Rapamycin Is Not Protective against Ischemic and Cisplatin-Induced Kidney Injury
N. V. Andrianova1,2, L. D. Zorova2,3, V. A. Babenko2,3, I. B. Pevzner2,3, V. A. Popkov2,3, D. N. Silachev2,3, E. Y. Plotnikov2,3,4,a*, and D. B. Zorov2,3,b*
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119992 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia
3Kulakov National Medical Research Center of Obstetrics, Gynecology and Perinatology, 117997 Moscow, Russia
4Sechenov First Moscow State Medical University, Institute of Molecular Medicine, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received June 24, 2019; Revised July 12, 2019; Accepted July 12, 2019
Autophagy plays an important role in the pathogenesis of acute kidney injury (AKI). Although autophagy activation was shown to be associated with an increased lifespan and beneficial effects in various pathologies, the impact of autophagy activators, particularly, rapamycin and its analogues on AKI remains obscure. In our study, we explored the effects of rapamycin treatment in in vivo and in vitro models of ischemic and cisplatin-induced AKI. The impact of rapamycin on the kidney function after renal ischemia/reperfusion (I/R) or exposure to the nephrotoxic agent cisplatin was assessed by quantifying blood urea nitrogen and serum creatinine and evaluating the content of neutrophil gelatinase-associated lipocalin, a novel biomarker of AKI. In vitro experiments were performed on the primary culture of renal tubular cells (RTCs) that were subjected to oxygen-glucose deprivation (OGD) or incubated with cisplatin under various rapamycin treatment protocols. Cell viability and proliferation were estimated by the MTT assay and real-time cell analysis using an RTCA iCELLigence system. Although rapamycin inhibited mTOR (mammalian target of rapamycin) signaling, it failed to enhance the autophagy and to ameliorate the severity of AKI caused by ischemia or cisplatin-induced nephrotoxicity. Experiments with RTCs demonstrated that rapamycin exhibited the anti-proliferative effect in primary RTCs cultures but did not protect renal cells exposed to OGD or cisplatin. Our study revealed for the first time that the mTOR inhibitor rapamycin did not prevent AKI caused by renal I/R or cisplatin-induced nephrotoxicity and, therefore, cannot be considered as an ideal mimetic of the autophagy-associated nephroprotective mechanisms (e.g., those induced by caloric restriction), as it had been suggested earlier. The protective action of such approaches like caloric restriction might not be limited to mTOR inhibition and can proceed through more complex mechanisms involving alternative autophagy-related targets. Thus, the use of rapamycin and its analogues for the treatment of various AKI forms requires further studies in order to understand potential protective or adverse effects of these compounds in different contexts.
KEY WORDS: rapamycin, acute kidney injury, ischemia, cisplatin, renal tubular cells, autophagy, nephroprotectionDOI: 10.1134/S0006297919120095
Abbreviations: AKI, acute kidney injury; BUN, blood urea nitrogen; DMSO, dimethyl sulfoxide; DPBS, Dulbecco’s phosphate-buffered saline; EGF, epidermal growth factor; FBS, fetal bovine serum; i.p., intraperitoneal (injection); I/R, ischemia/reperfusion; LC3, microtubule-associated protein 1A/1B, light chain 3; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium; NGAL, neutrophil gelatinase-associated lipocalin; OGD, oxygen-glucose deprivation; RTCs, renal tubular cells; SCr, serum creatinine.
Autophagy was originally defined as a catabolic process aimed at
clearing the cell interior from waste products, thus enabling better
survival of cells and organs under normal and adverse physiological
conditions. Nowadays, it is commonly believed that autophagy also
contributes to the pathogenesis of various diseases, such as cancer,
neurodegenerative disorders, myopathies, and infectious diseases [1, 2]. Recent data have
demonstrated that aging is associated with impairments in the autophagy
of mitochondria (mitophagy). Normalization of mitophagy in elderly
organisms in order to restore the pool of functional mitochondria is a
promising approach to the reversal of the age-related decline in organ
functioning that could be further used in the development of anti-aging
strategies [3, 4]. Autophagy
and mitophagy were found to play an important role in numerous renal
pathologies. However, it remains controversial whether autophagy has an
exclusively pro-survival role during acute kidney injury (AKI) or it
can also trigger the pro-apoptotic signaling under particular
conditions.
