Hypoxia as a Factor Involved in the Regulation of the apoA-1, ABCA1, and Complement C3 Gene Expression in Human Macrophages
A. M. Bogomolova1,2, V. S. Shavva2, A. A. Nikitin1,2, E. V. Nekrasova2, E. B. Dizhe2, E. E. Larionova2, I. V. Kudriavtsev2, and S. V. Orlov1,2,a,b*
1St. Petersburg State University, 199034 St. Petersburg, Russia2Institute of Experimental Medicine, 197376 St. Petersburg, Russia
* To whom correspondence should be addressed.
Received December 10, 2018; Revised January 29, 2019; Accepted January 29, 2019
Hypoxia plays a critical role in progression of atherosclerosis. Local oxygen deficiency in a plaque creates a specific microenvironment that alters the transcriptome of resident cells, particularly of macrophages. Reverse cholesterol transport from plaque to liver is considered a main mechanism for regression of atherosclerosis. Ubiquitously expressed ATP-binding cassette transporter A1 (ABCA1) and liver- and small intestine-derived apolipoprotein A-1 (ApoA-1) are two main actors in this process. We recently reported endogenous apoA-1 expression in human macrophages. While ABCA1 and ApoA-1 have antiatherogenic properties, the role of complement factor C3 is controversial. Plasma C3 level positively correlates with the risk of cardiovascular diseases. On the other hand, C3 gene knockout in a murine atherosclerosis model increases both plaque size and triglycerides level in blood. In the present study, we show for the first time that a hypoxia-mimicking agent, CoCl2, induces the upregulation of the apoA-1 and C3 genes and the accumulation of intracellular and membrane protein ApoA-1 in THP-1 macrophages. The MEK1/2-Erk1/2 and MKK4/7-JNK1/2/3 cascades are involved in upregulation of ABCA1 and C3 via activation of transcription factor NF-κB, which interacts with the HIF-1α subunit of hypoxia-inducible factor 1 (HIF-1). The three major MAP-kinase cascades (Erk1/2, JNK1/2/3, and p38) and the NF-κB transcription factor are involved in the hypoxia-induced expression of the apoA-1 gene in THP-1 macrophages.
KEY WORDS: atherosclerosis, hypoxia, macrophages, THP-1, gene expression regulation, apoA-1, ABCA1, C3DOI: 10.1134/S0006297919050079
Abbreviations: ABCA1, ATP-binding cassette transporter A1; ApoA-1, apolipoprotein A-1; HIF-1, hypoxia-inducible factor 1; LXR, liver X receptor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B-cells.
Atherosclerosis is a chronic inflammatory disease associated with the
formation of plaques in the arterial tunica intima. It is developed
mainly in distorted arteries under hemodynamic shear stress [1]. Shear stress can be caused by a laminar or
disturbed blood flow, with the latter leading to vessel damage and
subsequent endothelium activation and thickening of tunica media and
intima. Endotheliocytes trigger expression of adhesive agents,
integrins, selectins, interleukins, interferons, and cytokines.
Concurrent increase of vessel permeability leads to infiltration of the
subendothelial space by low density lipoproteins (LDL). These molecules
are bound in the intima by proteoglycans of the extracellular matrix
and undergo various modifications like oxidation, proteolysis, etc.
Modified LDL are absorbed by macrophages engaged during endothelium
activation, which induces accumulation of intracellular cholesterol and
leads to transformation of macrophages into foam cells [2].
Attraction of monocytes and thickening of intima by proliferation of smooth muscle cells leads to growth of atherosclerotic plaques and sparseness of capillaries that nourish the artery middle coat. An increased demand for oxygen triggers vascularization of the intima and plaque, but new vessels tend to originate at the perimeter of the plaque, with its center remaining under permanent hypoxia [3].
Transcription factor HIF-1 (hypoxia-inducible factor 1) is a key hypoxia mediator. It is a heterodimer of constitutively expressed subunit HIF-1β and inducible HIF-1α [4]. Under normoxia, proteins with a prolyl hydroxylase domain (PHD1, PHD2, PHD3) and asparaginyl hydroxylase FIH mediate hydroxylation-induced proteasome degradation of HIF-1α. Under low oxygen, no catalysis occurs, and HIF-1α is stabilized at the protein level [5]. HIF-1 induces proliferation of smooth muscle cells and recruitment of monocytes, other leukocytes, and T-lymphocytes to the plaque [6, 7], lowers the migration activity of macrophages, and detains them in the intima [3]. Thus, the population of macrophages and foam cells is constantly growing.
The core pathway of atherosclerosis regression is now considered to be the reverse transport of cholesterol in the form of high density lipoprotein (HDL) from plaque macrophages to the liver. The main proteins involved are apolipoprotein A-1 (ApoA-1) and ATP-binding cassette transporter A1 (ABCA1). ApoA-1 is the major protein component of antiatherogenic HDL, which regulates their plasma level. ApoA-1 is derived mainly from liver and small intestine and enhances ABCA1-mediated cholesterol efflux from peripheral tissues. ABCA1 is a ubiquitous cassette transporter that canalizes the membrane and mediates energy-dependent lipid efflux from the cell. Earlier, we reported endogenic ApoA-1 synthesis in human macrophages [8, 9]. The ApoA-1-homogeneous population of monocytes in human peripheral blood differentiates into ApoA-1-poor and ApoA-1-rich macrophages. This protein is localized in intracellular vesicles and at the external surface of the plasma membrane. Membrane ApoA-1 is either associated with ABCA1 or localizes unbound in lipid rafts [8]. Flow cytometry studies show that the membrane ABCA1 level is higher in ApoA-1-rich macrophages, and transfection with ApoA-1-specific small interfering RNA (siRNA) downregulates the transporter. Macrophage-derived ApoA-1 thus stabilizes ABCA1 in the plasma membrane. Also, ApoA-1 was demonstrated to affect proinflammatory status of the macrophage: its siRNA-induced knockdown results in upregulation of IL-1β and NOS2 in the presence of lipopolysaccharides [8].
While ApoA-1 and ABCA1 have atheroprotective properties, the role of complement C3 is highly controversial. Higher C3 levels in plasma are associated with atherosclerosis and cardiac infarctions, while the level of anaphylatoxin C3a, a hydrolysis derivate of C3, correlates with thickening of the intima and carotid middle coat and the development of acute coronary syndrome [10, 11]. Components of the complement system are present in atherosclerotic plaques arriving to the intima from blood and being synthesized by resident cells. Cascade activation in the plaque can follow three pathways. Classically, it is triggered by oxidized LDL-specific autoantibodies [12], and alternatively by cholesterol crystals, which ultimately induces the assembly of NLRP3 inflammasome and production of proinflammatory cytokines [13]. Plaque macrophages express receptors binding anaphylatoxins C3a and C5a, which trigger oxidative burst and synthesis of TNFα and IL-1. Also, C3a and C5a are chemoattractants for eosinophils, mast cells, monocytes, and B‐ and T‐lymphocytes; they increase the permeability of small blood vessels and induce contraction of smooth muscle cells [10].
