Methods for Correction of the Single-Nucleotide Substitution c.840C>T in Exon 7 of the SMN2 Gene
K. R. Valetdinova1,2,3,4,a*, V. S. Ovechkina1,2,3,4, and S. M. Zakian1,2,3,4
1Federal Research Center Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia2Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia
3Meshalkin National Medical Research Centre, Ministry of Healthcare of Russian Federation, 630090 Novosibirsk, Russia
4Novosibirsk State University, 630090 Novosibirsk, Russia
* To whom correspondence should be addressed.
Received April 30, 2019; Revised June 3, 2019; Accepted June 3, 2019
The CRISPR/Cas technology has a great potential in the treatment of many hereditary diseases. One of the prospective models for the CRISPR/Cas-mediated therapy is spinal muscular atrophy (SMA), a disease caused by deletion of the SMN1 gene that encodes the SMN protein required for the survival of motor neurons. SMA patients’ genomes contain either single or several copies of SMN2 gene, which is a paralog of SMN1. Exon 7 of SMN2 has the single-nucleotide substitution c.840C>T leading to the defective splicing and decrease in the amounts of the full-length SMN. The objective of this study was to create and test gene-editing systems for correction of the single-nucleotide substitution c.840C>T in exon 7 of the SMN2 gene in fibroblasts, induced pluripotent stem cells, and motor neuron progenitors derived from a SMA patient. For this purpose, we used plasmid vectors expressing CRISPR/Cas9 and CRISPR/Cpf1, plasmid donor, and 90-nt single-stranded oligonucleotide templates that were delivered to the target cells by electroporation. Although sgRNA_T2 and sgRNA_T3 guiding RNAs were more efficient than sgRNA_T1 in fibroblasts (p < 0.05), no significant differences in the editing efficiency of sgRNA_T1, sgRNA_T2, and sgRNA_T3 was observed in patient-specific induced pluripotent stem cells and motor neuron progenitors. The highest editing efficiency in induced pluripotent stem cells and motor neuron progenitors was demonstrated by the sgRNA_T1 and 90-nt single-stranded oligonucleotide donors.
KEY WORDS: spinal muscular atrophy, induced pluripotent stem cells, motor neuron progenitors, gene editingDOI: 10.1134/S0006297919090104
Abbreviations: iPSCs, induced pluripotent stem cells; MNP, motor neuron progenitor; SMA, spinal muscular atrophy; sgRNA, single guide RNA.
One of the approaches for the treatment of hereditary diseases is
correction of mutations in induced pluripotent stem cells (iPSCs)
derived from the patient’s somatic cells or regional stem cells
using modern gene-editing systems with subsequent autologous
transplantation of “corrected” progenitor cells or their
differentiated derivatives into the patient’s body. Currently,
CRISPR Therapeutics and Vertex Pharmaceuticals (USA) are conducting
phase I/II clinical trials of a similar ex vivo therapy for
β-thalassemia and sickle cell anemia and are expected to start
analogous trials for Duchenne muscular dystrophy and some other
diseases. However, the question whether a CRISPR-mediated system will
be efficient in the treatment of hereditary diseases of the central
nervous system remains open. Spinal muscular atrophy (SMA) occupies a
special place among such diseases. SMA is one of the most common
hereditary diseases that develop in childhood and one of the main
causes of early infant mortality. The most severe form of this disease,
SMA type I, develops during the first months of life and leads to the
patient’s death within two years [1]. In 95%
cases, SMA is caused by a homozygous deletion of the SMN1 gene
coding for the SMN protein that controls various aspects of RNA
metabolism [2]. SMN is also synthesized from the
SMN2 gene, a highly homologous copy of SMN1. Therefore,
SMA patients present a unique situation: SMN1 gene is deleted or
damaged by mutation; however, the paralogous SMN2 gene, which is
almost identical to SMN1 in its nucleotide sequence, is present
in the genome. The difference between these two genes comes to several
single-nucleotide substitutions. One of them, c.840C>T, is located in
exon 7 and results in the loss of exon 7 in most (~90%) mature
SMN2 transcripts that are translated with a formation of
truncated unstable protein unable to support its functions [3]. There are two hypotheses explaining the disruption
mechanism. The first one suggests that mutation in the SMN2 exon
7 disrupts the sequence of the exon splicing enhancer; as a result, the
splicing factors do not recognize and bind to the splicing sites, and
exon 7 is not included in the mature mRNA [4]. The
second hypothesis suggests that the exon splicing silencer is formed,
which prevents exon 7 inclusion in the mature mRNA [5]. Due to the instability of this region, the number
of SMN2 copies can vary considerably. The more copies of
SMN2 are in the genome, the more full-length protein is
synthesized, and therefore, the less are manifestations of disease
symptoms. It was shown that the number of SMN2 copies correlates
with the severity of SMA [6]. Most patients with
SMA type I have two SMN2 copies, patients with SMA type II have
three or four SMN2 copies, and patients with SMA type III have
three or four SMN2 copies. Healthy donors have 1-2 copies of
SMN2 on average.
Despite a large body of data on the pathogenesis and molecular genetics of SMA obtained during the last ten years, the pathogenetic therapy of SMA appeared relatively recently. In 2017, the first treatment for SMA, an antisense oligonucleotide modulating SMN2 splicing, was approved in the USA and EU [7]. However, the desired therapeutic effect requires regular intrathecal injections of the preparation because of its degradation by the cellular systems over time. Furthermore, the drug provides only a relief of disease symptoms and some increase in the patients’ life expectancy, and the long-term effects of treatment have not been studied. The only approach to the etiotropic therapy of SMA today is correction of the c.840C>T substitution in the paralogous gene.
