Analysis of Direct Effects of the CB1 Receptor Antagonist Rimonabant on Fatty Acid Oxidation and Glycogenolysis in Liver and Muscle Cells in vitro
G. A. Müller1,2,a*, S. Wied3, and A. W. Herling3
1Helmholtz Diabetes Center (HDC) at the Helmholtz Center for Health and Environment Munich, Institute for Diabetes and Obesity (IDO), 85764 Oberschleissheim, Germany2Ludwig-Maximilians-University of Munich, Department Biology I, Genetics, 82152 Planegg-Martinsried, Germany
3Sanofi Pharma Germany GmbH, Diabetes Research, 65926 Frankfurt am Main, Germany
* To whom correspondence should be addressed.
Received February 4, 2019; Revised June 24, 2019; Accepted June 25, 2019
Recent pharmacological findings regarding rimonabant, an anorectic and cannabinoid type 1 receptor (CB1R) antagonist, strongly suggest that some of its effects on the metabolic parameters and energy balance in rats are not related to the centrally mediated reduction in caloric intake. Instead, they may be associated with acute induction of glycogenolysis in the liver, in combination with transient increase in glucose oxidation and persistent increase in fat oxidation. It is possible that rimonabant produced direct short- or long-term stimulatory effect on these processes in primary and cultured rat cells. Rimonabant slightly stimulated β-oxidation of long-chain fatty acids in cultured rat myocytes overexpressing glucose transporter isoform 4, as well as activated phosphorylation of adenosine monophosphate-dependent protein kinase (AMPK) in primary rat hepatocytes upon long-term incubation. However, short-term action of rimonabant failed to stimulate β-oxidation in myocytes, myotubes, and hepatocytes, as well as to upregulate AMPK phosphorylation, glycogenolysis, and cAMP levels in hepatocytes. As a consequence, the acute effects of rimonabant on hepatic glycogen content (reduction) and total energy expenditure (increase) in rats fed with a standard diet cannot be explained by direct stimulation of glycogenolysis and fatty acid oxidation in muscles and liver. Rather, these effects seem to be centrally mediated.
KEY WORDS: AMP- and cAMP-dependent signaling, cannabinoid receptor 1, glucose and lipid metabolism, obesityDOI: 10.1134/S000629791908011X
Abbreviations: ACC, acetyl-CoA carboxylase; AGRP, agouti-related peptide; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; AMPK, adenosine monophosphate-dependent protein kinase; CART, cocaine- and amphetamine-regulated transcript; CB1R, cannabinoid type 1 receptor; CCK, cholecystokinin; EBSS, Earl’s balanced salt solution; (F)FA, (free) fatty acid; Glut4, glucose transport isoform 4; HBSS, Hank’s balanced salt solution; IBMX, 3-isobutyl-1-methylxanthine; MCH, melanin-concentrating hormone; NPY, neuropeptide Y; PKA, protein kinase A; POMC, proopiomelanocortin.
Endocannabinoids and cannabinoid receptors have been known for years as
key players in the control of food intake [1-4]. Other regulatory components are neuropeptide Y
(NPY), ghrelin, cholecystokinin (CCK), orexins, agouti-related peptide
(AGRP), melanin-concentrating hormone (MCH), leptin, insulin,
proopiomelanocortin (POMC), and CART (cocaine- and
amphetamine-regulated transcript) [5-7]. All the experimental data obtained so far indicate
that occupancy of the G-protein-coupled cannabinoid type 1 receptor
(CB1R) in the lateral hypothalamic area by endogenous ligands
(endocannabinoids) may lead to the hyperactivation of the downstream
endocannabinoid system. In combination with other orexigenic signals
[8], this stimulates food intake, possibly
contributing to overeating and obesity. The existence of this mechanism
in rodents and humans is supported by the effects of synthetic CB1R
selective antagonist rimonabant, which reduces the
endocannabinoid-induced hyperphagic effect, food intake, and body
weight gain when administered in diet-induced obese mice [9] and obese patients [10]. The
dependence of the food intake control by endocannabinoids and
rimonabant on the CB1R signaling has been proven by the facts that CB1R
knockout mice are lean, resistant to the diet-induced obesity [11], and do not respond to rimonabant in combination
with significantly reduced food intake when fed a high-fat diet [2, 12, 13].
In contrast to the apparently centrally and CB1R-mediated reduction in the food intake, which has been shown to be more pronounced in animals under re-fed conditions after temporary food deprivation than in animals with free access to food [2, 12, 13], the mechanism(s) involved in the body weight loss in response to rimonabant seem to be less clear. Recent observations have raised doubts as to whether transient reduction in the food intake provoked by rimonabant as an antagonist of hypothalamic CB1R is the only factor responsible for the sustained body weight loss. In fact, chip analysis of genes regulated at the transcriptional level by rimonabant in adipose tissues of diet-induced obese mice (mouse model of human obesity) [14], as well as pharmacological characterization of Wistar rats on a standard diet upon acute rimonabant challenge (unpublished data), strongly suggest that additional mechanisms contribute to the long-term anti-obesity effect of rimonabant. These putative mechanisms may be associated with the improvements in the metabolic parameters secondary to the reduced food intake, as exemplified by the decreased serum content of triglycerides and HDL-cholesterol in rimonabant-treated obese patients, which cannot be accounted for by the extent of weight loss alone [10]. Furthermore, as rimonabant-treated animals loose body weight more extensively than pair-fed controls [15], mechanisms causally unrelated to food intake seem to operate as well, which could include stimulation of energy expenditure in muscle and liver tissues during β-oxidation of fatty acids (FAs) released from the adipose tissue and/or oxidation of glucose released from the liver. This hypothesis is supported by the recent findings that (i) many alterations in gene expression induced by obesity in white and brown adipose tissues of obese mice and reversed by rimonabant treatment involve genes for proteins participating in β-oxidation, TCA cycle, and futile substrate cycling [14]; (ii) a single administration of rimonabant to Wistar rats leads to acute elevation in the serum levels of free fatty acids (FFAs), loss of liver glycogen and lipids, and increase in the total energy expenditure based on transient stimulation of glucose oxidation and persistent stimulation of FA oxidation (unpublished data).
