a larger number of conformations in a wider range of bonding energy more probable [32]. It is plausible that the π–hydrogen bonds do not provide noticeable functional advantages in comparison with the common hydrogen bonds (with respect to the effect on the redox properties of cofactors, for example), while the quite specific spatial organization of the π–hydrogen interaction and its potential heterogeneity with regards to bonding energy could be at least partial reason for the fact that this type of interaction between the protein and pigment and quinone cofactors is not realized in native RCs.
Funding
This work was financially supported by State Task No. AAAA-A17030110140-5 and partially supported by the Russian Foundation for Basic Research (grant No. 17-00-00207 KOMFI, FTIR measurements, femtosecond measurements; grant No. 17-44-500828, preparation of RC samples).
Conflict of Interest
The authors declare no conflict of interest.
Ethical Compliance
This work does not involve any experiments using humans and animals as study objects.
REFERENCES
1.Liao, S. M., Du, Q. S., Meng, J. Z., Pang, Z. W.,
and Huang, R. B. (2013) The multiple roles of histidine in protein
interactions, Chem. Cent. J., 7, 44-55, doi:
10.1186/1752-153X-7-44.
2.Fufina, T. Y., Vasilieva, L. G., Gabdulkhakov, A.
G., and Shuvalov, V. A. (2015) The L(M196)H mutation in Rhodobacter
sphaeroides reaction center results in new electrostatic
interactions, Photosynth. Res., 125, 23-29, doi:
10.1007/s11120-014-0062-0.
3.Lutz, M., and Mantele, W. (1991) Vibrational
spectroscopy of chlorophylls, in Chlorophylls (Scheer, H.,
ed.) CRC Press, Boca Raton, FL, pp. 855-902.
4.Breton, J., Nabedryk, E., and Parson, W. W. (1992)
A new infrared electronic transition of the oxidized primary electron
donor in bacterial reaction centers: a way to assess resonance
interactions between the bacteriochlorophylls, Biochemistry,
31, 7503-7510, doi: 10.1021/bi00148a010.
5.Gabdulkhakov, A. G., Fufina, T. Y., Vasilieva, L.
G., Mueller, U., and Shuvalov, V. A. (2013) Expression, purification,
crystallization and preliminary X-ray structure analysis of wild-type
and L(M196)H mutant Rhodobacter sphaeroides reaction centres,
Acta. Crystallogr. Sect. F, 69, 506-509, doi:
10.1107/S1744309113006398.
6.Zabelin, A. A., Fufina, T. Y., Vasilieva, L. G.,
Shkuropatova, V. A., Zvereva, M. G., Shkuropatov, A. Y., and Shuvalov,
V. A. (2009) Mutant reaction centers of Rhodobacter sphaeroides
I(L177)H with strongly bound bacteriochlorophyll a: structural
properties and pigment–protein interactions, Biochemistry
(Moscow), 74, 68-74, doi: 10.1134/S0006297909010106.
7.Khatypov, R. A., Khristin, A. M., Fufina, T. Yu.,
and Shuvalov, V. A. (2017) An alternative pathway of light-induced
transmembrane electron transfer in photosynthetic reaction centers of
Rhodobacter sphaeroides, Biochemistry (Moscow),
82, 692-697, doi: 10.1134/S0006297917060050.
8.Van Stokkum, I. H. M., Larsen, D. S., and van
Grondelle, R. (2004) Global and target analysis of time-resolved
spectra, Biochim. Biophys. Acta, 1657, 82-104, doi:
10.1016/j.bbabio.2004.04.011.
9.Snellenburg, J. J., Laptenok, S. P., Seger, R.,
Mullen, K. M., and van Stokkum, I. H. M. (2012) Glotaran: a Java-based
graphical user interface for the R package TIMP, J. Stat. Soft.,
49, 1-22, doi: 10.18637/jss.v049.i03.
10.Thompson, M. A., Zerner, M. C., and Fajer, J.
(1991) A theoretical examination of the electronic structure and
excited states of the bacteriochlorophyll b dimer from
Rhodopseudomonas viridis, J. Phys. Chem., 95,
5693-5700, doi: 10.1021/j100167a058.
11.Parson, W. W., and Warshel, A. (1987)
Spectroscopic properties of photosynthetic reaction centers. 2.
Application of the theory to Rhodopseudomonas viridis, J. Am.
Chem. Soc., 109, 6152-6163, doi: 10.1021/ja00254a040.
12.Naberdyk, E., Allen, J. P., Taguchi, A. K. W.,
Williams, J. C., Woodbury, N. W., and Breton, J. (1993) Fourier
transform infrared study of the primary electron donor in
chromatophores of Rhodobacter sphaeroides with reaction centers
genetically modified at residues M160 and L131, Biochemistry,
32, 13879-13885, doi: 10.1021/bi00213a017.
13.Reimers, J. R., and Hush, N. S. (2003) Modeling
the bacterial photosynthetic reaction center. VII. Full simulation of
the intervalence hole-transfer absorption spectrum of the special-pair
radical cation, J. Chem. Phys., 119, 3262-3277, doi:
10.1063/1.1589742.
14.Malferrari, M., Turina, P., Francia, F.,
Mezzetti, A., Leibl, W., and Venturoli, G. (2015) Dehydration affects
the electronic structure of the primary electron donor in bacterial
photosynthetic reaction centers: evidence from visible-NIR and
light-induced difference FTIR spectroscopy, Photochem. Photobiol.
Sci., 14, 238-251, doi:10.1039/c4pp00245h.