A series of studies report the systemic antiatherogenic role of C3. Atherosclerotic plaques in LDL-R- and C3-knockout mice contain more foam cells [14]. The same models also with knockout of ApoE exhibit higher levels of cholesterol and plasma triglycerides and have plaques 84% larger in size compared to C3-intact knockouts [15]. Inactivated C3b (iC3b) was also shown to play an important role in antiinflammatory clearance of apoptotic cells. The interaction of macrophages and dendrite cells with iC3b inhibits NF-κB signaling, lowers the TGF-β, and increases IL-10 secretion [16].
In this work we studied the expression mechanisms of the important for atherogenesis genes apoA-1, ABCA1, and C3 in THP-1 macrophages under hypoxic conditions.
MATERIALS AND METHODS
Cell cultures. Human acute monocytic leukemia cell line THP-1 was obtained from the Collection of Vertebrate Cell Cultures maintained by the Institute of Cytology of the Russian Academy of Sciences. The cells were cultivated under 5% CO2 at 37°C in RPMI medium (Biolot, Russia) containing 10% fetal calf serum (FCS, HyClone, USA). Differentiation of THP-1 monocytes to macrophages was induced by adding phorbol-12-myristate-13-acetate (PMA) to the concentration of 50 ng/ml (81 nM) for over 24 h. The cells were afterwards rinsed free of PMA and passaged in fresh RPMI medium with 10% FCS for over 48 h. Differentiating THP-1 macrophages were cultivated for three days in total.
Antibodies. Monoclonal murine antibodies against human β-actin (ab3280) were purchased from Abcam (GB). Intracellular ApoA-1 protein was detected with goat polyclonal antibodies against human ApoA-1 described earlier [17] and horseradish peroxidase-conjugated rabbit secondary antibodies against goat IgG (A5420; Sigma, USA). Surface ApoA-1 was detected with murine monoclonal antibodies against human ApoA-1 (0650-0050; Bio-Rad, USA) and Alexa Fluor 647-labeled rabbit secondary antibodies F(ab')2 against murine IgG (Abcam).
Signaling pathway inhibitors. Inhibitors of MAPK and NF-κB were purchased from Biomol (USA), and LXR agonist from Sigma. One hour prior to administration of CoCl2, the culture medium was supplemented with one of the following compounds: p38 inhibitor SB203580 (EI-286, 12.5 μΜ), JNK1/2/3 inhibitor SP600125 (EI-305, 10 μM), MEK1/2 inhibitor U0126 (EI-282, 10 μM), NF-κB inhibitor QNZ (EI-352, 10 nM), LXR agonist TO901317 (2.5 μM). The solvent dimethyl sulfoxide (DMSO) was used as a control in concentrations corresponding to the probes with inhibitors.
RNA extraction. For RNA extraction and subsequent RT-PCR, cells were seeded in 24-well plates with density 1·104 cells/cm2 and incubated under 5% CO2 at 37°C. After 16- or 24-h incubation with CoCl2, the cells were lysed with commercial RNA Extract Kit (Evrogen, Russia), and total RNA was extracted following the manufacturer’s protocol. Degradation level of total RNA was verified by electrophoresis in 1% agarose gel using 28S and 18S rRNA bands as relative markers. The concentration and purity of RNA was determined with an Avaspec-2048 spectrophotometer (Avantes, The Netherlands). Optical density ratios of RNA samples at 260 and 280 nm were considered as corresponding to pure RNA if they exceeded 2.0 and 1.7 at 260 and 230 nm, respectively.
Reverse transcription and real-time PCR. In this study, expression of the β-actin and GAPDH genes was considered as reference, and apoA-1, ABCA1, C3, IL-8 were the experimental genes. In RT-PCR assays, we used primers to the experimental genes and oligo-dT18; specific primers were used because of the low mRNA concentrations of the experimental genes and to avoid reverse transcription of long 3′-untranslated regions. The reaction mixture (25 µl) contained 2 µg RNA, specific primers (0.5 µl each) (2 pmol/µl), 0.5 µl oligo-dT18 (50 pmol/µl), reverse transcriptase M-MLV (50 U), nucleotides dATP, dGTP, dCTP, and dTTP (0.5 mM each), 5 µl 5× reverse transcription buffer (250 mM Tris-HCl, pH 8.3, at 25°C, 500 mM KCl, 15 mM MgCl2, 50 mM dithiothreitol). Immediately prior to the reaction, the mixture was preheated to 42°C for subsequent thermocycling (1.5 h – 42°C, 15 min – 70°C, hold at 4°C). cDNA obtained was stored at –20°C and used for RT-PCR assays.
A commercial kit from Syntol (Russia) was used for RT-PCR assays. Primers and hybridization probes for β-actin [18], GAPDH [19], apoA-1 [19], ABCA1 [20], and C3 [18] have been described earlier. The reaction mixture (25 µl) contained 0.5 μl primers (10 μM), 0.2 μl Taq polymerase (1.25 U), 2.5 μl 10× PCR buffer (100 mM Tris-HCl, pH 8.3, 500 mM KCl, 15 mM MgCl2), 1 µl cDNA (corresponds to reverse transcription reaction products obtained from 0.08 µg total RNA), and deionized water to 25 μl. Control wells contained the reaction mixture for PCR without cDNA. Assays were performed with a CFX-96 thermocycler (Bio-Rad) under the following conditions: 95°C – 5 min, 50 cycles: 95°C – 25 s, 59.5°C – 45 s (for TaqMan); 95°C – 5 min, 50 cycles: 95°C – 30 s, 60°C – 20 s, 72°C – 30 s (for SYBR Green).
The number of cycles (Ct) necessary to reach fluorescence threshold of 10 standard deviations from the background fluorescent signal was estimated with the built-in original software from Bio-Rad. Relative cDNA contents (in percent of control) were estimated as (2Ct control – Ct sample) × 100. The estimates for each gene were normalized based on the geometric mean of the two reference gene expression levels (GAPDH, β-actin) as described earlier [21] and represented as relative gene expression levels with the control expression set to 100%.
Western blotting. Cells were triple-rinsed in phosphate buffered saline (PBS), pH 7.6, and lysed in RIPA-50 buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 0.1% SDS, 0.01% NaN3, 1 mM phenylmethylsulfonylfluoride (PMSF), pH 7.4). Protein concentrations were measured by the Lowry assay. SDS electrophoresis was done in 12% or 20% polyacrylamide gels. Proteins were transferred to nitrocellulose membrane in 2 h. A 5% dried skim milk solution in PBS with 0.02% Tween 20 was used as the blocking buffer. The samples were incubated in the blocking buffer for 1 h, with primary antibodies for 12 h at 4°C, then with horseradish peroxidase-conjugated secondary antibodies for 1 h. Peroxidase was visualized by the ECL (enhanced chemiluminescence) method. Chemiluminescent signal was detected with the ChemiDoc MP Imaging System (Bio-Rad).