Here, we studied if rimonabant can directly affect peripheral target cells involved in the regulation of energy expenditure, as well as glycogenolysis and FA oxidation in cultured muscle and primary liver cells.
MATERIALS AND METHODS
Rat L6 muscle cell cultures. L6 myocytes and L6(Glut4) myocytes stably expressing myc-tagged GLUT4myc (L6-GLUT4myc cells) were acquired from Dr. Amira Klip (Program in Cell Biology, The Hospital for Sick Children, Toronto, ON, Canada M5G 1X8). L6 myocytes were cultured in 24-well plates in α-MEM containing 2% (v/v) fetal bovine serum (FBS) and 1% (v/v) antimycotic/antibiotic solution (100 units/ml penicillin, 100 μg/ml streptomycin, 250 ng/ml amphotericin B) at 37°C in 5% CO2 and then differentiated into L6 myotubes over 7 days using the same medium. Myotube cultures contained less than 20% mononucleated cells.
Rat primary hepatocyte cultures. Hepatocytes were isolated from adult male Sprague–Dawley rats (Moellgaard, Lille, Skensved, Denmark; fed ad libitum) according to the standard two-step perfusion protocol using Ca2+-free medium and collagenase, as described earlier [16]. Non-viable cells were removed by iso-density Percoll centrifugation [17]. Cell viability (> 95%) in the final hepatocyte preparation was assessed using Trypan blue.
Fatty acid oxidation. The rate of cellular β-oxidation of [9,10(n)-3H]palmitic acid was measured from 3H2O release [18]. Primary hepatocytes: the cells were seeded in 24-well plates (200,000 cells/well). After attachment, the cells were incubated overnight in 5 mM glucose and 10 nM dexamethasone. The cells were then washed once with phosphate buffered saline (PBS) and subsequently incubated in the presence of DMEM, 5 μM (0.4 µCi) [3H]palmitic acid, 0.02% BSA, 500 μM L-carnitine, 0.1% (v/v) DMSO in the presence or absence of glucose (25 mM), rimonabant, and soraphen, as indicated. Cultured myocytes: the cells were serum-starved for 5 h, washed with Earl’s balanced salt solution (EBSS; 130 mM NaCl, 26 mM NaHCO3, 5 mM KCl, 1.8 mM CaCl2, 1 mM NaH2PO4, 0.8 mM MgSO4, pH 7.4), and then sequentially incubated (30 min, 37°C) with EBSS containing 5 mM glucose, 2% BSA and with EBSS containing 0.1 mM palmitate (dissolved in ethanol at 10 mM and diluted with EBSS containing 2% BSA) and rimonabant, as indicated. β-Oxidation was initiated by adding 0.5 μCi [3H]palmitate to the incubation medium. After incubation for 4 h at 37°C, aliquots of cell supernatants were loaded onto Sep-Pak cartridges (Waters, Germany) and unbound radioactivity was measured by liquid scintillation counting.
First, we evaluated the parameters of [9,10(n)-3H]palmitate oxidation assay to maximize the activity in the control (in the absence of rimonabant) and the resolution of the rimonabant effect on FA oxidation. The sensitivity of the assay was found to depend on four major variables: substrate concentration, cell density, BSA concentration, and incubation time. Although the concentrations of [3H]palmitate employed routinely (5 and 100 μM, respectively) were not fully saturating, less than 0.2% of the substrate was converted to 3H2O by the control cells; therefore, the concentration of [3H]palmitate remained virtually constant throughout the incubation period. 3H2O production was linear up to 80 µg of cell protein per well, at which point the response leveled off. All experiments reported here were performed at the cell protein concentration from 20 to 60 μg/well. Initially, [3H]palmitate oxidation was assayed in a variety of media including MEM and Hank’s balanced salt solution (HBSS) supplemented with FBS and/or varying concentrations of BSA. The optimal activity was observed with HBSS containing 0.02 and 2% fatty acid-free BSA, respectively. Although the activity increased linearly from 1 to 4 h of incubation, the experiments were performed within a more convenient period of 2 h.
