Myeloperoxidase-Induced Oxidation of Albumin and Ceruloplasmin: Role of Tyrosines
I. I. Vlasova1,2,a*, A. V. Sokolov1,3,4, V. A. Kostevich1,3, E. V. Mikhalchik1, and V. B. Vasilyev3,4
1Federal Research and Clinical Center of Physical-Chemical Medicine, 119435 Moscow, Russia2I. M. Sechenov First Moscow State Medical University, Institute for Regenerative Medicine, Laboratory of Navigational Redox Lipidomics, 119991 Moscow, Russia
3Institute of Experimental Medicine, 197376 St. Petersburg, Russia
4St. Petersburg State University, 199034 St. Petersburg, Russia
* To whom correspondence should be addressed.
Received February 22, 2019; Revised March 14, 2019; Accepted March 14, 2019
Neutrophil myeloperoxidase (MPO) plays an important role in protecting the body against infections. MPO products – hypohalous acids and phenoxyl radicals – are strong oxidants that can damage not only foreign intruders but also host tissues, including blood plasma proteins. Here, we compared the MPO-induced oxidation of two plasma proteins with antioxidant properties – human serum albumin (HSA) and ceruloplasmin (CP). Incubation of both proteins with hypochlorite (NaOCl) or catalytically active MPO (MPO + H2O2), which synthesizes hypochlorous acid (HOCl) in the presence of chloride ions, resulted in the quenching of protein tryptophan fluorescence. Oxidation-induced changes in the structures of HSA and CP were different. HSA efficiently neutralized MPO-generated oxidants without protein aggregation, while CP oxidation resulted in the formation of large aggregates stabilized by strong covalent bonds between the aromatic amino acid residues. Tyrosine is present in the plasma as free amino acid and also as a component of the polypeptide chains of the proteins. The number of tyrosine residues in a protein does not determine its propensity for aggregate formation. In the case of CP, protein aggregation was primarily due to the high content of tryptophan residues in its polypeptide chain. MPO-dependent oxidation of free tyrosine results in the formation of tyrosyl radicals, that do not oxidize aromatic amino acid residues in proteins because of the high rate of recombination with dityrosine formation. At the same time, free tyrosine can influence MPO-induced protein oxidation due to its ability to modulate HOCl synthesis in the MPO active site.
KEY WORDS: hypochlorous acid, phenoxyl radicals, protein oxidation, tryptophan fluorescence, protein aggregationDOI: 10.1134/S0006297919060087
Abbreviations: CP, ceruloplasmin; MPO, myeloperoxidase; Tyr, tyrosine.
Myeloperoxidase (MPO) is the main enzyme in neutrophils. It possesses a
unique ability to generate HOCl, a potent oxidant, which is necessary
to protect the body from infections (redox potential, E0, of
the HOCl/Cl– pair is ~1.3 V) [1,
2]. H2O2 reaction with the
native ferri-form of the MPO heme results in the formation of a highly
reactive state of the active site – Compound I [3]. This form of MPO heme is able to oxidize halides
(Cl–, Br–) with the formation of
hypohalous acids (HOCl, HOBr), after which the enzyme active site
returns to its native form, completing the chlorination cycle. At the
same time, similar to any peroxidase, MPO oxidizes a number of
substances (peroxidase substrates) in the peroxidase cycle with the
formation of free radicals. In this case, after oxidation of the first
substrate molecule, Compound I is reduced to Compound II, and oxidation
of the next substrate molecule converts the active site of the enzyme
to its native state.
In the presence of high concentrations of H2O2 (>100 μM), native MPO heme is immediately converted to Compound II [4]. This form of the enzyme does not oxidize halides. Phenolic compounds can affect the synthesis of HOCl in the MPO active site, e.g., accelerate HOCl formation at high H2O2 concentrations due to their high affinity for Compound II and inhibit it at low H2O2 concentrations due to the competition with chloride ions for Compound I [5-7].
At the site of inflammation (usually characterized by an acidic pH shift), MPO mainly catalyzes HOCl formation. From the site of inflammation, the enzyme can enter the plasma, so that the MPO concentration in the plasma under a number of pathological conditions can reach several nanomoles per liter [8, 9]. At neutral pH values (typical for the plasma), the chlorinating activity of the enzyme is decreased, while oxidation of peroxidase substrates is activated [10]. Endogenous peroxidase substrates that are present in the plasma in micromolar concentrations include antioxidants (ascorbate, urate), phenolic compounds (tyrosine and phenolic xenobiotics), nitrite, tryptophan, serotonin, β-ketones, catecholamines, etc. [7].
High oxidative potential of MPO Compound I could cause serious damage to the macromolecules in the plasma. However, a unique structure of the enzyme active site, in which the heme is located at the bottom of the narrow pocket, limits its accessibility for the molecules with a size significantly larger than a dipeptide [11, 12]. Oxidation of macromolecules requires intermediate substrates, whose oxidized forms are strong oxidizing agents [13, 14]. Along with HOCl, macromolecules can be damaged by phenoxyl radicals formed by oxidation of some phenolic compounds in the MPO active site, for example, tyrosyl radicals that have a high redox potential [E0(Tyr•/Tyr) = 0.93 V] [15]. Free tyrosine is present in the plasma at a concentration of 80-200 μM.
Modification of proteins and lipids with hypohalous acids has been studied in detail [16, 17], with special attention to the oxidation of apoB-100, apoA-I, and lipoprotein lipids by MPO products. The significance of MPO-induced lipoprotein modification in atherosclerosis pathogenesis has been demonstrated [18, 19]. Oxidation of some proteins has been studied in model systems, and the rate constants for the HOCl reactions with amino acids were obtained. It was shown that in proteins, HOCl oxidizes cysteines, methionines, and tryptophans but chlorinates lysines, arginines, histidines, tyrosines, and terminal amino groups [16, 20]. Oxidation of amides in a polypeptide chain can cause its fragmentation, whereas oxidation of thiol groups and tyrosines results in the protein aggregation due to the formation of disulfide bonds and dityrosine cross-links between the protein globules. Aggregates of proteins containing complement C3, apoA1, fibrinogen, and albumin were detected in the plasma treated with high hypochlorite concentrations (0.5-5 mM) [21]. The content of protein aggregates with a characteristic dityrosine fluorescence was found to be higher in the plasma of patients with severe forms of urological diseases compared to healthy donors [22].
On the other hand, the role of MPO peroxidase substrates in the modification of macromolecules is rarely discussed in these studies [15, 23]. It was shown earlier that nitrite oxidation by MPO causes formation of nitrogen dioxide, a strong oxidant that can nitrate or nitrosylate amino acid residues in proteins [24, 25].
To study MPO-induced oxidation of plasma proteins, we chose two anionic proteins – ceruloplasmin (CP, pI ~ 4.4) and human serum albumin (HSA, pI ~ 4.7). Both proteins are known to possess the antioxidant properties; therefore, under conditions of oxidative stress, their modifications can be expected first. In addition, these proteins are present in the plasma at high concentrations (~600 μM HSA and 0.5-4 μM CP) and can bind to the positively charged MPO (pI ~ 10.7).