The purpose of this study was to create and test CRISPR-mediated systems for the correction of the single-nucleotide substitution c.840C>T in the SMN2 exon 7 in fibroblasts, induced pluripotent stem cells (IPSCs), and motor neuron progenitors. First, we had to introduce a double-strand break in the immediate vicinity of the substitution, since for efficient recombination, the distance between the DNA break and the region in which the target substitution is located should not exceed 20 nucleotides [8]. For this purpose, single guide RNAs (sgRNAs) for the CRISPR/Cas9 and CRISPR/Cpf1 systems were selected that introduced double-strand breaks as close as possible to the target substitution in order to increase the frequency of homologous recombination. Donor templates, including plasmid vector with a selection cassette and short single-stranded oligonucleotides, were also designed.
MATERIALS AND METHODS
Plasmid design. Plasmid vectors pX552_miCMV_Puro_SMN, pUC19_SMN2_contr, and pUC19_SMN2_mut were designed using the SnapGene software. DNA fragments amplified from the SMN1 and SMN2 genes, fragment 615-818 from the pcDNA 3.1(–) plasmid (Invitrogen, USA), and fragment 6775-7374 from the w-159-1 plasmid (Addgene plasmid #17481; kindly provided by Drs. E. Campeau and P. Kaufman) were used for the assembly. Plasmids pSpCas9(BB)-2A-GFP (pX458) and pY010 (Addgene plasmid #48138 and #69982, respectively) were a gift from Dr. F. Zhang; pTE4560 (Addgene plasmid #107526) was a gift from Dr. E. Welker. Amplification of DNA fragments was performed with Phusion Hot Start II polymerase (Thermo Fisher Scientific, USA) in a C1000 Touch Thermal Cycler (Bio-Rad, USA); the primers are listed in Table 1. Plasmid assembly was performed using standard molecular cloning methods and the Golden Gate technology (NEB, USA). Plasmid clones were sequenced with an ABI 3130XL Genetic Analyzer (Applied Biosystems, USA) at the Genomics Core Facility, Siberian Branch of the Russian Academy of Sciences. Plasmid DNA was isolated using PureLink High Pure Plasmid Midiprep kit (Invitrogen).
Table 1. Primers used in the study

Guide RNA and donor design. sgRNAs and donor sequences were designed using the online resource http://benchling.com.
Cell culture. We used f1SMA fibroblasts and two iPSC lines (ICGi005-A and ICGi005-B) from a patient with SMA type I [9]. Fibroblasts were cultured in DMEM/F-12 with 10% FBS, 0.1 mM non-essential amino acids (NEAA), 1 mM GlutaMax, 100× penicillin/streptomycin (all Gibco, USA). The cells were split at 1 : 3 every five days using TrypLE (Gibco). iPSCs were cultured in KnockOut DMEM with 15% KnockOut Serum Replacement, 0.1 mM NEAA, 1 mM GlutaMax, 100× penicillin/streptomycin (all Gibco), 0.25 mM 2-mercapthoethanol (Sigma, USA) and 10 ng/ml basic fibroblast growth factor (BioLegend, USA). iPSCs were split at 1 : 10 twice a week using TrypLE.
iPSC differentiation into motor neuron progenitors (MNPs). Differentiation of iPSCs into MNPs was carried out according to the protocol of Du et al. [10]. The cells were dissociated with Accutase (Gibco) and split at (1 : 3)-(1 : 4) twice a week.
Transfection of fibroblasts, iPSCs, and MNPs. iPSCs and fibroblasts were transfected with a Neon Transfection System (Thermo Fisher Scientific) according to the manufacturer’s instructions; 5 μg of plasmid DNA or 3 μg of plasmid DNA and 2 μl of 100 μM oligonucleotide donor were mixed with 5·105 cells.
Cell sorting. Cell sorting was performed with a S3e Cell Sorter (Bio-Rad) 48 h after transfection.
DNA isolation and PCR. Genomic DNA was isolated with a Quick-DNA Miniprep Kit (Zymo Research, USA). PCR products were purified with a Zymoclean Gel DNA Recovery Kit (Zymo Research). DNA fragments were amplified using a BioMaster HS-Taq PCR-Color 2x kit (Biolabmix, Russia) with a C1000 Touch Thermal Cycler. PCR conditions were: 95°C for 5 min, 35 cycles of 95°C for 30 s, 59°C for 30 s, 72°C for 30 s; then 72°C for 5 min. The primers are listed in Table 1.
RNA extraction and reverse transcription-quantitative PCR (RT-qPCR). RNA was isolated from the cells using TRIzol Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was synthesized with Super Script III Reverse Transcriptase (Thermo Fisher Scientific). qPCR was performed using BioMaster HS-qPCR SYBR Blue 2× (Biolabmix) in a LightCycler 480 system (Roche, Germany). PCR conditions were: 40 cycles of 95°C for 15 s, 60°C for 1 min. The CT values were normalized to GAPDH expression using the ΔΔCT method.
Immunocytochemistry. Immunofluorescent staining was performed according to the following protocol: fixation in 4% formaldehyde (Sigma) for 15 min at room temperature, permeabilization in 0.5% Triton X-100 (Sigma) for 30 min at room temperature, blocking in 1% BSA (Sigma) for 30 min. Incubation was carried out with primary anti-OLIG2 antibodies (AB9610, 1 : 500) (EMD Millipore, USA) for 16 h at 4°C and then with secondary antibodies Alexa Fluor 568 goat anti-rabbit IgG (H+L) (1 : 400; A11011, Thermo Fisher Scientific) for 1.5-2 h at room temperature. 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma) was used for nuclear staining. The images were analyzed with a NIS-Elements Eclipse Ti-E fluorescence microscope (Nikon, Japan).