Glucose release and glycogen content. Primary hepatocytes were seeded into 96-well plates (30,000 cells/well). After attachment, the cells were incubated overnight in William’s E-medium, then supplemented with 25 mM glucose and 100 nM insulin to accumulate glycogen. The cells were washed three times with prewarmed, oxygen-saturated KHH solution (20 mM HEPES, 115 mM NaCl, 4.5 mM KCl, 4.5 mM CaCl2, 1.1 mM KH2PO4, 1.1 mM MgSO4, pH 7.4), and subsequently incubated (30 min, 37°C) in a final volume of 100 μl of KHH containing 1% DMSO with either rimonabant or WIN55.212-2, as indicated. Where indicated, glycogenolysis was initiated by adding 10 μl glucagon (final concentration 100 nM). Glucose released into the medium was determined with an Amplex Red Glucose Assay Kit (Molecular Probes, USA) according to the manufacturer’s instructions. To assess glycogen accumulation, primary hepatocytes were seeded into 6-well plates (1,500,000 cells/well). The cells were incubated overnight in William’s E-medium and then supplemented with 25 mM glucose and 100 nM insulin in the presence or absence of rimonabant as indicated. The cells were then washed twice and lysed in 200 μl of 100 mM NaOH. Glycogen was isolated and analyzed as described previously [19].
Adenosine monophosphate-dependent protein kinase (AMPK) phosphorylation. Primary hepatocytes were incubated overnight in 5 mM glucose, 10 nM dexamethasone. The cells were washed once with PBS and subsequently incubated in the presence or absence of rimonabant and AICAR (5-aminoimidazole-4-carboxamide ribonucleotide). After 90 min, the cells were lysed in buffer B [50 mM Tris-HCl, pH 7.5, 0.1% (w/v) Triton X-100, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 10 mM sodium β-glycerophosphate, 1 mM Na3VO4, 5 mM NaPPi, 1 mM dithiothreitol and protease inhibitor tablets (Roche, Germany)], and cell debris was removed by centrifugation. Protein aliquots (20 μg) were fractionated by SDS-PAGE (4-12%) and transferred onto a PVDF membrane. The blot was incubated with the antibody against phosphoSer108-AMPKβ1 (Cell Signaling, USA). After washing, the blot was incubated (1 h) with peroxidase-conjugated anti-rabbit IgG. Detection was performed with LumiLIGHT (Roche).
Intracellular cAMP levels. Primary hepatocytes were seeded into 96-well plates (30,000 cells/well) and incubated overnight in William’s E-medium supplemented with 25 mM glucose and 100 nM insulin. Subsequently, the cells were incubated (30 min) in KHH supplemented with 1 mM IBMX in the presence or absence of glucagon, rimonabant, and CP55.940, as indicated. cAMP levels were determined using a HitHunter EFC cAMP chemiluminescence assay kit (Applied Biosystems, USA) according to the manufacturer’s instructions.
Miscellaneous. Rimonabant [9] was synthesized as described previously. Protein concentration was determined by the BCA method (Pierce, USA) with BSA as a calibration standard. SDS-PAGE (Novex 4-12%, Tris-glycine precast gels with (morpholino)propanesulfonic acid-SDS running buffer), immunoblotting, and subsequent enhanced chemiluminescent detection (ECL; Amersham-Buchler, Germany) and quantitative evaluation of the obtained images (Lumiimager and software from Roche) were carried out as reported earlier [20] unless indicated otherwise. Concentration response curves were fitted using the GraphPad Prism 4.03 software.
RESULTS
Rimonabant does not induce short-term activation of β-oxidation in cultured muscle and primary liver cells. In addition to the demonstrated rapid increase of FFA levels in the serum of Wistar rats after acute rimonabant administration, we also found by performing indirect calorimetry measurements that rimonabant causes a long-lasting increase in lipid oxidation and an acute decrease in hepatic lipid levels (unpublished data). To elucidate a putative direct effect of rimonabant on β-oxidation in the main oxidative tissues, muscle and liver, which could be fueled by FFA released from the adipose tissue in response to rimonabant, cultured rat myocytes and myotubes and primary rat hepatocytes were used. We found that short-term treatment of cultured L6 myocytes, L6 myotubes differentiated in vitro (and exhibiting higher β-oxidation rates) (Fig. 1), and primary rat hepatocytes (Fig. 2) with rimonabant did not significantly affect β-oxidation of exogenously added [3H]palmitate in these cells. As a control, palmitate oxidation was strongly stimulated in hepatocytes by both omission of glucose from the culture medium and addition of the acetyl-CoA carboxylase (ACC) inhibitor soraphen.