Albumin is the most abundant plasma protein that is often modified in various pathologies [26-28]. The only free cysteine in HSA (Cys34) provides about 50% of all reduced plasma thiols [29]. Nevertheless, only a few studies have been published on HSA binding to MPO and its oxidation by MPO-generated oxidants. It was shown that albumin binds to the heavy MPO chain with the dissociation constant of 20 ± 1.5 μM [30]. Salavej et al. studied albumin modification by MPO-produced hypohalous acids and nitrogen reactive species [24] and observed oxidation of Met147, Met353, Met572, and Trp214.
Similarly to albumin, CP is an efficient scavenger of free radicals [31]. CP binds near the MPO active site, forming a very strong complex (Kd ~ 0.13 μM) with a stoichiometry of 2 : 1 [32-34]. CP binding to MPO is of significant physiological importance, since it leads to the inhibition of both chlorinating and peroxidase activities of MPO, but at the same time, does not affect the ferroxidase and oxidase activity of CP toward most substrates [33, 35-37].
Here, we compared modification of CP and HSA by HOCl and phenoxyl radicals under conditions simulating local oxidative stress.
MATERIALS AND METHODS
All reagents used for the preparation of buffers, tyrosine, albumin, catalase, methionine, hydrogen peroxide, and hypochlorite were from Sigma-Aldrich (USA). Reagents for electrophoresis were from VWR Life Science Amresco (USA).
MPO was purified from leukocytes of healthy donors by heparin-Sepharose affinity chromatography, hydrophobic chromatography on phenyl-Sepharose, and gel filtration. The purity of MPO (A430/A280) was 0.85; the activity with guaiacol was ~1100 U/mg in 50 mM Na-phosphate buffer (pH 7.4) at 22°C [38]. Homogeneous non-proteolyzed CP was obtained from the plasma of healthy donors by UNOsphere Q ion-exchange chromatography and neomycin-agarose affinity chromatography [39]. Purified CP was characterized by A610/A280 = 0.049; about 50% of the preparation was unfragmented protein. When analyzed by SDS-PAGE in the presence of 2-mercaptoethanol, the protein was detected as 132- and 116-kDa bands of approximately equal intensity.
The proteins were oxidized in 50 mM Na-phosphate buffer at pH 7.4 or 6.8 in the presence of MPO and H2O2 or NaOCl. Phenol, tyrosine, 140 mM NaCl, or tyrosine and NaCl were added to the solution as MPO substrates. After addition of H2O2 (to samples with MPO) or NaOCl, fluorescence spectra of the solutions were recorded, or the samples were incubated for some time and then analyzed by electrophoresis. The concentrations of reagents are indicated in the figure legends.
Fluorescence of the samples was excited at 290 nm, and the spectra were recorded at 300-500 nm with a Hitachi F-4000 fluorescence spectrophotometer (Japan). Protein fluorescence intensity was measured at 340 nm; dityrosine fluorescence intensity was determined at 410 nm.
SDS-PAGE was run in the presence of 2-mercaptoethanol in 7.5% separating gel and 5% stacking gel. The gels were stained with Coomassie R-250 Brilliant Blue; the intensity of the protein band staining was evaluated with Paint.NET. The area of measurement for each lane was 51 × 51 px in the middle of the upper region of the separating gel.
All experimental data are presented as means of three replicates ± SD. Changes in variables were analyzed with the Student’s t-test. The differences were considered significant at p < 0.05. The results of protein fluorescence measurements and SDS-PAGE analysis presented in the figures are representative results of a typical experiment among at least three independent experiments.
RESULTS
Characterization of MPO-induced protein oxidation by the fluorescence method. A decrease in the fluorescence of proteins is known to be caused, first of all, by the oxidation of tryptophan residues in the polypeptide chain. Using fluorescence measurements, we compared the oxidation of tryptophans in HSA and CP after exposure of these proteins to HOCl and phenoxyl radicals formed in the MPO-catalyzed reaction.
HSA fluorescence is due mostly to its only tryptophan residue (Trp214). Figure 1a shows a characteristic fluorescence spectrum of HSA with the maximum near 340 nm. HSA incubation with MPO/Cl–/H2O2 resulted in the quenching of protein fluorescence, so that after 6 min, the spectrum amplitude decreased about 2 times.
Fig. 1. Decrease in the protein fluorescence upon MPO-induced protein oxidation in the presence of chloride ions. a) 20 nM MPO and 100 μM H2O2 were added to 4 μM HSA in 50 mM Na-phosphate buffer (pH 7.4) in the presence of 140 mM NaCl. The spectra were recorded at different time intervals after H2O2 addition. b) Time course of MPO-induced oxidation of HSA and CP. The experimental conditions are the same as in (a), except HSA concentration was 5 μM and CP concentration was 280 nM.
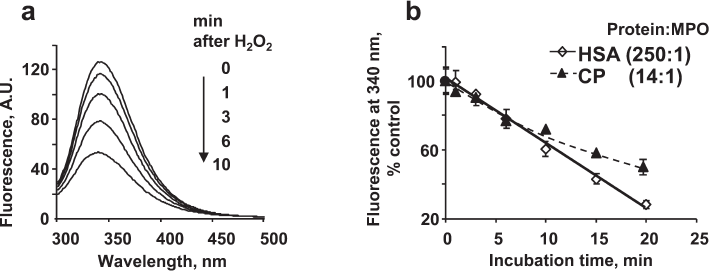
There are 18 tryptophans in the CP polypeptide chain (table); therefore, the concentration of CP in the experiments was 16-18 times lower than that of HSA. Figure 1b shows the kinetics of CP and HSA fluorescence quenching by MPO-generated HOCl. The kinetics of tryptophan oxidation in CP was nonlinear. Tryptophans located on the protein surface are oxidized first, while the amino acid residues inside the protein globule are less accessible to the oxidizing agents. Since the concentrations of HSA and CP in the experiments differed, whereas the concentrations of MPO and H2O2 were the same, the amount of oxidant per HSA molecule was lower. However, the kinetics of fluorescence decrease for the two proteins were identical within 10 min of incubation, which indicates a high propensity for oxidation of the single tryptophan residue in HSA.
Amino acid composition of HSA and CP and second-order rate constants of
HOCl reaction with some amino acids [50]
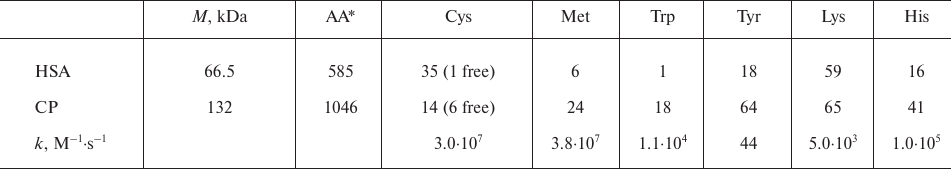
* AA, total number of amino acid residues.
Phenoxyl radicals formed in the peroxidase cycle of MPO are strong oxidants that can modify amino acid residues in proteins. In addition, phenoxyl radicals are able to recombine with the formation of diphenols, which have a characteristic fluorescence with the maximum at 410 nm. Usually, fluorescence of diphenols is excited at 325 nm [40]; however, we recorded the fluorescence spectra of diphenols at the excitation wavelength of 290 nm. Although at this excitation wavelength, the intensity of diphenol fluorescence is lower by 10-15%, this approach makes it possible to simultaneously characterize changes in the fluorescence of proteins and monitor formation of diphenols in a solution (Fig. 2a). Moreover, the fluorescence spectrum of diphenols does not contribute to the amplitude of the protein fluorescence spectrum.