15.Sun, C., Carey, A.-M., Gao B.-R., Wraight, C. A.,
Woodbury, N. W., and Lin, S. (2016) Ultrafast electron transfer
kinetics in the LM dimer of bacterial photosynthetic reaction center
from Rhodobacter sphaeroides, J. Phys. Chem. B,
120, 5395-5404, doi: 10.1021/acs.jpcb.6b05082.
16.Arlt, T., Schmidt, S., Kaiser, W., Lauterwasser,
C., Meyer, M., Scheer, H., and Zinth, W. (1993) The accessory
bacteriochlorophyll: a real electron carrier in primary photosynthesis,
Proc. Natl. Acad. Sci. USA, 90, 11757-11761, doi:
10.1073/pnas.90.24.11757.
17.Shkuropatov, A. Ya., and Shuvalov, V. A. (1993)
Electron transfer in pheophytin a-modified reaction centers from
Rhodobacter sphaeroides (R-26), FEBS Lett., 322,
168-172, doi: 10.1016/0014-5793(93)81561-D.
18.Kennis, J. T. M., Shkuropatov, A. Ya., van
Stokkum, I. H. M., Gast, P., Hoff, A. J., Shuvalov, V. A., and Aartsma,
T. J. (1997) Formation of a long-lived
P+BA– state in plant
pheophytin-exchanged reaction centers of Rhodobacter sphaeroides
R26 at low temperature, Biochemistry, 36, 16231-16238,
doi: 10.1021/bi9712605.
19.Zinth, W., and Wachtveitl, J. (2005) The first
picoseconds in bacterial photosynthesis – ultrafast electron
transfer for the efficient conversion of light energy, Chem. Phys.
Chem., 6, 871-880, doi: 10.1002/cphc.200400458.
20.Khatypov, R. A., Khmelnitskiy, A. Yu., Khristin,
A. M., Fufina, T. Yu., Vasilieva, L. G., and Shuvalov, V. A. (2012)
Primary charge separation within P870* in wild type and heterodimer
mutants in femtosecond time domain, Biochim. Biophys. Acta,
1817, 1392-1398, doi: 10.1016/j.bbabio.2011.12.007.
21.Vasilieva, L. G., Fufina, T. Y., Gabdulkhakov, A.
G., Leonova, M. M., Khatypov, R. A., and Shuvalov, V. A. (2012) The
site-directed mutation I(L177)H in Rhodobacter sphaeroides
reaction center affects coordination of PA and BB
bacteriochlorophylls, Biochim. Biophys. Acta, 1817,
1407-1417, doi: 10.1016/j.bbabio.2012.02.008.
22.DeLano, W. L. (2002) The PyMOL molecular graphics
system (http://www.pymol.org).
23.Lin, X., Murchison, H. A., Nagarajan, V., Parson,
W. W., Allen, J. P., and Williams, J. C. (1994) Specific alteration of
the oxidation potential of the electron donor in reaction centers from
Rhodobacter sphaeroides, Proc. Natl. Acad. Sci. USA,
91, 10265-10269, doi: 10.1073/pnas.91.22.10265.
24.Allen, J. P., and Williams, J. C. (1995)
Relationship between the oxidation potential of the bacteriochlorophyll
dimer and electron transfer in photosynthetic reaction centers, J.
Bioenerg. Biomembr., 27, 275-283, doi:
10.1007/BF02110097.
25.Williams, J. C., Alden, R. G., Murchison, H. A.,
Peloquin, J. M., Woodbury, N. W., and Allen, J. P. (1992) Effects of
mutations near the bacteriochlorophylls in reaction centers from
Rhodobacter sphaeroides, Biochemistry, 31,
11029-11037, doi: 10.1021/bi00160a012.
26.Kaupp, M. (2002) The function of photosystem I.
Quantum chemical insight into the role of tryptophan–quinone
interactions, Biochemistry, 41, 2895-2900, doi:
10.1021/bi0159783.
27.Wang, Y., Mao, L., and Hu, X. (2004) Insight into
the structural role of carotenoids in the photosystem I: a quantum
chemical analysis, Biophys. J., 86, 3097-3111, doi:
10.1016/S0006-3495(04)74358-1.
28.Allen, J. P., and Williams, J. C. (2006) The
influence of protein interactions on the properties of the
bacteriochlorophyll dimer in reaction centers, in Chlorophylls and
Bacteriochlorophylls: Biochemistry, Biophysics, Functions
and Applications (Grimm, B., Porra, R. J., Rudiger, W., and Scheer,
H., eds.) Springer, The Netherlands, pp. 283-295.
29.Bylina, E. J., Kirmaer, C., McDowell, L., Holten,
D., and Youvan, D. C. (1988) Influence of an amino acid residue on the
optical properties and electron transfer dynamics of a photosynthetic
reaction centre complex, Nature, 336, 182-184, doi:
10.1038/336182a0.
30.Flores, M., Isaacson, R., Abresch, E., Calvo, R.,
Lubitz, W., and Feher, G. (2007) Protein–cofactor interactions in
bacterial reaction centers from Rhodobacter sphaeroides R-26:
II. Geometry of the hydrogen bonds to the primary quinone
QA– by 1H and 2H
ENDOR spectroscopy, Biophys. J., 92, 671-682, doi:
10.1529/biophysj.106.092460.
31.Steiner, T., and Koellner, G. (2001) Hydrogen
bonds with π-acceptors in proteins: frequencies and role in
stabilizing local 3D structure, J. Mol. Biol., 305,
535-557, doi: 10.1006/jmbi.2000.4301.
32.Du, Q.-S., Wang, Q.-Y., Du, L.-Q., Chen, D., and
Huang, R.-B. (2013) Theoretical study on the polar hydrogen–π
(Hp-π) interactions between protein side chains, Chem. Cent.
J., 7, 92-99, doi: 10.1186/1752-153X-7-92.