Activation of autophagy and an increase in the number of autophagosomes have been shown in different models of AKI. For instance, autophagy activation in renal cells was demonstrated both in vitro and in vivo in the model of cisplatin-induced nephrotoxicity [5, 6]. Ischemia/reperfusion (I/R) is also accompanied by an increase in the number of autophagosomes in the kidney tissue [7, 8]. Autophagy activation in the septic AKI caused by lipopolysaccharide injection [9, 10] or intestinal ligation and perforation [11] has been well documented. Radiocontrast-induced AKI in mice was also accompanied by autophagy activation [12].
Results obtained in various experimental models using mice with a deficiency for the genes responsible for autophagosome formation in the kidney proximal tubules (e.g., Atg5 and Atg7) suggest that autophagy activation is a protective mechanism against ischemia-, sepsis-, and cisplatin-induced AKI or hyperuricemic AKI [7, 13, 14]. Recently, we have shown that caloric restriction in young rats is associated with the activation of autophagy, resulting in significant protection of the kidneys against I/R injury [15].
From the pharmacological viewpoint, it is important to develop drugs that would activate autophagy signaling and mimic the protective effects of caloric restriction or other autophagy-promoting mechanisms. It was found that some of the autophagy-stimulating molecules not only prolong the lifespan but also affect the pathogenesis of some diseases. In these cases, autophagy is seen as a protective mechanism [16]. One of such compounds is rapamycin, which is believed to extend healthy lifespan while preventing age-related diseases by slowing down the process of aging [17]. Theoretically, rapamycin could be used to protect kidney tissue from AKI via activation of autophagy in renal cells. However, the data on this possible application of rapamycin is limited and controversial, highlighting the need for further research.
In this study, we investigated the effects of rapamycin (Rapa) in the two most common models of AKI, renal I/R injury and cisplatin-induced nephrotoxicity, using in vivo (young rats) and in vitro (primary cultures of renal tubular cells) models.
MATERIALS AND METHODS
Animals. Outbred young male rats (age, 3-4 month; body weight, 300-400 g) were used in the study. The rats were randomly divided into the following experimental groups with 5-6 animals in each group: sham (control for I/R); vehicle control (for cisplatin treatment); Rapa; I/R; Rapa+I/R; Cisplatin; Rapa+cisplatin. The animals had unlimited access to food and water and were kept in cages in a temperature-controlled environment (20 ± 1°C) under the 12/12 h light/dark regime.
Kidney I/R protocol and cisplatin treatment. For I/R, the rats were anesthetized with chloral hydrate (300 mg/kg body weight, i.p.) and subjected to 40-min warm ischemia of the left kidney as previously described [18]. Briefly, the renal vascular bundle was occluded with a non-traumatic microvascular clip for 40 min. Circulation was restored by removing the clip; the lack of blood flow during ischemia and its restoration during reperfusion were assessed visually. Nephrectomy of the right kidney was performed simultaneously with ischemia of the left kidney. During the surgery, the rat body temperature was maintained at 37 ± 0.5°C. Sham-operated animals were subjected to the same procedures except the microvascular clamp was not applied.
Rats exposed to I/R were treated with rapamycin (Temsirolimus, Abcr GmbH, Germany) 1 h before the ischemia induction. Rapamycin was dissolved in dimethyl sulfoxide (DMSO, vehicle) at a concentration of 1 mg/ml. The in vivo concentration of rapamycin was 1 mg/kg body weight, which was achieved by i.p. injection of 0.1 ml rapamycin solution per 100 g body weight.
Cisplatin-induced nephrotoxicity was triggered by administering a single dose of cisplatin (15 mg/kg; TevaGuard, Netherlands) by i.p. injection. The rats were injected with the rapamycin solution in DMSO 3 h before and 24 h after cisplatin injection; in vivo concentration of rapamycin was 1 mg/kg body weight. The control group was injected with DMSO alone at the same time points.
Blood samples were taken from the carotid artery 48 h after I/R or cisplatin injection to determine the levels of blood urea nitrogen (BUN) and serum creatinine (SCr) using the AU480 Chemistry System (Beckman Coulter, USA). The rats were sacrificed by decapitation.
Another group of rats was exposed to a single i.p. injection of rapamycin solution in DMSO (1 mg/kg) either 3 or 24 h before the kidney excision. Control rats were injected with 0.1 ml DMSO per 100 g body weight (vehicle control).
Western blotting. Urine samples were obtained 24 h after I/R or 48 h after induction of cisplatin nephrotoxicity, centrifuged at 10,000g, and mixed with the sample buffer. Kidneys were excised from the rapamycin-treated rats 3 or 24 h after rapamycin injection and homogenized at 4°C with a glass-Teflon homogenizer in PBS containing 10 mM phenylmethylsulfonyl fluoride.