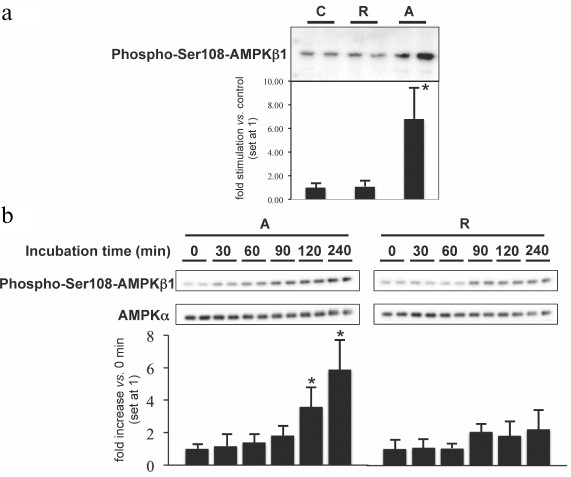
AMPK is one of the key regulators of FA oxidation and energy expenditure [21]. When phosphorylated and activated in response to the reduction in the cellular energy status, AMPK inactivates ACC and thereby stimulates FA oxidation. Short-term incubation with rimonabant failed to significantly induce AMPK phosphorylation in primary hepatocytes, whereas AICAR, a pharmacological activator of AMPK, caused strong increase in AMPK phosphorylation (Fig. 3a). Moreover, administration of CB1R agonists in rats produced no effect on the AMPK activity in skeletal muscle [22], which is consistent with the lack of short-term activation of FA oxidation in peripheral cells by rimonabant (Fig. 1). Overall, these data suggest that rimonabant does not directly and acutely affect metabolic and regulatory enzymes involved in β-oxidation of long-chain FAs in muscle and liver cells. In contrast, long-term (20 h) incubation of L6 myocytes overexpressing Glut4 with 0.3-1 μM rimonabant moderately (1.5-fold) stimulated FA oxidation above the basal level, and this effect was completely abrogated by the excess of CB1R agonist (Fig. 1). Incubation with rimonabant for more than 90 min led to a slight increase in the AMPK phosphorylation in hepatocytes (Fig. 3b), while the stimulatory effect of AICAR on both β-oxidation and the amount of phosphorylated AMPK was already detectable at 30 min (Fig. 3b and data not shown).
Fig. 1. Effect of rimonabant on β-oxidation in myocytes. Serum-starved cultured L6 myocytes (a), differentiated L6 myotubes (b), and L6 myocytes expressing Glut4 (c) were incubated at 37°C for various times in EBSS containing glucose and BSA and then in EBSS containing palmitate and BSA in the absence or presence of increasing concentrations of rimonabant. L6[Glut4] myocytes were incubated in the presence or absence of 10 μM CP55.940 as indicated. After addition of [3H]palmitate, the reaction mixture was incubated for 4 h. β-Oxidation was measured as 3H2O release (see “Materials and Methods”). The data are shown as mean ± S.D (each incubation was performed in triplicate; all measurements were performed in duplicate). The value of 5000 dpm for the water-soluble β-oxidation products under the assay conditions corresponded to the formation of 1149 pmol 3H2O/h per mg protein from [9,10(n)-3H]palmitate.
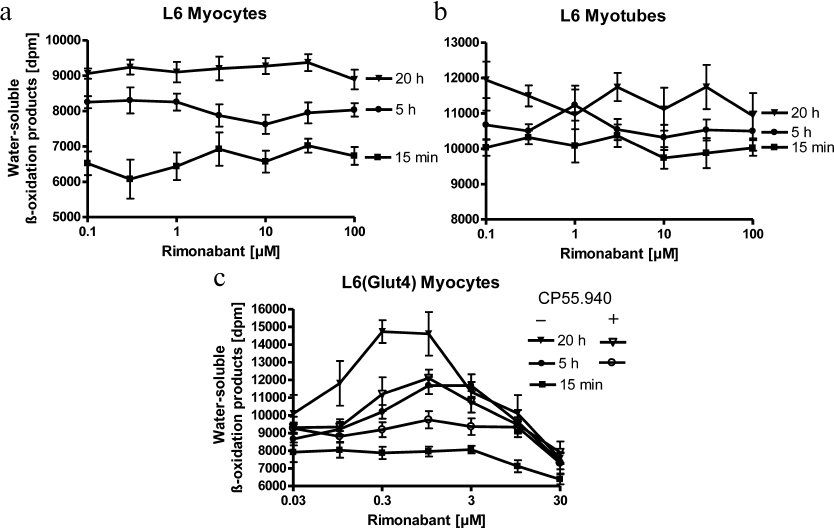
Fig. 2. Effect of rimonabant on β-oxidation in hepatocytes. The cells were incubated for 4 h with [3H]palmitic acid in the presence or absence of rimonabant or soraphen at the indicated glucose concentrations. β-Oxidation was measured as 3H2O release; * p < 0.05. Data are shown as mean ± S.D. (n = 3). The value of 1000 dpm for the water-soluble β-oxidation products under the assay conditions corresponded to the oxidation of 127 pmol of [9,10(n)-3H]palmitate/well per hour.
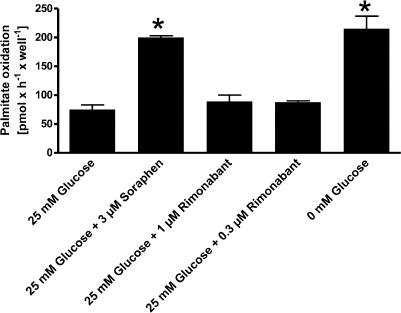
Fig. 3. Effect of rimonabant on AMPK phosphorylation in hepatocytes. The cells were incubated: (a) for 90 min in the absence (C) or presence of 1 μM rimonabant (R) or 0.5 mM AICAR (A) or (b) for the increasing periods of time in the presence of 3 μM rimonabant (R) or 0.5 mM AICAR (A). Aliquots containing 20 μg cell lysate protein were separated by SDS-PAGE and analyzed for Ser108-phosphorylated AMPKβ1 or AMPKα by immunoblotting. The experiments were repeated four times with different incubation times and different batches of cultured (and differentiated) cells. Immunoblotting for Ser108-phosphorylated AMPKβ1 or AMPKα was performed in duplicate. Representative immunoblots are shown. Quantitative evaluations of the extent of AMPKβ1 phosphorylation stimulation vs. control (C) (a) and increase in the AMPKβ phosphorylation (set at 1 for 0 min) vs. incubation time point 0 (b) are shown (n = 5; mean ± S.D.; * p < 0.05).