Flow cytometry. For detection of surface membrane ApoA-1, THP-1 macrophages were fixed in 4% formaldehyde/PBS solution for 20 min at 4°C and triple-rinsed in PBS. The samples were further blocked for 40 min at 22°C in the blocking buffer (1% BSA, 0.02% Tween-20/PBS). The cells were exposed to murine antibodies against human ApoA-1 in titer 1 : 250 in 1% BSA, 0.02% Tween-20/PBS at 25°C for 2 h. After double-rinsing in the blocking buffer, Alexa Fluor 647-labeled secondary antibodies were added in titer 1 : 1000 in 1% BSA, 0.02% Tween-20/PBS and incubated for 1 h at 25°C in the dark. The isotype controls used secondary antibodies only. After rinsing in PBS, the cells were transferred into tubes for an Epics Altra (Beckman Coulter, USA) flow cytometer and analyzed using the Beckman Coulter Navios system.
To detect toxic effects of CoCl2, THP-1 cells were washed off the plates with Versene solution and stained live with DNA-binding dyes propidium iodide and YO-PRO-1. Propidium iodide does not permeate intact cell membranes and stains dead cells, emitting in the red spectrum. YO-PRO-1 permeates cell membranes of apoptotic but not live cells and emits in the green spectrum. The analyses were done with the Beckman Coulter Navios system.
Statistical analysis. Results are presented as the mean ± mean standard error. Statistical confidence of differences between samples was estimated with Student’s unpaired t-test. Multiple comparisons were performed with Dunnett’s test. Differences were considered significant if p < 0.05. Statistics were calculated in Microsoft Excel.
RESULTS
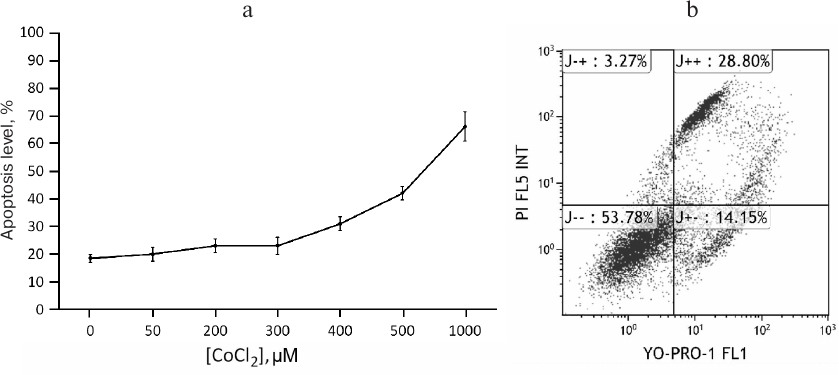
CoCl2 is a time- and dose-dependent regulator of apoA-1 and C3 genes and upregulates ApoA-1 protein in THP-1 macrophages. To study expression regulation of apoA-1 and C3 in THP-1 macrophages, we used the hypoxia-mimetic agent CoCl2. It stabilizes the HIF-1α protein by inhibiting PHD-containing enzymes that mediate proteasomal HIF-1α degradation in normoxia [5, 22]. Plausible cytotoxic effects of CoCl2 were revealed with flow cytometry. After incubation with CoCl2, the cells were stained with the DNA-binding dyes propidium iodide and YO-PRO-1 and analyzed by flow cytometry. CoCl2 at concentrations of 50-300 μM induces apoptosis in less than 10% of cells compared to the control. Higher concentrations are toxic (Fig. 1a). Figure 1b shows cell distribution by fluorescence levels of propidium iodide and YO-PRO-1 after incubation with 500 µM CoCl2. As a result, only 54% of the cells remain viable, and the rest go through different stages of apoptosis.
Fig. 1. Cytotoxic effects of CoCl2. a) Flow cytometry. Each dot represents a sum of quadrants J+– and J++. The graph is plotted based on three independent experiments. Error bars correspond to the mean standard error. b) Scatter dot plot showing the distribution of THP-1 macrophages over fluorescence levels of YO-PRO-1 and propidium iodide under 500 μM CoCl2. Axes YO-PRO-1 FL1 and PI FL5 INT represent YO-PRO-1 and propidium iodide fluorescence levels, respectively. Quadrants correspond to percentage of cells in different states: live cells (J– –); cells in early (J+–) and late (J++) apoptosis; dead cells (cell debris; J–+).
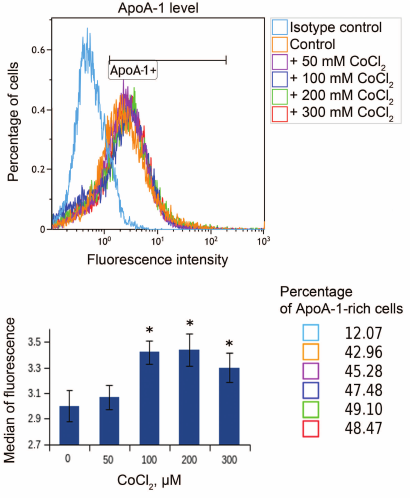
Hypoxia is known to positively regulate expression of IL-8 and ABCA1 in macrophages [6, 23, 24]. In this work, IL-8 induction in response to hypoxia was used as a positive control. Levels of IL-8 and ABCA1 mRNA reach their peak values under 100 μM CoCl2 during 16-h incubation (Fig. 2). These conditions are therefore optimal for hypoxia response induction in THP-1 macrophages. CoCl2 upregulates apoA-1 mRNA over the entire concentration range at incubation times of 16 and 24 h (Fig. 3a). The quantities of intracellular (Fig. 3b) and surface protein ApoA-1 (Fig. 4) also increase. Expression of C3 mRNA as well increases under 100 μM CoCl2 during 16-h-, but not 24-h incubation. On 24-h incubation, the gene expression lowers at concentrations of 200 μM and higher (Fig. 3c).
Fig. 2. CoCl2 is a dose- and time-dependent expression regulator of IL-8 (a) and ABCA1 (b) genes in THP-1 macrophages. Real-time PCR. Cells were incubated at the indicated CoCl2 concentrations for 16 and 24 h. Error bars correspond to the mean standard error. The data are derived from three independent experiments. Statistically significant differences between mRNA samples in the control and CoCl2 treatments are marked with asterisks (Student’s t test, * p < 0.05).