The samples were loaded onto 15% Tris-glycine polyacrylamide gel (20 µl/lane for urine samples and 10 μg protein/lane for kidney homogenates), fractionated by electrophoresis, and transferred onto PVDF membranes (Sigma-Aldrich, USA). The membranes were blocked by 5% fat-free milk in PBS with 0.05% Tween-20 and incubated with primary anti-NGAL (neutrophil gelatinase-associated lipocalin) monoclonal rabbit antibody (1 : 1000; Abcam, UK), anti-p70-S6K monoclonal rabbit antibody (1 : 1000; Cell Signaling, USA), anti-phospho (Thr389)-p70-S6K monoclonal rabbit antibody (1 : 1000; Cell Signaling), anti-LC3 A/B monoclonal rabbit antibody (1 : 1000; Cell Signaling), and anti-β-actin monoclonal mouse antibody (1 : 2000; Sigma-Aldrich). Then the membranes were incubated with anti-rabbit or anti-mouse IgG conjugated with horseradish peroxidase (1 : 7500; Jackson ImmunoResearch, UK); proteins were detected using a Chemidoc MP system (BioRad, USA). The intensity of protein bands was analyzed with ImageJ software (NIH, USA) [19]. Protein concentration in the kidney samples was determined by the bicinchoninic acid assay (Sigma-Aldrich).
For staining with the anti-phospho (Thr389)-p70-S6K antibody, the membranes were blocked with the membrane blocking agent RPN2125V (Amersham, UK). All antibodies and blocking solutions were prepared in TBS (Tris-buffered saline, pH 7.6).
Primary culture of renal tubular cells (RTCs). Rat kidneys were excised under aseptic conditions; kidney cortex was washed, ground, and dissociated with type II collagenase (0.25% w/v; Thermo Fisher Scientific, USA) for 30 min at 37°C. Pieces of undigested tissue were removed; the suspension was pipetted and centrifuged for 5 min at 100g to pellet kidney tubules. The pellet was resuspended in complete culture medium [DMEM/F12 supplemented with 10% fetal bovine serum (FBS; Invitrogen, USA) and epidermal growth factor (EGF, 10 ng/ml; Invitrogen)] and seeded onto culture dishes or 96-well plates at a density of 105 cells/ml. The cells were kept at 37°C in 5% CO2 in a humidified incubator; the medium was replaced after 48 h in order to eliminate non-adherent cells and residual cell fragments. RTCs formed a confluent monolayer on day 4; hence, all experiments were carried out on days 2-3 after plating.
To induce ischemia in vitro, the cells were subjected to oxygen-glucose deprivation (OGD) for 18 h. For this, the culture medium was replaced with Dulbecco’s phosphate-buffered saline (DPBS) saturated with N2, and the cells were placed in a Galaxy 170R multi-gas incubator (Eppendorf/NewBrunswick, UK) with 1% O2 content. After OGD, DPBS was replaced with complete culture medium under normal O2 incubation conditions. RTCs were treated with 0.5 μM rapamycin for 6 h before OGD, during OGD, or for 24 h after OGD. Control cells were treated with 0.5 μM rapamycin for 24 h before the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium] assay. Cells treated with DMSO added at the same final dilution (1 : 2000) were used as vehicle control.
To evaluate the cisplatin cytotoxicity in vitro, the cells were incubated for 24 h with various cisplatin concentrations (6.25, 12.5, and 25 μM) in DMEM/F12. The effects of rapamycin were evaluated under two experimental protocols: 1) pretreatment with 0.5 μM rapamycin for 3 h before 24-h cisplatin treatment, and 2) simultaneous treatment with 0.5 μM rapamycin and above-mentioned concentrations of cisplatin for 24 h.
Cell viability was evaluated by the MTT assay. Cells cultured in 96-well plates and exposed to various stimuli were incubated with the MTT solution (5 mg/ml in DMEM/F12) for 120 min at 37°C. Then MTT solution was removed, 50 μl DMSO was added to each well, and the absorbance at 540 nm was measured with a Zenyth universal microplate reader (Anthos Labtec, Austria).