Analysis of target and off-target activities of the CRISPR systems. CRISPR-mediated editing of target and off-target sites was evaluated using TIDE (for insertions/deletions) and TIDER (for the c.840C>T substitution) tools.
Statistical analysis. Statistical processing of the obtained data was carried out using the Wilcoxon test and ANOVA with the STATISTICA 10.0 software; p < 0.05 was considered as statistically significant.
RESULTS
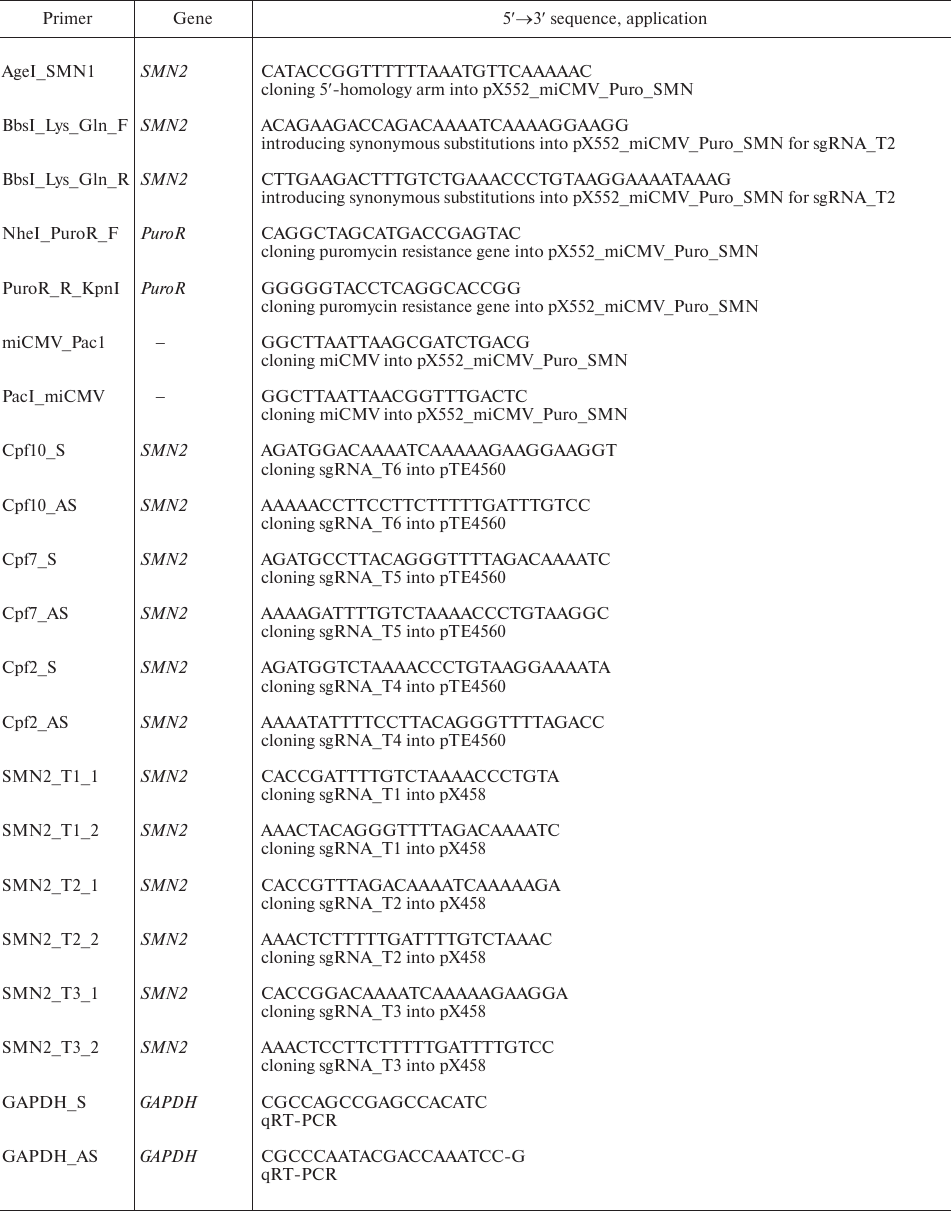
Tool design. Based on the bioinformatic analysis of the SMN2 gene intron 6 and exon 7 sequences, three protospacers for CRISPR/Cas9 (sgRNA_T1, sgRNA_T2, sgRNA_T3) and three protospacers for CRISPR/Cpf1 (sgRNA_T4, sgRNA_T5, sgRNA_T6) were selected, all of them located within a 20-nt distance from the c.840C>T substitution in exon 7 of SMN2 gene (Fig. 1). This choice was dictated by the fact that CRISPR/Cas9 introduces double-strand breaks with blunt ends, while CRISPR/Cpf1 cuts DNA with the formation of sticky ends. Therefore, the recombination efficiency of different types of DNA breaks may differ. In addition, the selected protospacers were complementary to different DNA strands, which is also important because the efficiency of non-homologous end joining was found to be different for guide RNAs complementary to the template and coding DNA strands [11].
Fig. 1. Design of guide RNAs for exon 7 of the SMN2 gene. a) Location of sgRNAs for CRISPR/Cas9. b) Location of sgRNAs for CRISPR/Cpf1. Single-nucleotide c.840C>T substitution in exon 7 of SMN2 gene is shown in red; protospacer adjacent motifs (PAMs) required for the target recognition by CRISPR/Cas9 and CRISPR/Cpf1 are shown in orange.