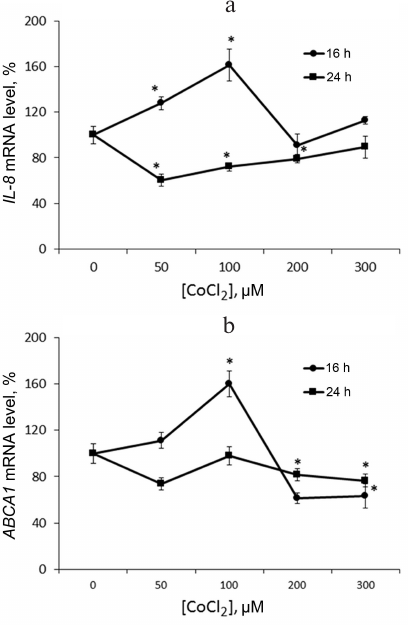
Fig. 3. CoCl2 is a dose- and time-dependent expression regulator of apoA-1 (a) and C3 (c) genes and a factor of intracellular ApoA-1 protein accumulation in THP-1 macrophages (b). a, c) Real-time PCR. Cells were incubated under the indicated CoCl2 concentrations for 16 and 24 h. Error bars correspond to the mean standard error. The data are derived from three independent experiments. The Y-axis shows the mRNA level in percent, with 100% being the level in intact cells. Statistically significant differences between mRNA levels in the samples are marked with asterisks (Student’s t test, * p < 0.05). b) Western blotting. Cells were incubated under the indicated CoCl2 concentrations for 16 h. The plot represents data from three immunoblots quantified with densitometry. The Y-axis shows relative ApoA-1 levels, with 100% corresponding to intact cells. Values are normalized to β-actin levels.
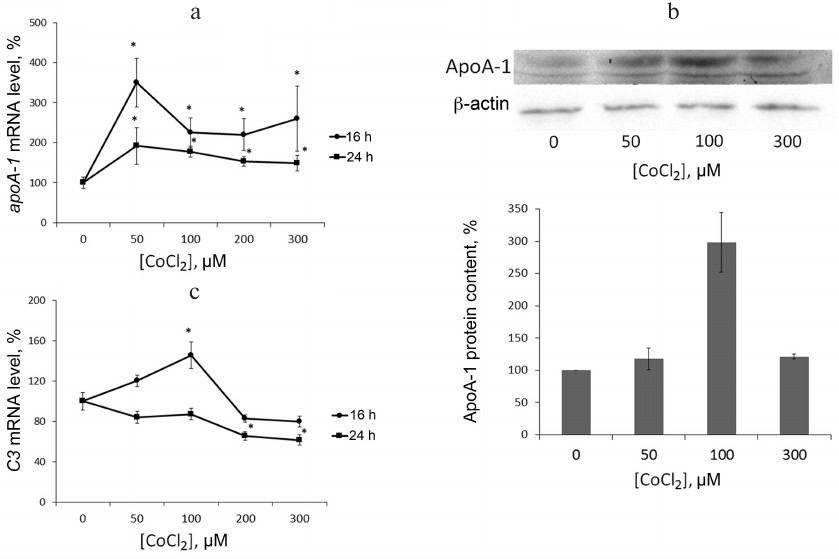
Fig. 4. CoCl2 induces accumulation of membrane surface ApoA-1 protein in THP-1 macrophages. Flow cytometry. Plots reflect changes in surface ApoA-1 quantities under the indicated CoCl2 treatments. ApoA1+ marks ApoA-1-rich macrophages. The plot shows medians of surface ApoA-1 content distributions in cells at different CoCl2 concentrations. Statistically significant deviations from the control (zero concentration of CoCl2) are marked with asterisks (Student’s t test, * p < 0.01). Cell percentage corresponds to that of ApoA-1-rich macrophages in different CoCl2 treatments (color-coded).
Role of signaling cascades MEK1/2-Erk1/2, MKK4/7-JNK1/2/3, and MKK3/6-p38 and transcription factors NF-κB and LXRs in CoCl2-dependent activation of ABCA1, C3, and apoA-1 genes in THP-1 macrophages. Transporter ABCA1, a key regulator of cholesterol efflux from macrophages, is known to be positively regulated by HIF-1α at the mRNA level [24]. However, the regulation mechanism remains unstudied. Transcription factor NF-κB plays a pivotal role in the adaptation of macrophage to hypoxia [25]. In a hypoxic environment, macrophages induce signaling that involves stress-activated MAP kinases Erk1/2 [26], p38 [27], and JNK [28]. Low oxygen-mediated activation of these kinases was also reported for other cell types [29-32].
To define the role of MAP kinases and transcription factor NF-κB in CoCl2-dependent expression of ABCA1, C3, and apoA-1 in THP-1 macrophages, inhibitors of p38 (SB203580), JNK1/2/3 (SP600125), and MEK1/2 kinases (U0126) and of transcription factor NF-κB (QNZ) were added to the culture medium 1 h prior to administration of CoCl2. The LXR agonist (TO901317) that strongly upregulates ABCA1 [24] and apoA-1 mRNA [9] in macrophages was used as a positive control.
Inhibitors of MEK1/2 and JNK1/2/3, but not of p38, arrest the CoCl2-dependent expression activation of ABCA1 (Fig. 5a) and C3 (Fig. 5b). Apart from abolishing the effect of CoCl2, inhibitor of NF-κB downregulates both genes in CoCl2-intact cells. The LXR agonist, as expected, strongly enhances ABCA1 expression by itself and in combination with CoCl2. The opposite effect of the agonist is observed for C3 gene; its expression lowers compared to the control in either presence or absence of CoCl2. The Co2+-mediated induction of apoA-1 expression is dependent on all three major MAP kinase cascades (Fig. 5c). Inhibition of JNK kinases dampens the effect of hypoxia on the apoA-1 expression, while inhibition of p38 and MEK1/2 kinases was associated with reversal of the impact of Co2+ (Fig. 5c). The inducing effect of Co2+ was also reversed by the inhibitor of NF-κB but not by the LXR agonist. Although the 5′-regulatory region of apoA-1 does not contain NF-κB-binding sites, NF-κB-dependent regulation of apoA-1 in macrophages was reported earlier [9]. NF-κB probably regulates apoA-1 via interaction with ligand-activated nuclear receptor PPARα (a negative regulator of apoA-1 in macrophages) leading to inactivation of both transcription factors.
Fig. 5. Role of MAP kinase cascades MEK1/2-Erk1/2, MKK4/7-JNK1/2/3, and MKK3/6-p38 and transcription factors NF-κB and LXRs in CoCl2-dependent activation of ABCA1 (a), C3 (b), and apoA-1 genes (c) in THP-1 macrophages. Real-time PCR. Cells were incubated with one of the following compounds – SB203580 (12.5 μM), SP600125 (10 μM), U0126 (10 μM), QNZ (10 nM), TO901317 (2.5 μM) – for 1 h prior to administration of CoCl2 (100 μM). The data are derived from three independent assays. The Y-axis shows mRNA levels in percent, with 100% corresponding to DMSO-treated cells. Error bars correspond to the mean standard error. Statistically significant differences between mRNA levels in compared groups are marked with asterisks (** p < 0.05, Dunnett’s test). Statistically significant differences from mRNA level in DMSO-treated cells are marked with # symbol (# p < 0.05, Student’s t-test).