Real-time monitoring of cell proliferation. Cell growth kinetics was analyzed with an RTCA iCELLigence™ instrument (ACEA, USA). The method is based on recording electrical impedance of cell-covered electrodes [20] and can be used in the studies of RTCs proliferation and death [21]. An RTCA iCELLigence apparatus was placed in a humidified incubator with 5% CO2 at 37°C. RTCs were seeded on 8-well plates with microelectrodes; the medium was changed after 48 h to remove unattached and dead cells. The cells were incubated with 0.5, 1, or 2 μM rapamycin dissolved in complete culture medium for 24 h and then subjected to OGD for 18 h. After OGD, DPBS was replaced with the culture medium with a normoxic O2 content. Cell proliferation rate was evaluated within 48 h after OGD completion.
Confocal microscopy. The number of autophagosomes was analyzed using an LSM510 inverted confocal microscope (Carl Zeiss, Germany). The RTCs culture was obtained as described above and incubated for 3 h with 0.5 μM rapamycin, 30 mM chloroquine (Sigma-Aldrich), or simultaneously with 0.5 μM rapamycin and 30 mM chloroquine dissolved in complete medium. DMSO at a corresponding dilution (1 : 2000) was used as vehicle control. Rapamycin, chloroquine, and DMSO were dissolved in complete culture medium (DMEM/F12 with 10% FBS and 10 ng/ml EGF). At the end of the incubation, RTCs were loaded with 2 μM Cyto-ID (Enzo Life Sciences, USA) and imaged at 500-530 nm (excitation, 488 nm) with a pinhole setting at 150 μm. The mean intensity of Cyto-ID fluorescence was calculated with the ImageJ software.
Statistics. The data are presented as mean ± SEM; the equality of variances was assessed with the Levene’s test. Comparisons between groups were made using the Mann–Whitney U-test. Statistical analysis was performed using the SciPy Python library and Microsoft Excel.
RESULTS
Effects of rapamycin on ischemic and cisplatin-induced AKI. We used kidney I/R as a conventional model of AKI. The severity of renal failure was evaluated based on BUN and SCr values. I/R caused almost a 6-fold increase in BUN (from 7.1 ± 0.2 to 42.2 ± 4.7 mM; n = 6) 48 h after reperfusion (Fig. 1a). Similar increase (from 47.0 ± 1.7 to 326.8 ± 54.3 μM) was observed for SCr (Fig. 1b). Moreover, Western blot analysis of the alternative AKI biomarker NGAL revealed its significant increase (approximately, 100-fold) in the urine 24 h after I/R (Fig. 1c). Administration of rapamycin 1 h before I/R did not affect the changes in the levels of BUN, SCr, and NGAL (n = 5, Fig. 1, a-c), indicating the absence of any protective effect of the drug.
Fig. 1. Evaluation of kidney function using serum and urine biomarkers in rats subjected to I/R or cisplatin treatment. a, b) Concentrations of BUN and SCr 48 h after I/R alone (I/R, n = 6) and I/R with rapamycin (1 mg/kg) pretreatment (Rapa + I/R, n = 5). Sham-operated rats were used as controls. c) Urine NGAL levels measured by Western blotting 24 h after I/R alone or I/R with rapamycin (1 mg/kg) pretreatment. d, e) Concentrations of BUN and SCr 48 h after injection of cisplatin (15 mg/kg) alone (Cisplatin, n = 6) or cisplatin with rapamycin (1 mg/kg) administration (Rapa + cisplatin) 3 h before and 24 h after cisplatin injection (n = 5). f) Urine NGAL levels measured by Western blotting 48 h after injection of cisplatin alone or cisplatin with rapamycin (1 mg/kg) treatment 3 h before and 24 h after cisplatin injection. Rats injected with DMSO only at the same time points were used as vehicle control; * p < 0.05 compared to sham-operated or vehicle control rats.