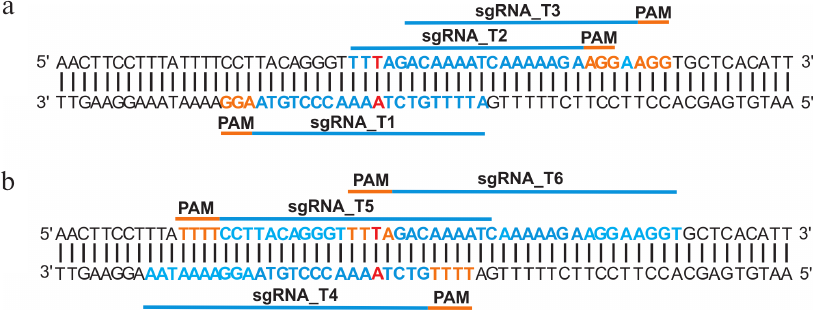
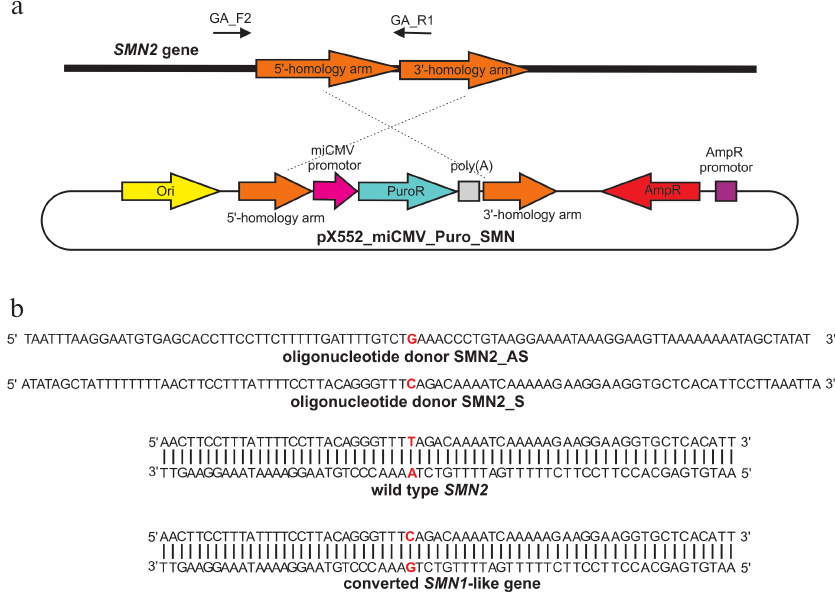
The pX552_miCMV_Puro_SMN donor plasmid for homologous recombination in the SMN2 gene was assembled as follows (Fig. 2a): sequences homologous to the regions flanking the DNA break (the so-called 5′- and 3′-homology arms) were placed on the outsides of the selection cassette carrying the puromycin resistance gene. The single-nucleotide Τ→C substitution was located in the 5′-homology arm, and the selection cassette was supposed to integrate in intron 7 of SMN2 gene after recombination. To prevent re-recognition and re-cutting by CRISPR/Cas9, additional synonymous single-nucleotide substitutions for the sgRNA_T1, sgRNA_T2, and sgRNA_T3 protospacers were introduced in the area immediately adjacent to PAM (protospacer 3′-region, “seed region”). When possible, codon usage frequencies were taken into account. Thus, the following additional substitutions were introduced into the 5′-homology arm in addition to the target Τ→C substitution:
- for T1: TCC→AGC (Ser; codon usage frequency, 17.7 and 19.4, respectively) and TTA→CTA (Leu; codon usage frequency, 7.6 and 7.1, respectively);
- for T2: AAA→AAG (Lys; codon usage frequency, 24.2 and 32.0, respectively) and CAA→CAG (Gln; codon usage frequency, 12.1 and 34.2, respectively);
- for T3: CGA→AGA (Arg; codon usage frequency, 6.2 and 11.9, respectively) and GGA→GGG (Gly; codon usage frequency, 16.4 and 16.5, respectively).
The amplified fragments were cloned into the plasmid vector; the length of the homology arms was ~500 bp (the optimal length for homologous recombination according to the published data [8]).
The following parameters were used for the design of oligonucleotide donors: 90-nt single-stranded oligonucleotides complementary to either the template (SMN1_S) or the coding (SMN1_AS) DNA strand. The T→C substitution was located approximately in the center of the donor oligonucleotide (Fig. 2b).
Fig. 2. a) Donor plasmid pX552_miCMV_Puro_SMN. GA_F2 and GA_R primers were used to verify insertion of the selection cassette. b) Single-stranded oligonucleotide donors; c.840C>T substitution in exon 7 of the SMN2 gene is highlighted in red.