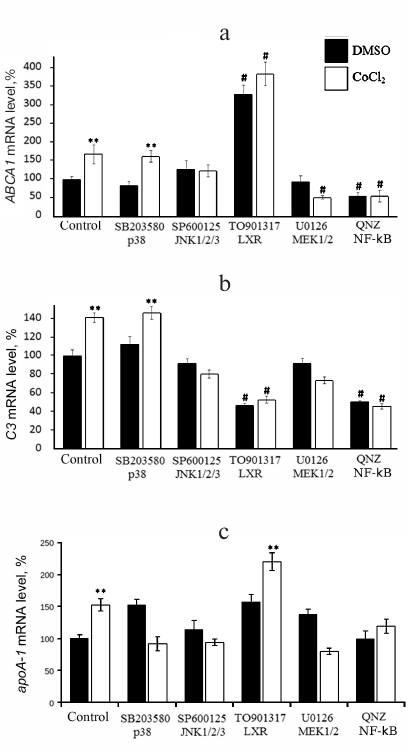
In summary, induction of the cell response to hypoxia activates the apoA-1, ABCA1, and C3 genes and also triggers accumulation of intracellular and surface-bound ApoA-1 protein in THP-1 macrophages. CoCl2-dependent regulation of ABCA1 and C3 gene expression engages signaling cascades NF-κB, MKK4/7-JNK1/2/3, and MEK1/2-Erk1/2, but not MKK3/6-p38. The CoCl2-dependent induction of apoA-1 expression is mediated by all three studied MAP kinase cascades and transcription factor NF-κB. Activation of nuclear receptor LXRα upregulates ABCA1 and apoA-1 in either the presence or absence of CoCl2 but has the opposite impact on the expression of C3.
DISCUSSION
Local hypoxia of atherosclerotic plaques has been described in the literature [33]. Hypoxic microenvironment is among the inducers of monocyte differentiation into macrophages, particularly via activation of proinflammatory genes [34]. Hypoxia-induced and inflammatory signalings are largely similar, which is particularly obvious from the hierarchical interaction of transcription factors NF-κB and HIF-1α, the major modulators of cell adaptation to hypoxia. NF-κB not only facilitates the cytokine-dependent induction of gene HIF1A under hypoxia, but also mediates its constitutive expression [35]. In turn, HIF-1α upregulates the p65 subunit of NF-κB and IKKα [3]. Interestingly, NF-κB activation under hypoxia may be partly mediated by a mechanism of HIF-1α stabilization. IKKβ kinase that induces dissociation of the NF-κB/IκBα complex contains conserved motif LxxLAP involved in proline hydroxylation. The same motif of HIF-1α is hydroxylated by PHD-containing enzymes in normoxia. Inhibiting of PHD1 and PHD2 leads to accumulation of IKKβ in the cell and subsequent activation of NF-κB [37].
In this work, we have shown that inhibition of NF-κB not only reverses the CoCl2-dependent upregulation of the ABCA1 and C3 genes, but also downregulates them without the induction of response to hypoxia. In the murine macrophage model, it was shown that expression of ABCA1 increases after treatment with TNFα in the NF-κB-dependent manner [38]. It was also shown that NF-κB knockdown in epithelium of small intestine precludes higher transcription of C3 gene in response to IL-1β [39]. Obviously, TNFα and IL-1β signalings can be considered hypoxia-specific, as in both cases NF-κB is the main modulator.
Another important element of hypoxia-induced signal cascades is MAP kinases. We demonstrated that kinases MEK1/2 and JNK1/2/3, but not p38, are involved in the regulation of the ABCA1 and C3 genes in THP-1 macrophages in response to hypoxia. Ample evidence suggests interactions between the MEK1/2-Erk1/2, HIF-1, and NF-κB pathways. HIF-1 activation is known to recruit the p300/CBP complex. MAPK inhibition was shown to disturb interaction with HIF-1α/p300. Overexpression of MEK1 results in transactivation of both proteins [40]. Apart from interaction with co-activators, the transcription activity of HIF-1α depends on certain posttranslational modifications. Phosphorylation of Ser641/643 residues in HIF-1α by Erk1/2 kinase arrests its exportin CRM1-geared efflux from the nucleus. This allows the buildup of the nuclear fraction of active HIF-1α [41]. Also, Erk1/2, but not p38, is involved in transactivation of NF-κB subunit p65. This interaction upregulates the MIP-2 gene in murine macrophages under hypoxia [42]. Activation of JNK1/2/3 under low O2 has been reported for macrophages [28] and other cell types [30]. Induction of apoA-1 in hypoxic macrophages depends on the activation of proinflammatory MAP kinase cascade p38, apart from the Erk1/2 and JNK1/2/3 cascades. Hypoxia is a known enhancement factor for phosphorylated p38 (in active form) in hippocampus cortex neurons [43]. Hypoxia enhances activation of p38 in macrophages in the presence of palmitic acid, which causes endoplasmic reticulum stress [44]. The arrest of p38 and Erk1/2 cascades leads to an increase of normal apoA-1 expression and reversal of the stimulating effect of Co2+. Previously [45, 46], we demonstrated enhancement of the apoA-1 gene under arrest of the Erk1/2 cascade in human hepatoma line HepG2. This effect was found to be associated with phosphorylation of transcription factor complex FOXO1/LXRβ interacting with the B and C sites of the hepatic enhancer of apoA-1 gene [45]. Moreover, damage of B or C sites of the hepatic enhancer of apoA-1 reverses the effect of hydrogen peroxide (oxidative stress) on activity of the 5′-regulatory region of the apoA-1 gene in HepG2 cells [46]. Considering (i) high similarity between proinflammatory cytokine-activated signaling pathways in hypoxia and in oxidative stress [25, 31-37, 44] and (ii) the interaction of transcription factors FOXO1 and LXRβ with B and C sites of the hepatic enhancer of the apoA-1 gene not only in HepG2 cells but in human macrophages as well [9], we suggest that the reversal of the effect of Co2+ on apoA-1 expression, when MAP kinase cascades p38 and Erk1/2 are blocked, is mediated by changes in phosphorylation level of transcription factors LXRβ and FOXO1. Further research is necessary to elucidate this question.
In our design, the synthetic LXR agonist TO901317 that strongly upregulates the ABCA1 gene was used as the positive control. As expected, it increased the ABCA1 mRNA level in THP-1 macrophages independently and in combination with CoCl2, thus suggesting independent action of LXR and NF-κB/HIF-1 reported earlier [38]. Of interest is the reverse effect of TO901317 on expression of the C3 gene. We previously reported the TO901317-dependent upregulation of the C3 gene in THP-1 macrophages after 5-day differentiation in culture [18]. In the present study, THP-1 macrophages were cultivated for 3 days. This effect can be compared with the TNFα-induced mechanism of apoA-1 expression regulation that we described earlier. The TNFα treatment results in more than a 5-fold increase of apoA-1 expression in THP-1 monocytes, while it grows only 1.5-fold in THP-1 macrophages. Time-dependent apoA-1 expression in THP-1 monocytes was also shown. A 5-fold increase in expression is observed after 24-h incubation with TNFα, while after 48-h incubation the apoA-1 expression drops to the control level [9].