To demonstrate the validity of the used method, we compared phosphorylation of ACC (at Thr72) as the downstream key regulatory enzyme for FA oxidation vs. phosphorylation of the upstream AMPK β1 subunit in a set of immunoblotting experiments. This comparison revealed only non-significant differences in the extent (folds) of stimulation of phosphorylation of AMPK β1 and ACC by rimonabant and AICAR, as well as a very moderate delay in the time courses of ACC vs. AMPK phosphorylation (data not shown). Therefore, phosphorylation of the AMPK β1 subunit can be used as a reliable surrogate marker for AMPK activation. Thus, the phosphorylation analysis of both AMPK and ACC unequivocally showed that rimonabant fails to induce AMPK activation as a molecular mechanism of upregulation of FA oxidation.
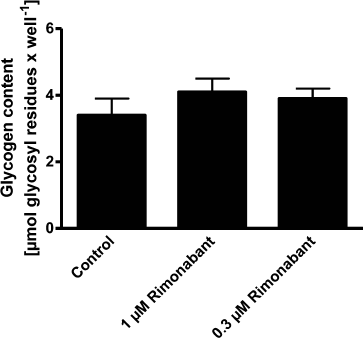
Rimonabant does not activate glycogenolysis by in primary liver cells. Administration of rimonabant acutely decreases the content of hepatic glycogen in Wistar rats (unpublished data). To investigate whether this decrease represents a direct influence of rimonabant on glucose release from endogenous glycogen stores was studied using glycogen-loaded primary rat hepatocytes (Fig. 4). Glucagon increased glucose release from these cells approximately 2-fold. AVE5688, an inhibitor of glycogen phosphorylase, blocked the effect of glucagon, thereby demonstrating that the increased glucose release was due to the stimulation of glycogen degradation. The CB1R agonist WIN55.212-2 had no effect on the glucagon-stimulated glucose-release. Rimonabant at concentrations up to 3 μM did not significantly increase the basal glucose release. This is in agreement with the measurements of glycogen content in primary hepatocyte cultures after addition of rimonabant, since this content was not significantly altered after incubation for 16 h in the presence of glucose and insulin (Fig. 5).
Fig. 4. Effect of rimonabant on glucose release in hepatocytes. Glucose release was determined after incubation of the cells in the absence (control) or presence of glucagon (see “Materials and Methods”), AVE5688 (Sanofi-Aventis, Germany), WIN55.212-2 (Tocris, UK), or rimonabant, as indicated; * p ≤ 0.05.
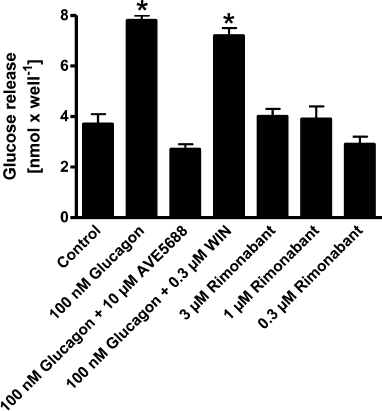
Fig. 5. Effect of rimonabant on glycogen content in hepatocytes. Glycogen content was determined after overnight incubation in the medium supplemented with 25 mM glucose and 100 nM insulin in the presence or absence of rimonabant (see “Materials and Methods”). Data are shown as mean ± S.D. (n = 3).
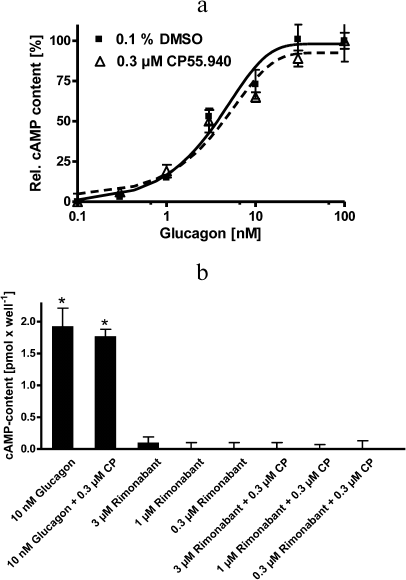
Overall, these data suggest that rimonabant does not exert direct effects on hepatic glycogen metabolism. This notion is further supported by measurements of the intracellular cAMP levels. cAMP mediates stimulation of glycogenolysis by glucagon. As expected, glucagon strongly increased intracellular cAMP levels in primary hepatocytes in a concentration-dependent manner, as measured in the presence of the phosphodiesterase inhibitor IBMX (Fig. 6a). The CB1R agonist CP55.940 failed to affect cAMP levels either alone or in a combination with glucagon. Rimonabant at concentrations up to 3 μM, either alone or in combination with CP55.940, had no significant effect on the cAMP content in the control cells, i.e., in the absence of glucagon (Fig. 6b). These data suggest that rimonabant does not act directly on primary rat hepatocytes via the cAMP second messenger system.