Fig. 2. Oxidation of proteins by the MPO-generated phenoxyl radicals as assayed by fluorescence: a) Spectra of protein (λem 340 nm) and dityrosine fluorescence (λem 410 nm) at the excitation wavelength of 290 nm; b) 100 μM phenol was added to the solution of 4 µM HSA in 50 mM Na-phosphate buffer (pH 7.4) and then 25 nM MPO was added. The spectra were acquired after addition of 100 μM H2O2; c) 100 μM tyrosine was added to the solution of 270 nM CP in 50 mM Na-phosphate buffer, pH 7.4; then, 25 nM MPO was added. The spectra were acquired after addition of 100 μM H2O2.
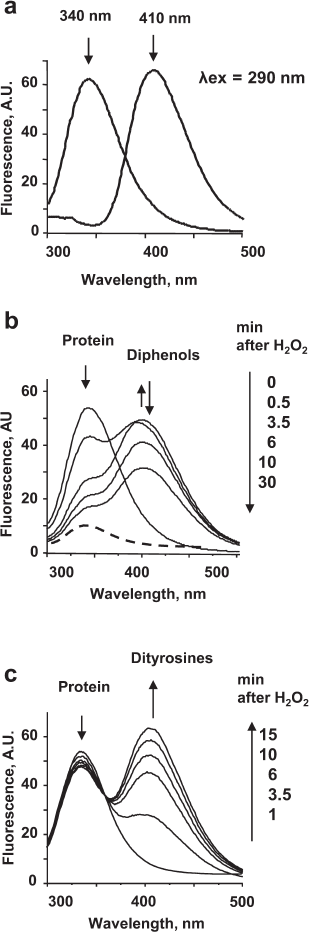
Changes in the spectra observed during incubation of CP or HSA with the catalytically active MPO in the presence of phenolic compounds were similar. Phenoxyl radicals formed in the MPO-catalyzed oxidation of phenol caused the quenching of the protein tryptophan fluorescence. The appearance of fluorescence in the 410-nm region indicated formation of diphenols in the solution (Fig. 2b). Rapid formation of diphenols (a fluorescence increase at 410 nm for 0.5 min) was followed by their disappearance accompanied by a decrease in the amplitude of protein fluorescence. Therefore, free phenol radicals can oxidize proteins, as well as form diphenols that can also be oxidized in the MPO active site. On the contrary, incubation of CP or HSA with MPO/Tyr/H2O2 was accompanied only by insignificant changes in the protein fluorescence, which may be caused by changes in the protein conformation as a result of oxidation of a number of amino acids (primarily, methionines and cysteines) by tyrosyl radicals (table). At the same time, rapid formation of dityrosines was observed in the solution, which was documented by the appearance of fluorescence at 410 nm (Fig. 2c).
These experiments showed that tyrosyl radicals do not oxidize aromatic amino acid residues in proteins. In particular, this can be explained by the high rate of dityrosine formation (k ~ 108 M–1·s–1) [41].
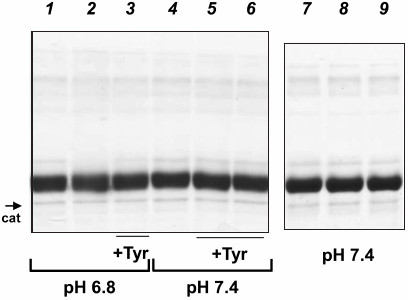
Electrophoresis of proteins incubated with catalytically active MPO. Protein oxidation can result in the formation of protein aggregates or fragmentation of polypeptide chains. Changes in the protein molecular weight can be observed by electrophoresis. The results of SDS-PAGE of HSA (M ~ 67 kDa, with ligands) and CP (M ~ 132 kDa, with carbohydrate chains and copper ions) in the presence of 2-mercaptoethanol are presented in Figs. 3 and 4, respectively. We compared the changes in proteins caused by the addition of NaOCl with the changes observed as a result of protein incubation with MPO/Cl–/H2O2. SDS-PAGE showed that the primary structure of HSA remained intact after its incubation with the catalytically active MPO at pH 6.8 or 7.0 or with exogenous hypochlorite – no formation of aggregates and low molecular weight protein fragments were observed (Fig. 3, lanes 1, 2, 4, and 7-9).
Fig. 3. SDS-PAGE of HSA oxidized by MPO-derived oxidants or NaOCl. MPO (25 nM) was added to the solutions of 4 μM HSA in 50 mM Na-phosphate, 140 mM NaCl (pH 7.4 or 6.8). Tyrosine was added to the indicated samples. H2O2 or NaOCl were added twice with a 12-min interval; 12 min after the second addition, the reaction was terminated by adding catalase (25 μg/ml) and then 2.5 mM methionine. Samples at pH 6.8: 1) MPO; 2) MPO + (100 μM) × 2 H2O2; 3) MPO + 100 μM tyrosine + (100 μM) × 2 H2O2. Samples at pH 7.4: 4) MPO + (100 μM) × 2 H2O2; 5) MPO + 100 μM tyrosine + (100 μM) × 2 H2O2; 6) MPO + 200 μM tyrosine + (100 μM) × 2 H2O2; 7) control (without MPO); 8) 100 μM NaOCl; 9) (100 μM) × 2 NaOCl. The band corresponding to catalase subunit is indicated (cat).
No MPO-induced oxidation of CP was observed at pH 7.4, which may be due to the formation of a strong complex between the two proteins and inhibition of MPO activity (compare lanes 5 and 6; Fig. 4a) [33]. CP can be oxidized by the catalytically active MPO at pH 6.8 or by exogenous hypochlorite (compare lanes 1 and 2, 9, and 10; Fig. 4a) [42]. SDS-PAGE evidences the formation of CP aggregates, including large agglomerates that did not enter the stacking gel (vertical arrows in Fig. 4a, lanes 2 and 10). Since electrophoresis was run in the presence of 2-mercaptoethanol, we concluded that the observed aggregates were formed due to the formation of strong cross-links between the protein molecules. CP molecules were similarly modified when incubated with MPO/Cl–/H2O2 or treated with hypochlorite, but in the latter case, CP oxidation was more significant when equal concentrations of the oxidants were added (Fig. 4b). In the case of exogenous hypochlorite addition, molecular fragments of CP with a mass of about 100 kDa were more pronounced, which indicates protein fragmentation and, therefore, oxidation of amides in the polypeptide chain.