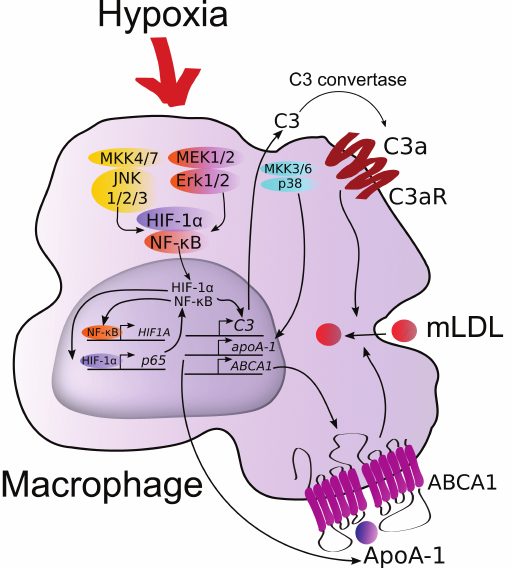
We can now consider the mechanism of positive regulation of expression of the apoA-1, ABCA1, and C3 genes in macrophages in response to hypoxia (Fig. 6). Elevated levels of C3 mRNA and plausibly its protein product result in the production of anaphylatoxin C3a. Binding to its receptor on the macrophage surface membrane, C3a induces expression of C3 and activates uptake of modified LDL (mLDL) by the cells [18]. Although endogenous ApoA-1 stabilizes ABCA1 and has antiinflammatory properties, its level does not correlate with the intensity of reverse cholesterol transport [8]. On the other hand, transmission of the expression vector of the human apoA-1 gene into macrophages of mice with apoA-1 knockout results in noticeable reduction of plaques [47]. Also, knockouts of ABCA1 and ABCG1 were shown to disturb chemotaxis in macrophages [48]. Considering the impaired migration activity of hypoxic macrophages, this effect is likely negated by endogenous ApoA-1 via stabilization of ABCA1. Therefore, if induction of C3 expression in hypoxia can be considered atherogenic, the role of highly expressed apoA-1 and ABCA1 remains controversial and demands further study.
Fig. 6. Hypothetical regulation of the ABCA1, C3, and apoA-1 genes in human macrophages under hypoxia. (For details see “Discussion” section.)
Funding
This work was supported by the Russian Science Foundation, grant no. 17-15-01326; expression regulation of ABCA1 and C3 genes under hypoxic conditions (Figs. 1, 2, 3b, 5a, 5b) and by the Russian Foundation for Basic Research, grant no. 17-04-01947; expression of apoA-1 gene (Figs. 3a, 3c, 4, 5c).
Conflict of Interest
The authors declare no conflict of interest.
Ethical Approval
This article does not contain any description of studies involving humans and animals as objects.
REFERENCES
1.Heo, K.-S., Fujiwara, K., and Abe, J. (2014) Shear
stress and atherosclerosis, Mol. Cells, 37, 435-440, doi:
10.14348/molcells.2014.0078.
2.Bonomini, F., Tengattini, S., Fabiano, A., Bianchi,
R., and Rezzani, R. (2008) Atherosclerosis and oxidative stress,
Histol. Histopathol., 23, 381-390, doi:
10.14670/HH-23.381.
3.Fong, G.-H. (2015) Potential contributions of
intimal and plaque hypoxia to atherosclerosis, Curr. Atheroscler.
Rep., 17, 32-41, doi: 10.1007/s11883-015-0510-0.
4.Parathath, S., Mick, S. L., Feig, J. E., Joaquin,
V., Grauer, L., Habiel, D. M., Gassmann, M., Gardner, L. B., and
Fisher, E. A. (2011) Hypoxia is present in murine atherosclerotic
plaques and has multiple adverse effects on macrophage lipid
metabolism, Circ. Res., 109, 1141-1152, doi:
10.1161/CIRCRESAHA.111.246363.
5.Epstein, A. C., Gleadle, J. M., McNeill, L. A.,
Hewitson, K. S., O’Rourke, J., Mole, D. R., Mukherji, M., Metzen,
E., Wilson, M. I., Dhanda, A., Tian, Y. M., Masson, N., Hamilton, D.
L., Jaakkola, P., Barstead, R., Hodgkin, J., Maxwell, P. H., Pugh, C.
W., Schofield, C. J., and Ratcliffe, P. J. (2001) C. elegans
EGL-9 and mammalian homologs define a family of dioxygenases that
regulate HIF by prolyl hydroxylation, Cell, 107, 43-54,
doi: 10.1016/S0092-8674(01)00507-4.
6.Hirani, N., Antonicelli, F., Strieter, R. M.,
Wiesener, M. S., Ratcliffe, P. J., Haslett, C., and Donnelly, S. C.
(2001) The regulation of interleukin-8 by hypoxia in human macrophages
– a potential role in the pathogenesis of the acute respiratory
distress syndrome (ARDS), Mol. Med. Camb. Mass, 7,
685-697.
7.Murdoch, C., Muthana, M., and Lewis, C. E. (2005)
Hypoxia regulates macrophage functions in inflammation, J.
Immunol., 175, 6257-6263, doi:
10.4049/jimmunol.175.10.6257.
8.Mogilenko, D. A., Orlov, S. V., Trulioff, A. S.,
Ivanov, A. V., Nagumanov, V. K., Kudriavtsev, I. V., Shavva, V. S.,
Tanyanskiy, D. A., and Perevozchikov, A. P. (2012) Endogenous
apolipoprotein A-I stabilizes ATP-binding cassette transporter A1 and
modulates Toll-like receptor 4 signaling in human macrophages, FASEB
J., 26, 2019-2030, doi: 10.1096/fj.11-193946.
9.Shavva, V. S., Mogilenko, D. A., Nekrasova, E. V.,
Trulioff, A. S., Kudriavtsev, I. V., Larionova, E. E., Babina, A. V.,
Dizhe, E. B., Missyul, B. V., and Orlov, S. V. (2018) Tumor necrosis
factor alpha stimulates endogenous apolipoprotein A-I expression and
secretion by human monocytes and macrophages: role of MAP-kinases,
NF-kB, and nuclear receptors PPARα and LXRs, Mol. Cell.
Biochem., 448, 211-223, doi: 10.1007/s11010-018-3327-7.
10.Speidl, W. S., Kastl, S. P., Huber, K., and
Wojta, J. (2011) Complement in atherosclerosis: friend or foe? J.
Thromb. Haemost., 9, 428-440, doi:
10.1111/j.1538-7836.2010.04172.x.
11.Hertle, E., van Greevenbroek, M. M., Arts, I. C.,
van der Kallen, C. J., Geijselaers, S. L., Feskens, E. J., Jansen, E.
H., Schalkwijk, C. G., and Stehouwer, C. D. (2014) Distinct
associations of complement C3a and its precursor C3 with
atherosclerosis and cardiovascular disease. The CODAM study, Thromb.