Fig. 6. Effect of rimonabant and CP55.940 on cAMP content in hepatocytes. a) Intracellular cAMP levels were determined after incubation (30 min) in the presence of the indicated glucagon concentration in the absence (filled squares, solid line) or presence of 0.3 μM CP55.940 (open triangles, dashed line). b) The cells were incubated (30 min) in the presence or absence of glucagon, CP55.940 (CP), and rimonabant, as indicated; * p < 0.05 vs. unstimulated cells. cAMP levels were determined by comparison to cAMP standard curve as described in “Materials and Methods”. Data are shown as mean ± S.D. (n = 3).
DISCUSSION
Here, we investigated the mechanisms underlying acute and subacute stimulatory effects of rimonabant on glycogenolysis in Wistar rats that were manifested as reduction of the hepatic glycogen content, upregulation of glucose and FA oxidation, and increased energy expenditure (unpublished data). In particular, we studied whether rimonabant exerts direct effects on cultured muscle cells and primary rat hepatocytes. Direct stimulation of lipolysis in isolated rat adipocytes [23] and the absence of stimulation of basal FA oxidation (and AMPK phosphorylation; Fig. 3) in cultured rat myocytes and myotubes (Fig. 1) and primary hepatocytes (Fig. 2) in response to the short-term action of rimonabant suggest that the rimonabant-induced direct release of FAs from the adipose tissue into the circulation in vivo drives FA oxidation in the muscle and liver tissues by the substrate flux alone. This combination of direct and indirect peripheral effects would be compatible with the acute and subacute effects of rimonabant on lipid metabolism in Wistar rats (unpublished data). Independently of indirect acute stimulation of FA oxidation in vivo, rimonabant seems to exert direct CB1R-dependent effect on the upregulation of β-oxidation of slow-onset (Fig. 1), which may be mediated through the AMPK activation (Fig. 3). Interestingly, previous analysis of gene expression in obese mice on a high-fat diet treated with rimonabant (10 mg/kg) daily for 40 days revealed induction of FA oxidation enzymes, as well as a 2-fold increase in Glut4 levels in white and brown adipose tissues compared to the vehicle-treated mice [14]. These data argue that rimonabant modulates expression of FA metabolism genes via CB1R. However, since stimulation of FA oxidation was not observed in L6 myocytes and myotubes not overexpressing Glut4, as well as required incubation for more than 5 h, the direct CB1R-mediated effect of rimonabant on muscle tissue upon single administration to rats is unlikely to explain the observed acute activation of FA oxidation. Hence, the primary site of rimonabant’s acute stimulatory action on FA oxidation and energy expenditure in vivo does not seem to be located in peripheral tissues.
Remarkably, no direct stimulation of basal glycogenolysis by rimonabant was observed in primary hepatocytes, as judged from the release of glucose into the incubation medium (Fig. 4), glycogen content (Fig. 5), and basal cAMP production (Fig. 6b), although it is well known that glycogenolysis in hepatocytes is regulated by cAMP, similarly to lipolysis in adipocytes. The failure to detect CB1R-dependent glycogenolytic activity of rimonabant may be explained by lower expression or less efficient coupling of CB1R to the downstream effectors in liver vs. adipose cells. Expression of CB1R has been demonstrated so far in primary rat hepatocytes [24] and mouse and rat adipocytes [25], but it appears to be highly dependent on the age of donor animals. Alternatively, the ability of the anticipated occupancy of CB1R by endocannabinoids (which compete with rimonabant) to stimulate glycogenolysis and lipolysis is less pronounced or even absent in primary hepatocytes vs. adipocytes. The only direct effect of rimonabant on hepatocytes reported so far is the inhibition of basal and CB1R agonist-stimulated FA synthesis [24]. This may correspond to subtle changes in AMPK and/or protein kinase A (PKA) signaling with greater sensitivity compared to the FA oxidation or rely on different signaling cascades engaged by the hepatic CB1R, but not coupled to the FA oxidation. In any case, the described acute and subacute effects of rimonabant on glucose and FA oxidation in rats resulting in the observed acute elevation of the energy expenditure (unpublished data) seem to be the result of its central CB1R-mediated action or be secondary to the increased lipolysis and glycogenolysis driven by the substrate flux.
The situation may become different upon long-term treatment with rimonabant, as seen in obesity therapy. With regard to the putative mechanisms of direct long-term effects, it may be of relevance that rimonabant at higher concentrations (>10 μM) and applied for longer incubation periods is capable of stimulating lipolysis via a mechanism that does not depend on functional CB1R or alternative receptors coupled to the cAMP/PKA axis [23]. Strikingly, incubation with rimonabant of native lipid droplets prepared from isolated rat adipocytes, but not of synthetic emulsified neutral lipids, increases their affinity for association with and cleavage by the hormone-sensitive lipase. The mechanism engaged by rimonabant in cell-free lipolysis or reactions in lipid droplets prepared from rimonabant-treated adipocytes is apparently mimicked by the PKA-dependent phosphorylation of lipid droplets rather than the action of hormone-sensitive lipase. This primary site of rimonabant action in lipid droplets is in agreement with the current view on the crucial role of lipid droplet-associated proteins in the regulation of lipolysis [26, 27].