Fig. 4. SDS-PAGE of CP oxidized by MPO-derived oxidants or NaOCl. MPO (25 nM) was added to the solutions of 2 μM CP in 50 mM Na-phosphate, 140 mM NaCl (pH 7.4 or 6.8). Tyrosine was added to the indicated samples. H2O2 or NaOCl were added twice with a 12-min interval; 12 min after the second addition, the reaction was terminated by adding catalase (25 μg/ml) and then 2.5 mM methionine. a) Gel electrophoresis of native and oxidized CP. Samples at pH 6.8: 1) MPO; 2) MPO + (100 μM) × 2 H2O2; 3) MPO + 100 μM tyrosine + (100 μM) × 2 H2O2; 4) MPO + 200 μM tyrosine + (100 μM) × 2 H2O2. Samples at pH 7.4: 5) MPO; 6) MPO + (100 μM) × 2 H2O2; 7) + MPO + 100 μM tyrosine + (100 μM) × 2 H2O2; 8) MPO + 200 μM tyrosine + (100 μM) × 2 H2O2; 9) 100 μM NaOCl; 10) (100 μM) × 2 NaOCl. Arrows indicate protein agglomerates on the top of the stacking gel. Protein band at ~60 kDa corresponds to catalase subunits. Blue square (lane 8) indicates the area of color density measurements. b) Color density in the upper area of the running gel in each lane.
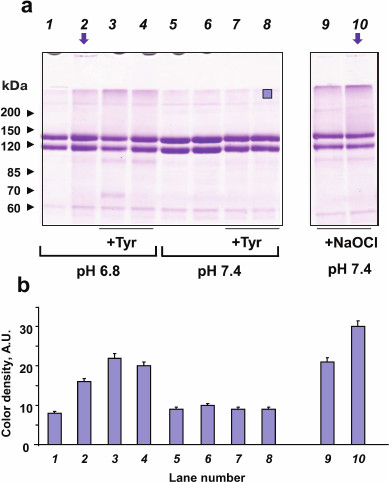
No fragmentation into smaller polypeptides was registered for CP and HSA when the proteins were incubated with MPO/Tyr/H2O2 in the absence of chloride ions (data not shown).
Effect of tyrosine on the MPO-induced oxidation of HSA. We examined the effect of tyrosine on the fluorescence of HSA incubated with MPO/Cl–/H2O2 using different experimental protocols: (i) single addition of high H2O2 concentrations (100 μM) as a model of oxidative burst in the case of neutrophil activation at the site of inflammation (Fig. 5, a and c); (ii) multiple additions of low H2O2 concentrations (10 μM; 10 additions every 3 min) as a model of oxidative stress in the blood plasma (Fig. 5, b and d).
Fig. 5. Effect of tyrosine on the MPO-induced oxidative modification of HSA. Reaction mixture: 5 μM HSA and 20 nM MPO were added to 50 mM Na-phosphate (pH 7.4) in the presence of 140 mM NaCl (diamonds), or 140 mM NaCl + 100 μM tyrosine (circles). HSA fluorescence spectra were recorded before H2O2 addition and then each 3-5 min after a single addition of 100 µM H2O2 (a, c) or after each addition of 10 µM H2O2 made with 3-min intervals (b, d). a, b) Fluorescence at 340 nm; c, d) fluorescence spectra of HSA solution.
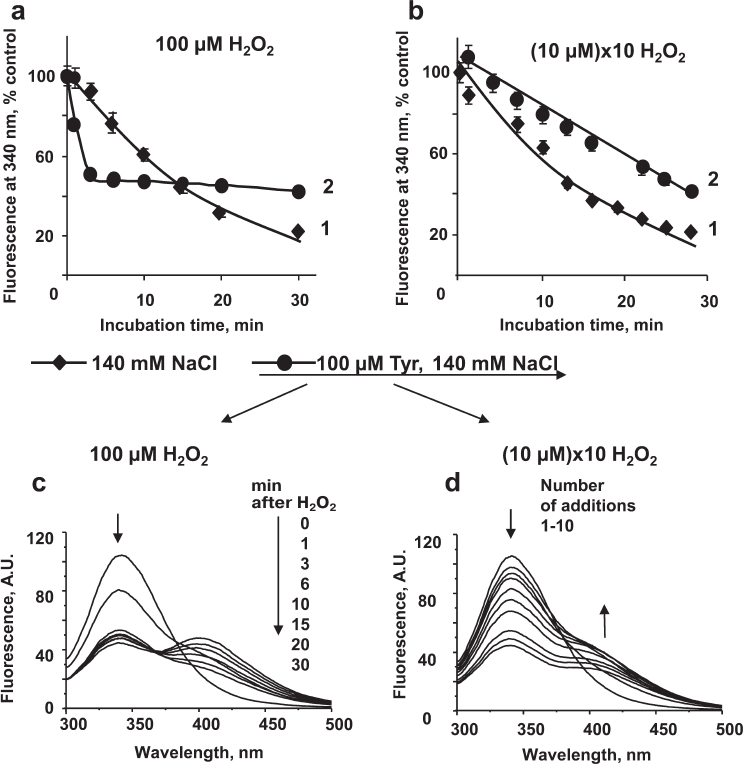
HSA fluorescence slowly decreased after a single addition of 100 μM H2O2. This might be due to a slow production of HOCl, because at high H2O2 concentrations, the active site of MPO is converted into Compound II which is inactive toward HOCl synthesis (Fig. 5a, curve 1). In the presence of tyrosine, H2O2 addition resulted in a rapid decrease in HSA fluorescence (curve 2), which can be explained by the acceleration of HOCl synthesis [7]. As shown earlier, under these conditions, tyrosine interacts with Compound II and increases the enzyme turnover rate. Accordingly, more HOCl was simultaneously synthesized, which caused rapid oxidation of amino acid residues in HSA, including tryptophan (within the first 2 min of incubation) until H2O2 in solution was spent (linear portion of curve 2). With longer incubation times, the presence of tyrosine in the solution reduced the extent of HSA modification. After 15 min of incubation, HSA fluorescence was higher in the solution containing tyrosine (Fig. 5a). Similar acceleration of the MPO-induced fluorescence quenching in the presence of tyrosine and 100 μM H2O2 was observed for CP, IgG, and fibrinogen (data not shown). However, in the case of these proteins, the effect was less significant, because not all tryptophan residues were available for the oxidation by the MPO-generated HOCl (the maximum difference in the magnitude between the spectra without and with tyrosine did not exceed 17%).
When low H2O2 concentrations (10 μM × 10 times) were added to the HSA solution containing MPO and Cl–, the kinetics of HSA fluorescence decrease was similar to that observed after a single addition of 100 μM H2O2 (curves 1 in Fig. 5, a and b). Under our experimental conditions, the rate of HOCl synthesis was the same in the two systems: at high H2O2 concentrations, most enzyme molecules were in the Compound II state that does not synthesize HOCl, so the rate of HOCl generation in the solution was low, whereas upon multiple additions of small doses of H2O2, the rate of HSA oxidation was determined by the frequency of additions. In the latter case, the presence of tyrosine in the incubation mixture inhibited protein oxidation (Fig. 5b).
At the same time, no changes in the molecular weight of HSA after its incubation with MPO/Cl–/Tyr/H2O2 were registered (Fig. 3, lanes 3, 5, and 6).