Haemost., 111, 1102-1111, doi: 10.1160/TH13-10-0831.
12.Binder, C. J., Chang, M.-K., Shaw, P. X., Miller,
Y. I., Hartvigsen, K., Dewan, A., and Witztum, J. L. (2002) Innate and
acquired immunity in atherogenesis, Nat. Med., 8,
1218-1226, doi: 10.1038/nm1102-1218.
13.Samstad, E. O., Niyonzima, N., Nymo, S., Aune, M.
H., Ryan, L., Bakke, S. S., Lappegard, K. T., Brekke, O.-L., Lambris,
J. D., Damas, J. K., Latz, E., Mollnes, T. E., and Espevik, T. (2014)
Cholesterol crystals induce complement-dependent inflammasome
activation and cytokine release, J. Immunol., 192,
2837-2845, doi: 10.4049/jimmunol.1302484.
14.Buono, C., Come, C. E., Witztum, J. L., Maguire,
G. F., Connelly, P. W., Carroll, M., and Lichtman, A. H. (2002)
Influence of C3 deficiency on atherosclerosis, Circulation,
105, 3025-3031.
15.Persson, L., Boren, J., Robertson, A.-K. L.,
Wallenius, V., Hansson, G. K., and Pekna, M. (2004) Lack of complement
factor C3, but not factor B, increases hyperlipidemia and
atherosclerosis in apolipoprotein E–/–
low-density lipoprotein receptor–/– mice,
Arterioscler. Thromb. Vasc. Biol., 24, 1062-1067, doi:
10.1161/01.ATV. 0000127302.24266.40.
16.Amarilyo, G., Verbovetski, I., Atallah, M., Grau,
A., Wiser, G., Gil, O., Ben-Neriah, Y., and Mevorach, D. (2010)
iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory
response and transcriptional NF-κB-dependent blockade, Eur. J.
Immunol., 40, 699-709, doi: 10.1002/eji.200838951.
17.McVicar, J. P., Kunitake, S. T., Hamilton, R. L.,
and Kane, J. P. (1984) Characteristics of human lipoproteins isolated
by selected-affinity immunosorption of apolipoprotein A-I, Proc.
Natl. Acad. Sci. USA, 81, 1356-1360.
18.Mogilenko, D. A., Kudriavtsev, I. V., Trulioff,
A. S., Shavva, V. S., Dizhe, E. B., Missyul, B. V., Zhakhov, A. V.,
Ischenko, A. M., Perevozchikov, A. P., and Orlov, S. V. (2012) Modified
low density lipoprotein stimulates complement C3 expression and
secretion via liver X receptor and Toll-like receptor 4 activation in
human macrophages, J. Biol. Chem., 287, 5954-5968, doi:
10.1074/jbc.M111.289322.
19.Orlov, S. V., Mogilenko, D. A., Shavva, V. S.,
Dizhe, E. B., Ignatovich, I. A., and Perevozchikov, A. P. (2010) Effect
of TNFα on activities of different promoters of human
apolipoprotein A-I gene, Biochem. Biophys. Res. Commun.,
398, 224-230, doi: 10.1016/j.bbrc.2010.06.064.
20.Mogilenko, D. A., Shavva, V. S., Dizhe, E. B.,
Orlov, S. V., and Perevozchikov, A. P. (2010) PPARγ activates
ABCA1 gene transcription but reduces the level of ABCA1 protein in
HepG2 cells, Biochem. Biophys. Res. Commun., 402,
477-482, doi: 10.1016/j.bbrc.2010.10.053.
21.Mogilenko, D. A., Kudriavtsev, I. V., Shavva, V.
S., Dizhe, E. B., Vilenskaya, E. G., Efremov, A. M., Perevozchikov, A.
P., and Orlov, S. V. (2013) Peroxisome proliferator-activated receptor
alpha positively regulates complement C3 expression but inhibits
TNFα-mediated activation of C3 gene in mammalian hepatic derived
cells, J. Biol. Chem., 288, 1726-1738, doi:
10.1074/jbc.M112.437525.
22.Vengellur, A., and LaPres, J. J. (2004) The role
of hypoxia inducible factor 1α in cobalt chloride induced cell
death in mouse embryonic fibroblasts, Toxicol. Sci., 82,
638-646, doi: 10.1093/toxsci/kfh278.
23.Rydberg, E. K., Salomonsson, L., Hulten, L. M.,
Noren, K., Bondjers, G., Wiklund, O., Bjornheden, T., and Ohlsson, B.
G. (2003) Hypoxia increases 25-hydroxycholesterol-induced interleukin-8
protein secretion in human macrophages, Atherosclerosis,
170, 245-252, doi: 10.1016/S0021-9150(03)00302-2.
24.Schmitz, G., and Langmann, T. (2005)
Transcriptional regulatory networks in lipid metabolism control ABCA1
expression, Biochim. Biophys. Acta, 1735, 1-19, doi:
10.1016/j.bbalip.2005.04.004.
25.Marsch, E., Sluimer, J. C., and Daemen, M. J. A.
P. (2013) Hypoxia in atherosclerosis and inflammation, Curr. Opin.
Lipidol., 24, 393-400, doi:
10.1097/MOL.0b013e32836484a4.
26.Li, R. C., Haribabu, B., Mathis, S. P., Kim, J.,
and Gozal, D. (2011) Leukotriene B4 receptor-1 mediates intermittent
hypoxia-induced atherogenesis, Am. J. Respir. Crit. Care Med.,
184, 124-131, doi: 10.1164/rccm.201012-2039OC.
27.Ortiz-Masia, D., Diez, I., Calatayud, S.,
Hernandez, C., Cosin-Roger, J., Hinojosa, J., Esplugues, J. V., and
Barrachina, M. D. (2012) Induction of CD36 and thrombospondin-1 in
macrophages by hypoxia-inducible factor 1 and its relevance in the
inflammatory process, PloS One, 7, e48535, doi:
10.1371/journal.pone.0048535.
28.Fuhrmann, D. C., Tausendschon, M., Wittig, I.,
Steger, M., Ding, M. G., Schmid, T., Dehne, N., and Brune, B. (2015)
Inactivation of tristetraprolin in chronic hypoxia provokes the
expression of cathepsin B, Mol. Cell. Biol., 35, 619-630,
doi: 10.1128/MCB.01034-14.
29.Li, Q., Yu, B., and Yang, P. (2015)
Hypoxia-induced HMGB1 in would tissues promotes the osteoblast cell
proliferation via activating ERK/JNK signaling, Int. J. Clin. Exp.
Med., 8, 15087-15097.
30.Chiu, C.-Z., Wang, B.-W., and Shyu, K.-G. (2014)
Angiotensin II and the JNK pathway mediate urotensin II expression in
response to hypoxia in rat cardiomyocytes, J. Endocrinol.,
220, 233-246, doi: 10.1530/JOE-13-0261.