These data argue for rimonabant interaction with the lipid droplet surface (at high concentrations) and CB1R of adipocytes, leading to the increased accessibility of lipid droplets for the hormone-sensitive lipase in phosphorylation-independent and dependent fashion, respectively. The two mechanisms may lead to direct and acute stimulation of lipolysis and, consequently, FA oxidation upon rimonabant administration and represent potential targets for future obesity therapy bypassing hypothalamic CB1R.
Furthermore, it is tempting to speculate that the direct effect of rimonabant on lipolysis is not restricted to the adipose tissue. Hormone-sensitive lipase is almost ubiquitously expressed. Regulated mobilization of endogenous fat stores is crucial for many cell types, including myocytes and hepatocytes [28]. Interestingly, 6 h after a single administration of rimonabant, elevation in the rat serum FFA levels associated with the increased levels of intramyocellular lipid was observed, which subsequently declined to the levels below those observed in pair-fed control animals at 20 h [29]. This release of FA from intramyocyte/hepatocellular lipids for subsequent FA oxidation in muscle and liver tissues could be related to the direct stimulation of lipolysis in myocytes and hepatocytes by the CB1R-dependent and/or independent mechanisms, similar to those known to be effective in rat adipocytes [23].
Taken together, our results add new insights to the present knowledge of the CB1R-dependent and independent molecular and physiological mechanisms of regulation of lipid and glycogen degradation by endocannabinoids and their antagonists, in particular, to the action mechanism of the anorectic drug rimonabant, which may support future drug discovery in obesity therapy [30, 31].
Conflict of interest. The authors declare no conflict of interest neither in financial nor in any other area.
Compliance with ethical standards. This article does not contain any studies involving animals or human participants performed by any of the authors.
REFERENCES
1.Sack, N., Hutcheson, J. R., Watts, J. M., and Webb,
R. E. (1990) Case report: the effect of tetrahydrocannabinol on food
intake during chemotherapy, J. Am. Coll. Nutr., 9,
630-632.
2.Di Marzo, V., Goparaju, S. K., Wang, L., Liu, J.,
Batkai, S., Jarai, Z., Fezza, F., Miura, G. I., and Palmiter, R. D.
(2001) Leptin-regulated endocannabinoids are involved in maintaining
food intake, Nature, 410, 822-825.
3.Di Marzo, V., and Matias, I. (2005) Endocannabinoid
control of food intake and energy balance, Nat.
Neurosci., 8, 585-589.
4.Cohen, K., Weizman, A., and Weinstein, A. (2019)
Positive and negative effects of cannabis and cannabinoids on health,
Clin. Pharmacol. Ther., 105, 1139-1147, doi:
10.1002/cpt.1381.
5.Benardis, L. L., and Bellinger, L. L. (1996) The
lateral hypothalamic area revisited: ingestive behavior, Neurosci.
Biobehav. Rev., 20, 189-287.
6.Spiegelman, B. M., and Flier, J. S. (2001) Obesity
and the regulation of energy balance, Cell, 104,
531-543.
7.Barsh, G. S., and Schwartz, M. W. (2002) Genetic
approaches to studying energy balance, Nat. Rev. Genet.,
3, 589-600.
8.Hilairet, S., Bouaboula, M., Carriere, D., Le Fur,
G., and Casellas, P. (2003) Hypersensitization of the orexin 1 receptor
by the CB1 receptor: evidence for cross-talk blocked by the specific
CB1 antagonist, SR141716, J. Biol. Chem., 278,
23731-23737.
9.Rinaldi-Carmona, M., Barth, F., Heaulme, M.,
Alonso, R., Shire, D., Congy, C., Soubrie, P., Breliere, J. C., and Le
Fur, G. (1995) Biochemical and pharmacological characterization of
SR141716, the first potent and selective brain cannabinoid receptor
antagonist, Life Sci., 56, 1941-1947.
10.Van Gaal, L. F., Rissanen, A. M., Scheen, A. J.,
Ziegler, O., and Rossner, S. (2005) Effects of the cannabinoid-1
receptor blocker rimonabant on weight reduction and cardiovascular risk
factors in overweight patients: 1-year experience from the RIO-Europe
study, Lancet, 365, 1389-1397.
11.Ravinet, T. C., Delgorge, C., Menet, C., Arnone,
M., and Soubrie, P. (2004) CB1 cannabinoid receptor knockout in mice
leads to leanness, resistance to diet-induced obesity and enhanced
leptin sensitivity, Int. J. Obes. Relat. Metab. Disord.,
28, 640-648.
12.Gomez, R., Navarro, M., Ferrer, B., Trigo, J. M.,
Bilbao, A., Del, A., Cippitelli, A., Nava, F., Piomelli, D., and
Rodriguez, D. F. (2002) A peripheral mechanism for CB1 cannabinoid
receptor-dependent modulation of feeding, J. Neurosci.,
22, 9612-9617.
13.Freedland, C. S., Poston, J. S., and Porrino, L.
J. (2000) Effects of SR141716A, a central cannabinoid receptor
antagonist, on food-maintained responding, Pharmacol. Biochem.
Behav., 67, 265-270.