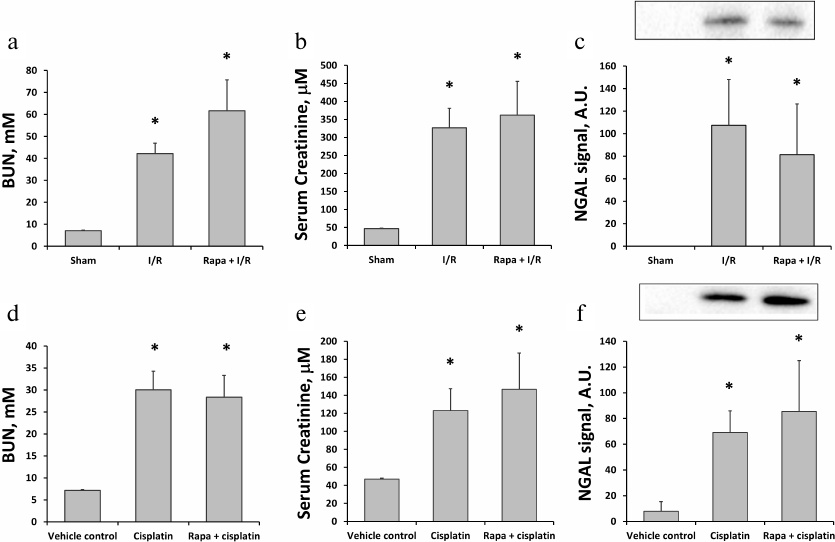
To address the effect of rapamycin on the cisplatin nephrotoxicity, we examined cisplatin-induced AKI with and without rapamycin treatment. Single cisplatin administration at a dose of 15 mg/kg induced moderate AKI within 48 h indicated by the elevated BUN (n = 6; Fig. 1d) and SCr levels (Fig. 1e). Changes in the levels of BUN and SCr were not affected by the treatment with rapamycin: on day 2 after cisplatin injection, the concentration of BUN in untreated rats increased to 30.1 ± 4.2 mM, which was similar to the BUN concentration in the rapamycin-treated animals (28.3 ± 5.0 mM, n = 5; Fig. 1d). Similarly, SCr levels increased to 123.0 ± 24.3 and 146.6 ± 40.3 μM in cisplatin and cisplatin plus rapamycin groups, respectively (Fig. 1e). Moreover, we have observed the increase in the NGAL level in the urine of animals with the cisplatin-induced AKI, which positively correlated with the functional markers (Fig. 1f).
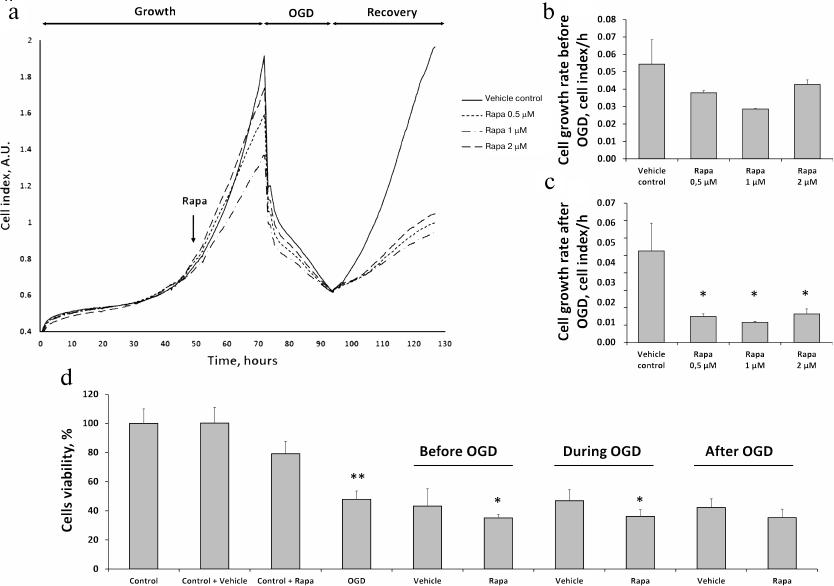
Effects of rapamycin on the primary culture of RTCs. To model I/R in vitro, we subjected primary RTCs culture to OGD with the following reoxygenation. Real-time evaluation of the RTCs growth using an iCELLigence device revealed that rapamycin at the concentrations of 0.5, 1, and 2 μM slightly reduced cell proliferation rate before the OGD (Fig. 2, a and b) and significantly diminished it during the recovery phase after the OGD (Fig. 2, a and c). These observations were supported by the results of the MTT assay. Incubation with 0.5 μM rapamycin slightly reduced cell viability vs. vehicle control. Cell viability decreased to less than 50% after OGD, while incubation with rapamycin 6 h before the OGD decreased cell viability even to a greater extent (Fig. 2d). Rapamycin treatment of the RTCs cultures during OGD or immediately after reoxygenation for 24 h reduced cell viability compared to OGD or appropriate vehicle control (Fig. 2d).
Fig. 2. Effects of different doses of rapamycin on the proliferation of RTCs in primary culture after OGD. a) Kinetics of RTCs proliferation evaluated with an RTCA iCELLigence device, including cell growth under normoxic conditions, growth after addition of different doses of rapamycin (0.5, 1, and 2 μM), cell death during OGD, and recovery after the restoration of oxygen supply and addition of complete culture medium. b) RTCs growth rate after rapamycin (Rapa) addition, but before OGD. c) RTCs growth rate in the recovery phase after OGD. d) Cell viability (according to MTT assay) after 0.5 μM rapamycin addition: 6 h before, during, and after OGD for 24 h with appropriate vehicle controls (DMSO); * p < 0.05 compared to the corresponding vehicle control, ** p < 0.05 compared to control cells.