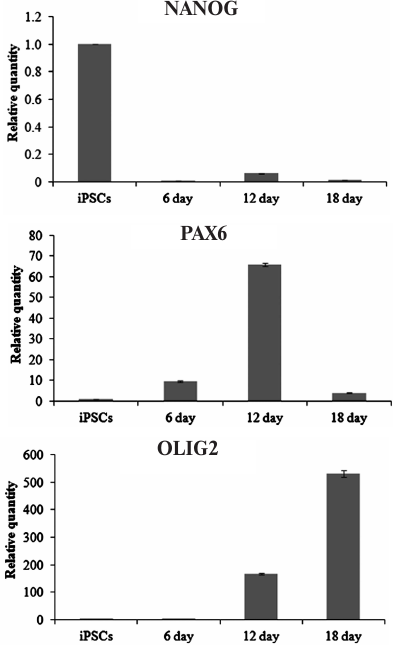
Generation of MNPs. Differentiation of ICGi005-A iPSCs (Fig. 3a) towards MNPs (Fig. 3b) was accompanied by the reduction in the expression of the NANOG gene (one of the main markers of pluripotent stem cells). At the same time, expression of the PAX6 gene (neuroepithelial cell marker) increased on days 6 and 12 of differentiation and expression of the OLIG2 gene (coding for the MNP transcription factor) increased on days 12 and 18 of differentiation (Fig. 4). On day 18, PAX6 expression decreased, which can be explained by the fact that by this time, most of the neuroepithelial cells reached the stage of MNPs, as evidenced by a more than a 500-fold increase in the OLIG2 expression. Immunocytochemical analysis confirmed expression of the major neuroepithelial markers PAX6 and SOX1, as well as the MNP marker OLIG2 (Fig. 3, c and d).
The advantage of MNPs is that these cells can be cultured for 10 passages or more without losing high proliferative activity, OLIG2 expression, and therefore, the ability to further differentiate toward mature motor neurons, which are the major type of cells suffering in SMA.
Fig. 3. Differentiation of iPSCs into MNPs. a) iPSC ICGi005-A on day 0 of differentiation. b) MNPs on day 12 of differentiation. c) Expression of neuroepithelial precursor markers PAX6 (red) and SOX1 (green) on day 6 of differentiation. d) Expression of the MNP marker OLIG2 (red) on day 12 of differentiation. Nuclei are stained with DAPI (blue). Scale bar, 200 μm.
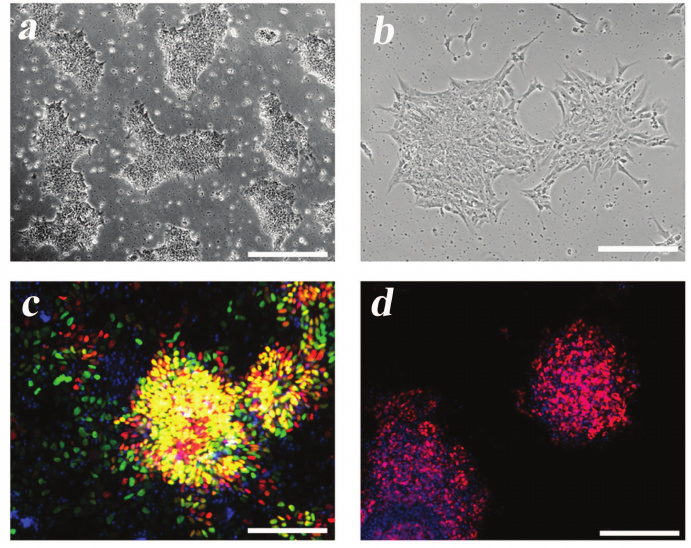
Fig. 4. Expression of NANOG, PAX6, and OLIG2 during iPSC differentiation into MNPs.
Comparative evaluation of the SMN2 gene editing efficiency by CRISPR-mediated systems. To assess the efficiency of CRISPR/Cas9 in the SMN2 gene editing, f1SMA fibroblasts from a patient with SMA type I, iPSCs ICGi005-A derived from the f1SMA fibroblasts, and MNPs obtained by differentiation of ICGi005-A cells were transfected with the pX458 plasmid expressing Cas9 protein, one of the guide RNAs (sgRNA_T1, sgRNA_T2, or sgRNA_T3), and GFP (green fluorescent protein).
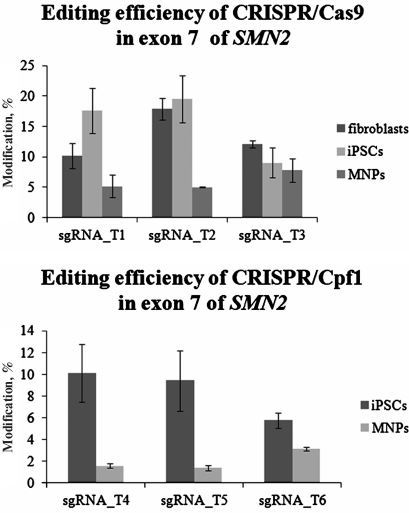
The repair of double-strand DNA breaks by the non-homologous end joining generates small insertions and deletions, the number of which can be used to analyze the editing efficiency of a CRISPR system. One of the simplest and most reliable approaches for quantitative assessment of genome editing is the use of the TIDE web tool for analysis of the sequencing patterns of PCR products obtained after delivery of the CRISPR system into the cells [12]. PCR products obtained from all experimental DNA samples were Sanger sequenced and analyzed using TIDE (Fig. 5). In fibroblasts, the editing activity of sgRNA_T2 was higher than the editing activities of sgRNA_T1 (p < 0.05) and sgRNA_T3 (p < 0.05). However, no significant differences in the editing efficiencies of all three guide RNAs were observed in the patient-specific iPSCs and MNPs. Comparison of the total CRISPR/Cas9 activity for the SMN2 gene exon 7 in different cell types showed that the editing efficiency was higher in fibroblasts and iPSCs than in MNPs (p < 0.05). The highest CRISPR/Cas9 activity in fibroblasts, iPSCs, and MNPs was found with the sgRNA_T2 (trend at p = 0.097).