14.Jbilo, O., Ravinet-Trillou, C., Arnone, M.,
Buisson, I., Bribes, E., Peleraux, A., Penarier, G., Soubrie, P., Le
Fur, G., Galiegue, S., and Casellas, P. (2005) The CB1 receptor
antagonist rimonabant reverses the diet-induced obesity phenotype
through the regulation of lipolysis and energy balance, FASEB
J., 19, 1567-1569.
15.Ravinet, T. C., Arnone, M., Delgorge, C.,
Gonalons, N., Keane, P., Maffrand, J. P., and Soubrie, P. (2003)
Anti-obesity effect of SR141716, a CB1 receptor antagonist, in
diet-induced obese mice, Am. J. Physiol. Regul. Integr. Comp.
Physiol., 284, R345-353.
16.Seglen, P. O. (1976) Preparation of isolated rat
liver cells, Methods Cell Biol., 13, 29-83.
17.Kreamer, B. L., Steacker, J. L., Sawada, N.,
Sattler, G. L., Hsia, M. T., and Pitot, H. C. (1986) Use of a
low-speed, iso-density Percoll centrifugation method to increase the
viability of isolated rat hepatocyte preparations, In Vitro Cell
Dev. Biol., 22, 201-211.
18.Minnich, A., Tian, N., Byan, L., and Bilder, G.
(2001) A potent PPARalpha agonist stimulates mitochondrial fatty acid
beta-oxidation in liver and skeletal muscle, Am. J. Physiol.
Endocrinol. Metab., 280, E270-279.
19.Schmoll, D., Fuhrmann, E., Gebhardt, R., and
Hamprecht, B. (1995) Significant amounts of glycogen are synthesized
from 3-carbon compounds in astroglial primary cultures from mice with
participation of the mitochondrial phosphoenolpyruvate carboxy kinase
isoenzyme, Eur. J. Biochem., 227, 308-315.
20.Muller, G., Jung, C., Wied, S., Welte, S., and
Frick, W. (2001) Insulin-mimetic signaling by the sulfonylurea
glimepiride and phosphoinositolglycans involves distinct mechanisms for
redistribution of lipid raft components, Biochemistry,
40, 14603-14620.
21.Kahn, B. B., Alquier, T., Carling, D., and
Hardie, D. G. (2005) AMP-activated protein kinase: ancient energy gauge
provides clues to modern understanding of metabolism, Cell
Metab., 1, 15-25.
22.Kola, B., Hubina, E., Tucci, S. A., Kirkham, T.
C., Garcia, E. A., Mitchell, S. E., Williams, L. M., Hawley, S. A.,
Hardie, D. G., Grossman, A. B., and Korbonits, M. (2005) Cannabinoids
and ghrelin have both central and peripheral metabolic and cardiac
effects via AMP-activated protein kinase, J. Biol. Chem.,
280, 25196-25201.
23.Müller, G. A., Herling, A. W., and Wied, S.
(2019) Upregulation of phosphorylation of lipid droplet-associated
proteins in primary rat adipocytes by the cannabinoid receptor 1
antagonist rimonabant, Arch. Physiol. Biochem., in press.
24.Osei-Hyiaman, D., DePetrillo, M., Pacher, P.,
Liu, J., Radaeva, S., Batkai, S., Harvey-White, J., Mackie, K.,
Offertaler, L., Wang, L., and Kunos, G. (2005) Endocannabinoid
activation at hepatic CB1 receptors stimulates fatty acid
synthesis and contributes to diet-induced obesity, J. Clin.
Invest., 115, 1298-1305.
25.Bensaid, M., Gary-Bobo, M., Esclangon, A.,
Maffrand, J. P., Le Fur, G., Oury-Donat, F., and Soubrie, P. (2003) The
cannabinoid CB1 receptor antagonist SR141716 increases Acrp30 mRNA
expression in adipose tissue of obese fa/fa rats and in cultured
adipocyte cells, Mol. Pharmacol., 63, 908-914.
26.Brown, D. A. (2001) Lipid droplets: proteins
floating on a pool of fat, Curr. Biol., 11,
R446-R449.
27.Blanchette-Mackie, E. J., Dwyer, N. K., Barber,
T., Coxey, R. A., Takeda, T., Rondinone, C. M., Theodorakis, J. L.,
Greenberg, A. S., and Londos, C. (1995) Perilipin is located on the
surface layer of intracellular lipid droplets in adipocytes, J.
Lipid Res., 36, 1211-1226.
28.Yeaman, S. J. (2004) Hormone-sensitive lipase
– new roles for an old enzyme, Biochem. J., 379,
11-22.
29.Herling, A. W., Kilp, S., Juretschke, H. P.,
Neumann-Haefelin, C., Gerl, M., and Kramer, W. (2008) Reversal of
visceral adiposity in candy-diet fed female Wistar rats by the CB1
receptor antagonist rimonabant, Int. J. Obes., 32,
1363-1372.
30.Lu, Y., and Anderson, H. D. (2017) Cannabinoid
signaling in health and disease, Can. J. Physiol. Pharmacol.,
95, 311-327.
31.Simon, V., and Cota, D. (2017) Mechanisms in
endocrinology: endocannabinoids and metabolism: past, present and
future, Eur. J. Endocrinol., 176, R309-R324.