Effect of tyrosine on the MPO-induced oxidation of CP. CP oxidation by the catalytically active MPO was studied in the absence of tyrosine and at tyrosine concentrations of 100 and 200 μM (Fig. 4a). Tyrosine is a small molecule; therefore, the binding of CP to MPO does not prevent tyrosine oxidation in the MPO active site [43]. Under our experimental conditions, 100 μM tyrosine promoted the disappearance of the native CP band and formation of high-molecular-weight protein aggregates at pH 6.8. At the same time, tyrosine completely abolished formation of large protein agglomerates sticking on the top of the stacking gel (Fig. 4a, compare lanes 2 and 3). Tyrosine addition to the incubation mixture promoted CP fragmentation: the intensity of the low-molecular-weight CP band in the 100-kDa region increased (similar effect was observed with NaOCl; lanes 9 and 10), together with the appearance of an additional band of low-molecular-weight in the 65-kDa region. In the presence of 200 μM tyrosine (lane 4), CP was oxidized to a greater extent than in the absence of tyrosine (lane 2), and the amount of high-molecular-weight aggregates was comparable to that observed at 100 µM tyrosine (Fig. 4b). Further increasing the tyrosine concentration did not make sense since in this case, its concentration would have exceeded physiologically reasonable values.
Tyrosine did not significantly affect CP stability upon MPO-induced oxidation at pH 7.4 (Fig. 4, lanes 7 and 8).
DISCUSSION
Protein oxidation is accompanied by changes in the protein structure and physicochemical properties, including spectral characteristics. Oxidation of cysteines and tyrosines in the polypeptide chain leads to the formation of disulfide bonds and dityrosine bridges between the molecules, resulting in protein aggregation. Formation of aggregates of oxidized proteins underlies the emergence and development of various pathologies. MPO involvement in the formation of protein aggregates has been demonstrated for cardiovascular disorders [2, 18], Alzheimer’s disease [44], kidney diseases [45], and other socially significant pathologies.
In this work, we compared MPO-induced oxidation of two proteins with antioxidant properties, namely CP and HSA. As demonstrated earlier, these proteins form complexes with MPO in vivo [30, 33, 36]. Our studies have shown that CP and HSA undergo oxidative modification when incubated with hypochlorite or catalytically active MPO in the presence of Cl–, but the effects of modification are different for the two proteins.
The binding of CP near the MPO active site causes a decrease in the chlorinating activity of the enzyme [33, 35]. We failed to detect any MPO-induced modification of CP at neutral pH values. At pH 7.4, the protein can be oxidized after its incubation with exogenous NaOCl, while in the presence of MPO/Cl–/H2O2, CP is modified only at acidic pH values (pH ≤ 7.0) [42]. This may be due to the weakening of the CP–MPO complex because of changes in the protein charge at lower pH.
As for most proteins, CP oxidation leads to the formation of high-molecular-weight products (protein aggregates) and/or, under certain conditions, to the protein fragmentation. In both cases, the amount of the native protein decreases. Our experiments showed that formation of CP aggregates after incubation with the catalytically active MPO (100 + 100 μM H2O2) was comparable to that observed after protein treatment with exogenous hypochlorite (100 µM NaOCl) (Fig. 4a, lanes 3 and 9, 10). Large aggregates with M ~ 200-300 kDa formed in the solution, as well as protein agglomerates that did not enter the stacking gel, were observed. Formation of CP aggregates occurs due to a large number of SH-groups and tyrosine residues in the protein (table). Dityrosine cross-links, unlike disulfide bonds, cannot be reduced; they are strong intermolecular bonds that can initiate formation of large covalently bound protein aggregates in vivo. Aouffen et al. showed formation of CP aggregates after protein treatment with high H2O2 concentrations [46].
We have previously shown that the area of contact between CP and MPO includes CP loops between the 4th and 5th domains (a.a. 699-710) and the 5th and 6th domains (a.a. 883-892) [47]. It can be assumed that the appearance of low-molecular-weight fragments of CP after its incubation with catalytically active MPO is due to the hydrolysis of peptide bonds in the CP regions located at the surface of protein globule.
CP oxidation results in the formation of protein aggregates that might also contain MPO. Similar interactions have been shown earlier for the formation of hemoglobin and haptoglobin aggregates; such aggregates were more efficiently engulfed by macrophages than the complexes of hemoglobin with haptoglobin [48]. One may assume that formation of the MPO-containing CP aggregates can promote removal of potentially dangerous peroxidase from the blood plasma.
On the contrary, HSA incubation with NaOCl or catalytically active MPO did not result in the formation of protein aggregates (Fig. 3). Albumin is the major antioxidant plasma protein that can neutralize oxidants such as products of the MPO-catalyzed reactions. It was shown earlier that HSA can efficiently neutralize oxidants while preserving its globular shape and functions of some active sites [49]. HSA has only one free SH-group (Cys34); therefore, the probability of the intermolecular disulfide bond formation is low, given the existence of steric hindrances for such interactions [29]. On the other hand, HSA has 18 tyrosine residues, but nevertheless, does not form aggregates upon oxidation. The fact is that the reaction of tyrosine with HOCl results in tyrosine chlorination with the formation of chlorotyrosines, which are stable markers of active MPO. The contribution of tyrosine chlorination to the overall pattern of protein oxidation was not significant in our experiments, since the rate constant of the HOCl reaction with tyrosine is several orders of magnitude lower than that for most other amino acids (k ~ 44 M–1·s–1) [50] (table).
At the same time, tryptophan oxidation by hypochlorous acid is characterized by a relatively high reaction rate (k ~ 1.1·104 M–1·s–1) [50]. Among the products of the reaction, tryptophan derivatives (products of tryptophan ionic oxidation) and tryptophan radicals (formed by the free-radical oxidation of its indole ring) were detected [51, 52]. Tryptophan oxidation can also produce compounds with the antioxidant properties [53]. Tryptophan radicals are long-lived (up to a few minutes) and can recombine to form covalent bonds between proteins (k ~ 6·108 M–1·s–1) [54]. The redox potential of the tryptophanyl radical is relatively high and depends on pH and amino acid local environment within a protein molecule (E0 up to 1 V) [15]. Migration of the oxidative equivalent from tryptophan to other oxidizable amino acids of the polypeptide chain can result in tyrosine oxidation and, consequently, formation of dityrosine cross-links between proteins.
The only tryptophan in HSA was rapidly oxidized by the MPO-generated oxidants (Figs. 1 and 2b), while the polypeptide chain of the protein was stable during oxidation – HSA retained its primary structure when incubated with catalytically active MPO (Fig. 3). The relatively high content of lysines and histidines in HSA can also attenuate HOCl-induced protein damage (table).
The results of studies on the oxidation-induced HSA aggregation are contradictory. Disulfide bond formation between albumin molecules was detected in the blood plasma after treatment with various hydroperoxides and in the plasma of patients after hemodialysis [55]. Formation of dityrosine bonds between protein molecules was shown by mass spectrometry for delipidated albumin oxidized in a buffer in the presence of a 10-fold (and higher) excess of NaOCl [56]. Colombo et al. found no significant formation of dityrosines after HSA treatment with hypochlorite (contrary to fibrinogen) [57].