To assess the editing efficiency of the CRISPR/Cpf1 system in iPSCs ICGi005-A and MNPs, the cells were transfected with the pY010 plasmid expressing Cpf1 protein and the pTE4560 plasmid expressing one of the guide RNAs (sgRNA_T4, sgRNA_T5, sgRNA_T6) and mCherry red fluorescent protein. We found no significant differences in the editing efficiencies of sgRNA_T4, sgRNA_T5, and sgRNA_T6 in iPSCs and MNPs. However, the editing efficiency of CRISPR/Cpf1 was higher in iPSCs than in MNPs (p < 0.05).
Fig. 5. Editing efficiency of CRISPR/Cas9 and CRISPR/Cpf1 in the SMN2 gene exon 7 as analyzed by TIDE.
Comparative evaluation of the off-target activity of selected protospacers. To identify protospacers with the smallest number of off-target sites in the genome, we used bioinformatic analysis. Modern bioinformatic tools for identification of potential off-target sites for CRISPR/Cas9 use different algorithms. Comparison of these algorithms revealed that, although the protospacers suggested by different tools for a given genome region are the same, the number of identified potential off-target sites may vary [13]. It should be also noted that most of these tools reveal off-target sites containing single nucleotide substitutions relative to the target site; however, several studies have shown that off-target double-strand breaks can occur in sites containing insertions or deletions [14, 15]. Hence, we used several currently available bioinformatic tools to identify potential off-target sites: Cas-OFFinder and COSMID. The sequences of sgRNA_T1, sgRNA_T2, sgRNA_T3, sgRNA_T4, sgRNA_T5, and sgRNA_T6 protospacers, as well as complete human genome, were used as the input information.
Each algorithm identifies off-target sites containing nucleotide substitutions, insertions, and deletions relative to the sequence of the analyzed protospacer; however, Cas-OFFinder shows a total number of off-target sites without ranking the substitutions according to the probability of their occurrence. It is known that CRISPR/Cas9 most likely binds to the sites with substitutions located in the protospacer 5′-region, while substitutions in the 3′-region prevent the recognition of the DNA segment as a potential target [16]. Cas-OFFinder is more suitable for quick identification and comparison of the total number of off-target sites and substitutions in them. It also allows to evaluate the specificity of the selected CRISPR/Cas9 protospacers [13]. Using this tool, we determined the total number of off-target sites with the minimal number of replacements equal to 3: 24, 72 and 115 off-target sites for sgRNA_T1, sgRNA_T2, and sgRNA_T3, respectively. Therefore, it seemed more reliable to use sgRNA_T1, because the total number of off-target sites for this protospacer was less than for sgRNA_T2 and sgRNA_T3.
COSMID assigns a rank to each off-target site – the lower the rank, the higher the probability of the off-target effect (the target site has a rank of 0.00). After analysis, we obtained a list of the most likely off-target sites containing no more than four substitutions and/or insertions/deletions of no more than 2 nt. The smallest number of the off-target sites was found for sgRNA_T1, the largest – for sgRNA_T3. However, the majority of potential off-target sites for CRISPR/Cas9 had a very high rank and were located in the non-coding regions and intergenic gaps. Therefore, only those sites that were located in the coding regions were selected from the obtained list (Table 2). Then, we analyze the activity of CRISPR/Cas9 at these sites in fibroblasts, iPSCs, and MNPs using TIDE (as described above). As a result, no insertions/deletions were detected in these DNA sites.
Table 2. Off-target sites for CRISPR/Cas9 in
genome coding regions
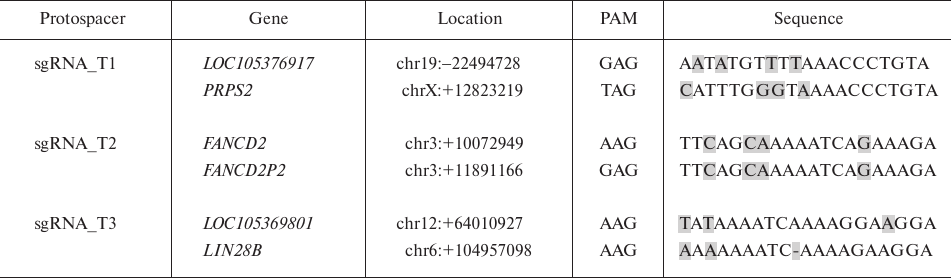
Note: Substitutions in the off-target sites are highlighted in grey.
Similar analysis was carried out to assess potential off-target activity of CRISPR/Cpf1, which was found to be significantly lower than that of CRISPR/Cas9, since the guide RNA for CRISPR/Cpf1 is longer. Using the Cas-OFFinder software, three potential off-target sites were found for sgRNA_T4, 0 sites– for sgRNA_T5, and 5 sites – for sgRNA_T6. No off-target sites were found in genome coding regions; hence, no experimental assessment of the CRISPR/Cpf1 off-target activity in the most probable genome sites was performed.
Therefore, all tested protospacers for the CRISPR/Cpf1 system and sgRNA_T1 and sgRNA_T2 for CRISPR/Cas9 were found to be optimal in terms of potential off-target activity.