31.Zhang, J., Liu, Q., Fang, Z., Hu, X., Huang, F.,
Tang, L., and Zhou, S. (2016) Hypoxia induces the proliferation of
endothelial progenitor cells via upregulation of Apelin/APLNR/MAPK
signaling, Mol. Med. Rep., 13, 1801-1806, doi:
10.3892/mmr.2015.4691.
32.Wu, Y., Yang, Y., Yang, P., Gu, Y., Zhao, Z.,
Tan, L., Zhao, L., Tang, T., and Li, Y. (2013) The osteogenic
differentiation of PDLSCs is mediated through MEK/ERK and p38 MAPK
signaling under hypoxia, Arch. Oral Biol., 58, 1357-1368,
doi: 10.1016/j.archoralbio.2013.03.011.
33.Mayr, M., Sidibe, A., and Zampetaki, A. (2008)
The paradox of hypoxic and oxidative stress in atherosclerosis, J.
Am. Coll. Cardiol., 51, 1266-1267, doi:
10.1016/j.jacc.2008.01.005.
34.Strehl, C., Fangradt, M., Fearon, U., Gaber, T.,
Buttgereit, F., and Veale, D. J. (2014) Hypoxia: how does the
monocyte-macrophage system respond to changes in oxygen availability?
J. Leukoc. Biol., 95, 233-241, doi:
10.1189/jlb.1212627.
35.Taylor, C. T., and Cummins, E. P. (2009) The role
of NF-κB in hypoxia-induced gene expression, Ann. N. Y. Acad.
Sci., 1177, 178-184, doi:
10.1111/j.1749-6632.2009.05024.x.
36.Walmsley, S. R., Print, C., Farahi, N.,
Peyssonnaux, C., Johnson, R. S., Cramer, T., Sobolewski, A., Condliffe,
A. M., Cowburn, A. S., Johnson, N., and Chilvers, E. R. (2005)
Hypoxia-induced neutrophil survival is mediated by
HIF-1α-dependent NF-κB activity, J. Exp. Med.,
201, 105-115, doi: 10.1084/jem.20040624.
37.Cummins, E. P., Berra, E., Comerford, K. M.,
Ginouves, A., Fitzgerald, K. T., Seeballuck, F., Godson, C., Nielsen,
J. E., Moynagh, P., Pouyssegur, J., and Taylor, C. T. (2006) Prolyl
hydroxylase-1 negatively regulates IκB kinase-β, giving
insight into hypoxia-induced NFκB activity, Proc. Natl. Acad.
Sci. USA, 103, 18154-18159, doi:
10.1073/pnas.0602235103.
38.Gerbod-Giannone, M.-C., Li, Y., Holleboom, A.,
Han, S., Hsu, L.-C., Tabas, I., and Tall, A. R. (2006) TNFα
induces ABCA1 through NF-κB in macrophages and in phagocytes
ingesting apoptotic cells, Proc. Natl. Acad. Sci. USA,
103, 3112-3117, doi: 10.1073/pnas.0510345103.
39.Moon, M. R., Parikh, A. A., Pritts, T. A.,
Fischer, J. E., Cottongim, S., Szabo, C., Salzman, A. L., and
Hasselgren, P. O. (1999) Complement component C3 production in
IL-1β-stimulated human intestinal epithelial cells is blocked by
NF-κB inhibitors and by transfection with ser 32/36 mutant
IκBα, J. Surg. Res., 82, 48-55, doi:
10.1006/jsre.1998.5503.
40.Sang, N., Stiehl, D. P., Bohensky, J.,
Leshchinsky, I., Srinivas, V., and Caro, J. (2003) MAPK signaling
up-regulates the activity of hypoxia-inducible factors by its effects
on p300, J. Biol. Chem., 278, 14013-14019, doi:
10.1074/jbc.M209702200.
41.Mylonis, I., Chachami, G., Samiotaki, M.,
Panayotou, G., Paraskeva, E., Kalousi, A., Georgatsou, E., Bonanou, S.,
and Simos, G. (2006) Identification of MAPK phosphorylation sites and
their role in the localization and activity of hypoxia-inducible
factor-1α, J. Biol. Chem., 281, 33095-33106, doi:
10.1074/jbc.M209702200.
42.Zampetaki, A., Mitsialis, S. A., Pfeilschifter,
J., and Kourembanas, S. (2004) Hypoxia induces macrophage inflammatory
protein-2 (MIP-2) gene expression in murine macrophages via
NF-κB: the prominent role of p42/p44 and PI3 kinase pathways,
FASEB J., 18, 1090-1092, doi: 10.1096/fj.03-0991fje.
43.Haddad, J. J., and Hanbali, L. B. (2014) Hypoxia
upregulates MAPK(p38)/MAPK(ERK) phosphorylation in vitro:
neuroimmunological differential time-dependent expression of MAPKs,
Protein Pept. Lett., 21, 444-451, doi:
10.2174/092986652105140218112521.
44.Snodgrass, R. G., Boss, M., Zezina, E., Weigert,
A., Dehne, N., Fleming, I., Brune, B., and Namgaladze, D. (2016)
Hypoxia potentiates palmitate-induced pro-inflammatory activation of
primary human macrophages, J. Biol. Chem., 291, 413-424,
doi: 10.1074/jbc.M115.686709.
45.Shavva, V. S., Bogomolova, A. M., Nikitin, A. A.,
Dizhe, E. B., Tanyanskiy, D. A., Efremov, A. M., Oleinikova, G. N.,
Perevozchikov, A. P., and Orlov, S. V. (2017) Insulin-mediated
downregulation of apolipoprotein A-I gene in human hepatoma cell line
HepG2: the role of interaction between FOXO1 and LXRβ
transcription factors, J. Cell. Biochem., 118, 382-396,
doi: 10.1002/jcb.25651.
46.Shavva, V. S., Bogomolova, A. M., Nikitin, A. A.,
Dizhe, E. B., Oleinikova, G. N., Lapikov, I. A., Tanyanskiy, D. A.,
Perevozchikov, A. P., and Orlov, S. V. (2017) FOXO1 and LXRα
downregulate the apolipoprotein A-I gene expression during hydrogen
peroxide-induced oxidative stress in HepG2 cells, Cell Stress
Chaperon., 22, 123-134, doi: 10.1007/s12192-016-0749-6.
47.Ishiguro, H., Yoshida, H., Major, A. S., Zhu, T.,
Babaev, V. R., Linton, M. F., and Fazio, S. (2001) Retrovirus-mediated
expression of apolipoprotein A-I in the macrophage protects against
atherosclerosis in vivo, J. Biol. Chem., 276,
36742-36748, doi: 10.1074/jbc.M106027200.
48.Pagler, T. A., Wang, M., Mondal, M., Murphy, A.
J., Westerterp, M., Moore, K. J., Maxfield, F. R., and Tall, A. R.
(2011) Deletion of ABCA1 and ABCG1 impairs macrophage migration because
of increased Rac1 signaling, Circ. Res., 108, 194-200,
doi: 10.1161/CIRCRESAHA.110.228619.