We have previously shown that phenolic compounds are preferred MPO peroxidase substrates at neutral pH in both the model system and suspension of lysed neutrophils [10]. The active site of MPO is not available for large molecules; however, oxidation of small phenolic compounds can produce highly reactive free radicals that mediate macromolecule oxidation. Tyrosine is an important endogenous molecule and a specific peroxidase substrate of MPO. Its oxidation results in the formation of tyrosyl radicals (E0 = 0.93 V) [15] that can oxidize proteins and lipids, as well as form dityrosines. Dityrosines have a characteristic fluorescence with the maximum at 410 nm. Due to the high rate of dityrosine formation (k ~ 108 M–1·s–1), oxidation of free tyrosine by MPO did not cause oxidation of tryptophan residues in the proteins (Fig. 2c), whereas free radicals of phenol oxidized protein tryptophans (Fig. 1c). This may be due to the higher redox potential of phenol radicals and their lower capacity for recombination as compared to tyrosyl radicals (Fig. 2). Protein oxidation by phenoxyl radicals indicates the danger of phenolic xenobiotics that can be oxidized by MPO under inflammatory conditions.
Our previous study on the effect of tyrosine on the chloride oxidation by MPO at neutral pH revealed the following trends [7]: (i) tyrosine competes with chloride ions for Compound I even though its plasma concentration is three orders of magnitude lower than the NaCl concentration; (ii) at high H2O2 concentrations capable of converting MPO to Compound II, tyrosine by interacting with this stable form of the enzyme heme can increase the enzyme turnover and thus accelerate HOCl synthesis.
In this paper, we showed that tyrosine affects MPO-induced protein oxidation by modulating enzyme’s chlorinating activity: it accelerates protein oxidation at high H2O2 concentrations due to acceleration of HOCl synthesis by the enzyme, and inhibits it at low H2O2 concentrations or high tyrosine concentrations due to direct competition of the peroxidase substrate with chloride ions for Compound I (Fig. 5). Free tyrosine also inhibits formation of large CP aggregates (Fig. 4). Unlike the effects of antioxidants, the action of tyrosine is highly specific and involves only regulation of the MPO activity. It is possible that tyrosine or other phenolic compounds can be used to regulate MPO-induced protein oxidation, and first of all, to reduce formation of covalently bound protein aggregates in the plasma during inflammation.
Funding. This study was supported by the Russian Foundation for Basic Research (project no. 16-14-00873) and Russian Academic Excellence project 5-100.
Conflict of interest. The authors declare no conflict of interest in financial or any other sphere.
Ethical approval. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Arnhold, J. (2004) Free radicals – friends or
foes? Properties, functions, and secretion of human myeloperoxidase,
Biochemistry (Moscow), 69, 4-9, doi:
10.1023/B:BIRY.0000016344.59411.ee.
2.Davies, M. J., Hawkins, C. L., Pattison, D. I., and
Rees, M. D. (2008) Mammalian heme peroxidases: from molecular
mechanisms to health implications, Antioxid. Redox Signal.,
10, 1199-1234, doi: 10.1089/ars.2007.1927.
3.Arnhold, J., Furtmuller, P. G., and Obinger, C.
(2003) Redox properties of myeloperoxidase, Redox Rep.,
8, 179-186, doi: 10.1179/135100003225002664.
4.Furtmuller, P. G., Burner, U., Jantschko, W.,
Regelsberger, G., and Obinger, C. (2000) Two-electron reduction and
one-electron oxidation of organic hydroperoxides by human
myeloperoxidase, FEBS Lett., 484, 139-143.
5.Kirchner, T., Flemmig, J., Furtmüller, P. G.,
Obinger, C., and Arnhold, J. (2010) (–)-Epicatechin enhances the
chlorinating activity of human myeloperoxidase, Arch. Biochem.
Biophys., 495, 21-27, doi: 10.1016/j.abb.2009.12.013.
6.Flemmig, J., Remmler, J., Rohring, F., and Arnhold,
J. (2014) (–)-Epicatechin regenerates the chlorinating activity
of myeloperoxidase in vitro and in neutrophil granulocytes,
J. Inorg. Biochem., 130, 84-91, doi:
10.1016/j.jinorgbio.2013.10.002.
7.Vlasova, I. I., Sokolov, A. V., and Arnhold, J.
(2012) The free amino acid tyrosine enhances the chlorinating activity
of human myeloperoxidase, J. Inorg. Biochem., 106, 76-83,
doi: 10.1016/j.jinorgbio.2011.09.018.
8.Tzikas, S., Schlak, D., Sopova, K., Gatsiou, A.,
Stakos, D., Stamatelopoulos, K., Stellos, K., and Laske, C. (2014)
Increased myeloperoxidase plasma levels in patients with
Alzheimer’s disease, J. Alzheimer’s Dis., 39,
557-564, doi: 10.3233/JAD-131469.
9.Baldus, S., Heeschen, C., Meinertz, T., Zeiher, A.
M., Eiserich, J. P., Munzel, T., Simoons, M. L., and Hamm, C. W. (2003)
Myeloperoxidase serum levels predict risk in patients with acute
coronary syndromes, Circulation, 108, 1440-1445, doi:
10.1161/01.CIR.0000090690.67322.51.
10.Vlasova, I. I., Arnhold, J., Osipov, A. N., and
Panasenko, O. M. (2006) pH-dependent regulation of myeloperoxidase
activity, Biochemistry (Moscow), 71, 667-677.
11.Furtmuller, P. G., Zederbauer, M., Jantschko, W.,
Helm, J., Bogner, M., Jakopitsch, C., and Obinger, C. (2006) Active
site structure and catalytic mechanisms of human peroxidases, Arch.
Biochem. Biophys., 445, 199-213, doi:
10.1016/j.abb.2005.09.017.
12.Ramos, D. R., Garcia, M. V., Canle, L. M.,
Santaballa, J. A., Furtmuller, P. G., and Obinger, C. (2008)
Myeloperoxidase-catalyzed chlorination: the quest for the active
species, J. Inorg. Biochem., 102, 1300-1311, doi:
10.1016/j.jinorgbio.2008.01.003.
13.Zhang, R., Brennan, M. L., Shen, Z., MacPherson,
J. C., Schmitt, D., Molenda, C. E., and Hazen, S. L. (2002)
Myeloperoxidase functions as a major enzymatic catalyst for initiation
of lipid peroxidation at sites of inflammation, J. Biol. Chem.,
277, 46116-46122, doi: 10.1074/jbc.M209124200.
14.Vlasova, I. I., Feng, W.-H., Goff, J. P.,
Giorgianni, A., Do, D., Gollin, S. M., Lewis, D. W., Kagan, V. E., and
Yalowich, J. C. (2011) Myeloperoxidase-dependent oxidation of etoposide
in human myeloid progenitor CD34+ cells, Mol. Pharmacol.,
79, 479-487, doi: 10.1124/mol.110.068718.
15.Jantschko, W., Furtmuller, P. G., Zederbauer, M.,
Neugschwandtner, K., Lehner, I., Jakopitsch, C., Arnhold, J., and
Obinger, C. (2005) Exploitation of the unusual thermodynamic properties
of human myeloperoxidase in inhibitor design, Biochem.
Pharmacol., 69, 1149-1157, doi:
10.1016/j.bcp.2005.02.006.
16.Pattison, D. I., and Davies, M. J. (2006)
Reactions of myeloperoxidase-derived oxidants with biological
substrates: gaining chemical insight into human inflammatory diseases,
Curr. Med. Chem., 13, 3271-3290, doi:
10.2174/092986706778773095.