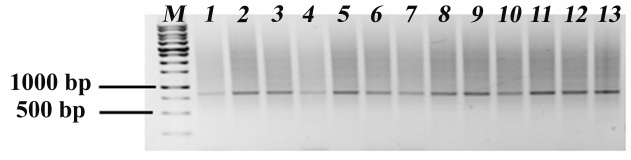
Evaluation of the efficiency of the CRISPR-mediated systems in the correction of the single-nucleotide c.840C>T substitution in exon 7 of the SMN2 gene. In order to evaluate the efficiency of the editing system to make the single-nucleotide T>C substitution in exon 7 of the SMN2 gene, f1SMA fibroblasts were transfected with the pX458 plasmid expressing Cas9 and sgRNA_T2 and the donor plasmid pX552_miCMV_Puro_SMN1. Forty-eight hours after transfection, GFP-positive (and therefore, expressing components of the CRISPR/Cas9 system) cells were selected using a cell sorter. Forty-eight hours after this procedure, puromycin was added to the culture medium at a concentration of 850 ng/ml for 4 days to select recombinant cells with the integrated selection cassette, i.e., cell that underwent homologous recombination. Insertion of the selection cassette was verified by PCR: in the case of insertion, the product of PCR with the primers, one of which was complementary to the sequence outside the 5′-homology arm and the other one – to the sequence within the 3′-homology arm, should be longer by the size of the selection cassette (1664 vs. 850 bp). However, PCR analysis of the mixed cell population obtained after selection revealed only the 850-bp (wild-type) products (Fig. 6). TIDE analysis of these products demonstrated that the editing efficiency was 27.7% (as estimated from the number of insertions/deletions).
Taking into account the obtained data, similar experiment was carried out in iPSCs ICGi005-A. In this case, selection was performed in two rounds using puromycin concentration of 250 ng/ml. The resulting subclones and mixed cell population were analyzed separately. According to the PCR of mixed cell population and individual clones obtained by selection, only wild-type products were detected (Fig. 6). TIDE analysis of the mixed population showed that the editing efficiency was 23%.
Fig. 6. PCR analysis for the presence of selection cassette. Lanes: M) markers; 1) mixed population of f1SMA fibroblasts; 2) mixed population of iPSCs ICGi005-A; 3-13) individual iPSC ICGi005-A clones.
Because of ambiguous results obtained in the experiments with the plasmid donor, we decided to use single-stranded oligonucleotide donors containing the target single-nucleotide substitution. Analogous experiment was carried out using a vector expressing sgRNA_T1 and SMN_AS/SMN_S oligonucleotides. According to TIDER (modified version of TIDE), the average number of T>C substitutions in the target locus with the SMN_S oligonucleotide was 13.7 ± 1.7%; the number of targeted substitutions with the SMN_AS antisense sequence of the donor oligonucleotide was 15.25 ± 0.35%. Comparable editing efficiency values were obtained in a similar experiment with MNPs: 15.9 ± 2.40% for the sense sequence and 16.35 ± 1.34% for the antisense sequence.
DISCUSSION
In this work, we analyzed the activity of CRISPR/Cas9 and CRISPR/Cpf1 in exon 7 of the SMN2 gene in fibroblasts, iPSCs, and MNPs obtained from a patient with SMA. Despite the fact that sgRNA_T1 and sgRNA_T2/sgRNA_T3 were complementary to the template and coding DNA strands, respectively, significant differences in their editing efficiency were found only in the fibroblasts. No significant differences in the repair efficiency by the non-homologous end joining with different guide RNAs were observed in iPSCs and MNPs, which are of most interest as precursors of motor neurons. However, comparison of the editing efficiency of CRISPR/Cas9 and CRISPR/Cpf1 in different cell types showed that the editing efficiency in MNPs was significantly lower than in iPSCs. In all likelihood, this might be explained by the specific properties of these cells or by the non-optimal delivery of the plasmid construct to MNPs.
Evaluation of potential off-target sites showed that sgRNA_T3 had the greatest risk of the off-target effects: the genome contains two sites in the LIN28B and LOC105369801 genes that differ only by three bases from the target sequence, with two bases located in the 5′-region of the protospacer. For sgRNA_T1 and sgRNA_T2, the probability of damage to the coding regions is rather low, which was experimentally confirmed. In the case of CRISPR/Cpf1 system, the probability of the off-target site damage was much lower. In addition, reduction of off-target activity can be achieved by introducing modified variants of Cas9, as well as using short-lived RNA–protein complexes of sgRNA-Cas9 instead of plasmid vectors [8].
Since application of human genome editing tools is related to personalized medicine, selection of potential targets for CRISPR/Cas9 should take into account genetic polymorphisms. Analysis of the protospacers used in this work with the CRISPOR resource (http://crispor.tefor.net/) and the hg19 version of human genome found that one of the polymorphisms (G substitution with C) eliminates PAM adjacent to the most efficient protospacer sgRNA_T2 and, being located in the critical region, most likely makes sgRNA_T3 inactive. The occurrence frequency of this polymorphism is ~1%. Therefore, only sgRNA_T1 can be used in this case for correction of the c.840C>T substitutions.
The use of plasmid donor for correction of the single-nucleotide c.840C>T substitution in exon 7 of the SMN2 gene demonstrated a high probability of off-target insertions with the low probability of target insertions, which might be related to the circular shape of the plasmid molecule. A number of studies have demonstrated a higher efficiency of linearized plasmid donors containing only homology arms and selection cassette and lacking other elements of the donor plasmid [17]. However, we found that the use of single-stranded oligonucleotide donors ensures efficient introduction of the target T>C substitution into exon 7 of the SMN2 gene in iPSC and MNPs, which is consistent with the evidence that short oligonucleotide donors are preferable for recombination aimed to introduce single-nucleotide substitutions [8].
Funding. This work was supported by the Russian Science Foundation (project 17-75-10041).
Acknowledgements. The authors are grateful to A. A. Nemudryi for help in assembling pX552_miCMV_Puro_SMN.
Conflict of interest. The authors declare no conflict of interest.
Ethical approval. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
REFERENCES
1.Verhaart, I. E., Robertson, A., Leary, R.,
McMacken, G., Konig, K., Kirschner, J., Jones, C. C., Cook, S. F., and
Lochmuller, H. (2017) A multi-source approach to determine SMA
incidence and research ready population, J. Neurol., 264,
1465-1473, doi: 10.1007/s00415-017-8549-1.