17.Senthilmohan, R., and Kettle, A. J. (2006)
Bromination and chlorination reactions of myeloperoxidase at
physiological concentrations of bromide and chloride, Arch. Biochem.
Biophys., 445, 235-244, doi: 10.1016/j.abb.2005.07.005.
18.Brennan, M. L., and Hazen, S. L. (2003) Amino
acid and protein oxidation in cardiovascular disease, Amino
Acids, 25, 365-374, doi: 10.1007/s00726-003-0023-y.
19.Shao, B., Tang, C., Sinha, A., Mayer, P. S.,
Davenport, G. D., Brot, N., Oda, M. N., Zhao, X. Q., and Heinecke, J.
W. (2014) Humans with atherosclerosis have impaired ABCA1 cholesterol
efflux and enhanced high-density lipoprotein oxidation by
myeloperoxidase, Circ. Res., 114, 1733-1742, doi:
10.1161/CIRCRESAHA.114.303454.
20.Arnhold, J., Hammerschmidt, S., Wagner, M.,
Mueller, S., Arnold, K., and Grimm, E. (1990) On the action of
hypochlorite on human serum albumin, Biomed. Biochim. Acta,
49, 991-997.
21.Colombo, G., Clerici, M., Altomare, A., Rusconi,
F., Giustarini, D., Portinaro, N., Garavaglia, M. L., Rossi, R.,
Dalle-Donne, I., and Milzani, A. (2017) Thiol oxidation and di-tyrosine
formation in human plasma proteins induced by inflammatory
concentrations of hypochlorous acid, J. Proteomics, 152,
22-32, doi: 10.1016/j.jprot.2016.10.008.
22.Colombo, G., Reggiani, F., Cucchiari, D.,
Portinaro, N. M., Giustarini, D., Rossi, R., Garavaglia, M. L., Saino,
N., Milzani, A., Badalamenti, S., and Dalle-Donne, I. (2017) Plasma
protein-bound di-tyrosines as biomarkers of oxidative stress in end
stage renal disease patients on maintenance haemodialysis, BBA
Clin., 7, 55-63, doi: 10.1016/j.bbacli.2016.12.004.
23.Meotti, F. C., Jameson, G. N. L., Turner, R.,
Harwood, D. T., Stockwell, S., Rees, M. D., Thomas, S. R., and Kettle,
A. J. (2011) Urate as a physiological substrate for myeloperoxidase:
implications for hyperuricemia and inflammation, J. Biol. Chem.,
286, 12901-12911, doi: 10.1074/jbc.M110.172460.
24.Salavej, P., Spalteholz, H., and Arnhold, J.
(2006) Modification of amino acid residues in human serum albumin by
myeloperoxidase, Free Radic. Biol. Med., 40, 516-525,
doi: 10.1016/j.freeradbiomed.2005.09.007.
25.Carr, A. C., McCall, M. R., and Frei, B. (2000)
Oxidation of LDL by myeloperoxidase and reactive nitrogen species:
reaction pathways and antioxidant protection, Arterioscler. Thromb.
Vasc. Biol., 20, 1716-1723, doi:
0.1161/01.ATV.20.7.1716.
26.Dobretsov, G. E., Syrejshchikova, T. I., Smolina,
N. V., and Uzbekov, M. V. (2015) CAPIDAN, a fluorescent reporter for
detection of albumin drug-binding site changes, in Human Serum
Albumin (HSA) (Stokes T., ed.) Nova Science Publisher, Inc., pp.
129-171.
27.Colombo, G., Clerici, M., Giustarini, D., Rossi,
R., Milzani, A., and Dalle-Donne, I. (2012) Redox albuminomics:
oxidized albumin in human diseases, Antioxid. Redox Signal.,
17, 1515-1527, doi: 10.1089/ars.2012.4702.
28.Sozarukova, M. M., Proskurnina, E. V., and
Vladimirov, Yu. A. (2016) Serum albumin as a sourse of and a target for
free radicals in pathology, Bull. RSMU, 1, 56-61.
29.Torres, M. J., Turell, L., Botti, H., Antmann,
L., and Carballal, S. (2012) Modulation of the reactivity of the thiol
of human serum albumin and its sulfenic derivative by fatty acids,
Arch. Biochem. Biophys., 521, 102-110, doi:
10.1016/j.abb.2012.03.011.
30.Tiruppathi, C., Naqvi, T., Wu, Y., Vogel, S. M.,
Minshall, R. D., and Malik, A. B. (2004) Albumin mediates the
transcytosis of myeloperoxidase by means of caveolae in endothelial
cells, Proc. Natl. Acad. Sci. USA, 101, 7699-7704, doi:
10.1073/pnas.0401712101.
31.Atanasiu, R. L., Stea, D., Mateescu, M. A.,
Vergely, C., Dalloz, F., Briot, F., Maupoil, V., Nadeau, R., and
Rochette, L. (1998) Direct evidence of caeruloplasmin antioxidant
properties, Mol. Cell. Biochem., 189, 127-135.
32.Barinov, N. A., Vlasova, I. I., Sokolov, A. V.,
Kostevich, V. A., Dubrovin, E. V., and Klinov, D. V. (2018)
High-resolution atomic force microscopy visualization of
metalloproteins and their complexes, Biochim. Biophys. Acta Gen.
Subj., 1862, 2862-2868, doi:
10.1016/j.bbagen.2018.09.008.
33.Sokolov, A., Ageeva, K., Pulina, M., Cherkalina,
O., Samygina, V., Vlasova, I. I., Panasenko, O., Zakharova, E., and
Vasilyev, V. (2008) Ceruloplasmin and myeloperoxidase in complex affect
the enzymatic properties of each other, Free Radic. Res.,
42, 989-998, doi: 10.1080/10715760802566574.
34.Griffin, S. V., Chapman, P. T., Lianos, E. A.,
and Lockwood, C. M. (1999) The inhibition of myeloperoxidase by
ceruloplasmin can be reversed by anti-myeloperoxidase antibodies,
Kidney Int., 55, 917-925, doi:
10.1046/j.1523-1755.1999.055003917.x.
35.Park, Y. S., Suzuki, K., Mumby, S., Taniguchi,
N., and Gutteridge, J. M. (2000) Antioxidant binding of caeruloplasmin
to myeloperoxidase: myeloperoxidase is inhibited, but oxidase,
peroxidase and immunoreactive properties of caeruloplasmin remain
intact, Free Radic. Res., 33, 261-265.
36.Chapman, A. L. P., Mocatta, T. J., Shiva, S.,
Seidel, A., Chen, B., Khalilova, I., Paumann-Page, M. E., Jameson, G.
N. L., Winterbourn, C. C., and Kettle, A. J. (2013) Ceruloplasmin is an
endogenous inhibitor of myeloperoxidase, J. Biol. Chem.,
288, 6465-6477, doi: 10.1074/jbc.M112.418970.
37.Segelmark, M., Persson, B., Hellmark, T., and
Wieslander, J. (1997) Binding and inhibition of myeloperoxidase (MPO):
a major function of ceruloplasmin? Clin. Exp. Immunol.,
108, 167-174.
38.Sokolov, A. V., Pulina, M. O., Ageeva, K. V.,
Ayrapetov, M. I., Berlov, M. N., Volgin, G. N., Markov, A. G.,
Yablonsky, P. K., Kolodkin, N. I., Zakharova, E. T., and Vasilyev, V.
B. (2007) Interaction of ceruloplasmin, lactoferrin, and
myeloperoxidase, Biochemistry (Moscow), 72, 409-415.
39.Sokolov, A. V., Kostevich, V. A., Romanico, D.
N., Zakharova, E. T., and Vasilyev, V. B. (2012) Two-stage method for
purification of ceruloplasmin based on its interaction with neomycin,
Biochemistry (Moscow), 77, 631-638, doi:
10.1134/S0006297912060107.
40.Marquez, L. A., and Dunford, H. B. (1995)
Kinetics of oxidation of tyrosine and dityrosine by myeloperoxidase
compounds I and II, J. Biol. Chem., 270, 30434-30440,
doi: 10.1074/jbc.270.51.30434.
41.Pfeiffer, S., Schmidt, K., and Mayer, B. (2000)
Dityrosine formation outcompetes tyrosine nitration at low steady-state
concentrations of peroxynitrite: implications for tyrosine modification
by nitric oxide/superoxide in vivo, J. Biol. Chem.,
275, 6346-6352, doi: 10.1074/jbc.275.9.6346.
42.Sokolov, A. V., Kostevich, V. A., Varfolomeeva,
E. Y., Grigorieva, D. V., Gorudko, I. V., Kozlov, S. O., Kudryavtsev,
I. V., Mikhalchik, E. V., Filatov, M. V., Cherenkevich, S. N.,
Panasenko, O. M., Arnhold, J., and Vasilyev, V. B. (2018) Capacity of
ceruloplasmin to scavenge products of the respiratory burst of
neutrophils is not altered by the products of reactions catalyzed by
myeloperoxidase, Biochem. Cell Biol., 96, 457-467, doi:
10.1139/bcb-2017-0277.
43.Panasenko, O. M., Chekanov, A. V., Vlasova, I.
I., Sokolov, A. V., Ageeva, K. V., Pulina, M. O., Cherkalina, O. S.,
and Vasil’ev, V. B. (2008) Influence of ceruloplasmin and
lactoferrin on the chlorination activity of leukocyte myeloperoxidase
assayed by chemiluminescence, Biophysics, 53, 268-272,
doi: 10.1134/S0006350908040052.
44.Green, P. S., Mendez, A. J., Jacob, J. S.,
Crowley, J. R., Growdon, W., Hyman, B. T., and Heinecke, J. W. (2004)
Neuronal expression of myeloperoxidase is increased in
Alzheimer’s disease, J. Neurochem., 90, 724-733,
doi: 10.1111/j.1471-4159.2004.02527.x.
45.Malle, E., Buch, T., and Grone, H.-J. (2003)
Myeloperoxidase in kidney disease, Kidney Int., 64,
1956-1967, doi: 10.1046/j.1523-1755.2003.00336.x.
46.Aouffen, M., Paquin, J., Furtos, A., Waldron, K.
C., and Mateescu, M.-A. (2004) Oxidative aggregation of ceruloplasmin
induced by hydrogen peroxide is prevented by pyruvate, Free Radic.
Res., 38, 19-26.
47.Samygina, V. R., Sokolov, A. V., Bourenkov, G.,
Petoukhov, M. V., Pulina, M. O., Zakharova, E. T., Vasilyev, V. B.,
Bartunik, H., and Svergun, D. I. (2013) Ceruloplasmin: macromolecular
assemblies with iron-containing acute phase proteins, PLoS One,
8, e67145, doi: 10.1371/journal.pone.0067145.
48.Kapralov, A., Vlasova, I. I., Feng, W., Maeda,
A., Walson, K., Tyurin, V. A., Huang, Z., Aneja, R. K., Carcillo, J.,
Bayir, H., and Kagan, V. E. (2009) Peroxidase activity of
hemoglobin–haptoglobin complexes. Covalent aggreation and
oxidative stress in plasma and macrophages, J. Biol. Chem.,
284, 30395-30407, doi: 10.1074/jbc.M109.045567.
49.Anraku, M., Yamasaki, K., Maruyama, T.,
Kragh-Hansen, U., and Otagiri, M. (2001) Effect of oxidative stress on
the structure and function of human serum albumin, Pharm. Res.,
18, 632-639.
50.Hawkins, C. L., Pattison, D. I., and Davies, M.
J. (2003) Hypochlorite-induced oxidation of amino acids, peptides and
proteins, Amino Acids, 25, 259-274, doi:
10.1007/s00726-003-0016-x.
51.Potsch, S., Lendzian, F., Ingemarson, R.,
Hornberg, A., Thelander, L., Lubitz, W., Lassmann, G., and Graslund, A.
(1999) The iron–oxygen reconstitution reaction in protein
R2-Tyr177 mutants of mouse ribonucleotide reductase: EPR and electron
nuclear double resonance studies on a new transient tryptophan radical,
J. Biol. Chem., 274, 17696-17704, doi:
10.1074/jbc.274.25.17696.
52.Carvalho, L. C., Estevao, M. S., Ferreira, L. M.,
Fernandes, E., and Marques, M. M. B. (2010) A new insight on the
hypochlorous acid scavenging mechanism of tryptamine and tryptophan
derivatives, Bioorg. Med. Chem. Lett., 20, 6475-6478,
doi: 10.1016/j.bmcl.2010.09.067.
53.Polimova, A. M., Vladimirova, G. A., Proskurnina,
E. V., and Vladimirov, Y. A. (2011) Aromatic amino acid oxidation
products as antioxidants, Biophysics, 56, 585-589, doi:
10.1134/S000635091104021X.
54.Carroll, L., Pattison, D. I., Davies, J. B.,
Anderson, R. F., Lopez-Alarcon, C., and Davies, M. J. (2018) Superoxide
radicals react with peptide-derived tryptophan radicals with very high
rate constants to give hydroperoxides as major products, Free Radic.
Biol. Med., 118, 126-136, doi:
10.1016/j.freeradbiomed.2018.02.033.
55.Ogasawara, Y., Namai, T., Togawa, T., and Ishii,
K. (2006) Formation of albumin dimers induced by exposure to peroxides
in human plasma: a possible biomarker for oxidative stress, Biochem.
Biophys. Res. Commun., 340, 353-358, doi:
10.1016/j.bbrc.2005.11.183.
56.Annibal, A., Colombo, G., Milzani, A.,
Dalle-Donne, I., Fedorova, M., and Hoffmann, R. (2016) Identification
of dityrosine cross-linked sites in oxidized human serum albumin, J.
Chromatogr. B Anal. Technol. Biomed. Life Sci., 1019,
147-155, doi: 10.1016/j.jchromb.2015.12.022.
57.Colombo, G., Clerici, M., Giustarini, D.,
Portinaro, N., Badalamenti, S., Rossi, R., Milzani, A., and
Dalle-Donne, I. (2015) A central role for intermolecular dityrosine
cross-linking of fibrinogen in high molecular weight advanced oxidation
protein product (AOPP) formation, Biochim. Biophys. Acta Gen.
Subj., 1850, 1-12, doi: 10.1016/j.bbagen.2014.09.024.