2.Lefebvre, S., Burglen, L., Reboullet, S., Clermont,
O., Burlet, P., Viollet, L., Benichou, B., Cruaud, C., Millasseau, P.,
Zeviani, M., Le Paslier, D., Frezal, J., Cohen, D., Weissenbach, J., Munnich, A., and Melki, J. (1995) Identification and
characterization of a spinal muscular atrophy-determining gene,
Cell, 80, 155-165, doi: 0092-8674(95)90460-3.
3.Monani, U. R., Lorson, C. L., Parsons, D. W.,
Prior, T. W., Androphy, E. J., Burghes, A. H., and McPherson, J. D.
(1999) A single nucleotide difference that alters splicing patterns
distinguishes the SMA gene SMN1 from the copy gene SMN2,
Hum. Mol. Genet., 8, 1177-1183.
4.Cartegni, L., and Krainer, A. R. (2002) Disruption
of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes
spinal muscular atrophy in the absence of SMN1, Nat.
Genet., 30, 377-384, doi: 10.1038/ng854.
5.Kashima, T., and Manley, J. L. (2003) A negative
element in SMN2 exon 7 inhibits splicing in spinal muscular
atrophy, Nat. Genet., 34, 460-463, doi:
10.1038/ng1207.
6.McAndrew, P. E., Parsons, D. W., Simard, L. R.,
Rochette, C., Ray, P. N., Mendell, J. R., Prior, T. W., and Burghes, A.
H. (1997) Identification of proximal spinal muscular atrophy carriers
and patients by analysis of SMNT and SMNC gene copy
number, Am. J. Hum. Genet., 60, 1411-1422, doi:
10.1086/515465.
7.Gidaro, T., and Servais, L. (2019) Nusinersen
treatment of spinal muscular atrophy: current knowledge and existing
gaps, Dev. Med. Child Neurol., 61, 19-24, doi:
10.1111/dmcn.14027.
8.Liang, X., Potter, J., Kumar, S., Ravinder, N., and
Chesnut, J. D. (2017) Enhanced CRISPR/Cas9-mediated precise genome
editing by improved design and delivery of gRNA, Cas9 nuclease, and
donor DNA, J. Biotechnol., 241, 136-146, doi:
10.1016/j.jbiotec.2016.11.011.
9.Valetdinova, K. R., Maretina, M. A., Kuranova, M.
L., Grigor’eva, E. V., Minina, Y. M., Kizilova, E. A., Kiselev,
A. V., Medvedev, S. P., Baranov, V. S., and Zakian, S. M. (2019)
Generation of two spinal muscular atrophy (SMA) type I patient-derived
induced pluripotent stem cell (iPSC) lines and two SMA type II
patient-derived iPSC lines, Stem Cell Res., 34, 101376,
doi: 10.1016/j.scr.2018.101376.
10.Du, Z. W., Chen, H., Liu, H., Lu, J., Qian, K.,
Huang, C. L., Zhong, X., Fan, F., and Zhang, S. C. (2015) Generation
and expansion of highly pure motor neuron progenitors from human
pluripotent stem cells, Nat. Commun., 6, 6626, doi:
10.1038/ncomms7626.
11.Clarke, R., Heler, R., MacDougall, M. S., Yeo, N.
C., Chavez, A., Regan, M., Hanakahi, L., Church, G. M., Marraffini, L.
A., and Merrill, B. (2018) Enhanced bacterial immunity and mammalian
genome editing via RNA-polymerase-mediated dislodging of Cas9 from
double strand DNA breaks, Mol. Cell, 71, 42-55, doi:
10.1016/j.molcel.2018.06.005.
12.Brinkman, E. K., Chen, T., Amendola, M., and van
Steensel, B. (2014) Easy quantitative assessment of genome editing by
sequence trace decomposition, Nucleic Acids Res., 42,
e168, doi: 10.1093/nar/gku936.
13.Ishida, K., Gee, P., and Hotta, A. (2015)
Minimizing off-target mutagenesis risks caused by programmable
nucleases, Int. J. Mol. Sci., 16, 24751-24771, doi:
10.3390/ijms161024751.
14.Ran, F. A., Cong, L., Yan, W. X., Scott, D. A.,
Gootenberg, J. S., Kriz, A. J., Zetsche, B., Shalem, O., Wu, X.,
Makarova, K. S., Koonin, E. V., Sharp, P. A., and Zhang, F. (2015)
In vivo genome editing using Staphylococcus aureus Cas9,
Nature, 520, 186-191, doi: 10.1038/nature14299.
15.Lin, Y., Cradick, T. J., Brown, M. T., Deshmukh,
H., Ranjan, P., Sarode, N., Wile, B. M., Vertino, P. M., Stewart, F.
J., and Bao, G. (2014) CRISPR/Cas9 systems have off-target activity
with insertions or deletions between target DNA and guide RNA
sequences, Nucleic Acids Res., 42, 7473-7485, doi:
10.1093/nar/gku402.
16.Nemudryi, A. A., Valetdinova, K. R., Medvedev, S.
P., and Zakian, S. M. (2014) TALEN and CRISPR/Cas genome editing
systems: tools of discovery, Acta Naturae, 6, 19-40.
17.Song, F., and Stieger, K. (2017) Optimizing the
DNA donor template for homology-directed repair of double-strand
breaks, Mol. Ther. Nucleic Acids, 7, 53-60, doi:
10.1016/j.omtn.2017.02.006.