Incubation of RTCs with 6.25, 12.5, and 25 μM cisplatin for 24 h induced cell death in a concentration-dependent manner, as evidenced by the results of MTT assay (Fig. 3, a and b). Treatment by rapamycin together with cisplatin either had no effect on the cisplatin-induced changes in the RTCs viability (at 6.25 and 25 μM cisplatin) or aggravated cisplatin-induced cell death (Fig. 3a). Similar results were obtained when rapamycin was added before cisplatin (Fig. 3b).
Fig. 3. The proliferation of the RTCs primary culture after incubation with cisplatin and effect of rapamycin (Rapa) on cell viability. a, b) Cell viability of RTCs incubated with different concentrations of cisplatin as determined by the MTT assay and effects of rapamycin added (a) 3 h before cisplatin treatment or (b) simultaneously with cisplatin. c) RTCs proliferation kinetics evaluated with RTCA iCELLigence, including growth under standard conditions and cell death induced by incubation with different cisplatin concentrations. d) The proliferation of RTCs at different cisplatin concentrations. e, f) RTCs proliferation kinetics after addition of 12.5 (e) or 25 μM (f) cisplatin alone, with rapamycin pretreatment 3 h before cisplatin addition, and after simultaneous incubation with cisplatin and rapamycin; * p < 0.05 compared to 12.5 μM cisplatin.
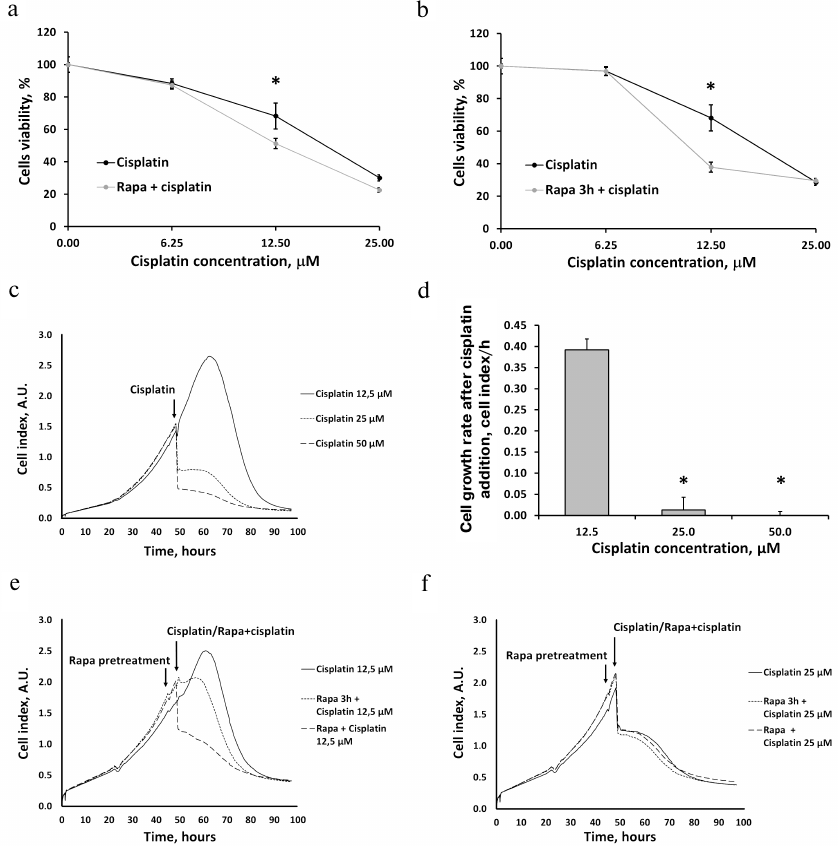
We also monitored the proliferation of RTCs following incubation with different cisplatin concentrations after rapamycin pretreatment and found that cisplatin significantly decreased the growth rate of RTCs (Fig. 3, c, e, f). As measured by the MTT assay, cisplatin caused substantial cell death (Fig. 3, a and b) and almost completely inhibited cell proliferation at 25 and 50 μM concentration (Fig. 3d). Real-time estimation of the RTCs growth showed that rapamycin treatment did not protect cells from cell death or even aggravated it (Fig. 3, e and f).
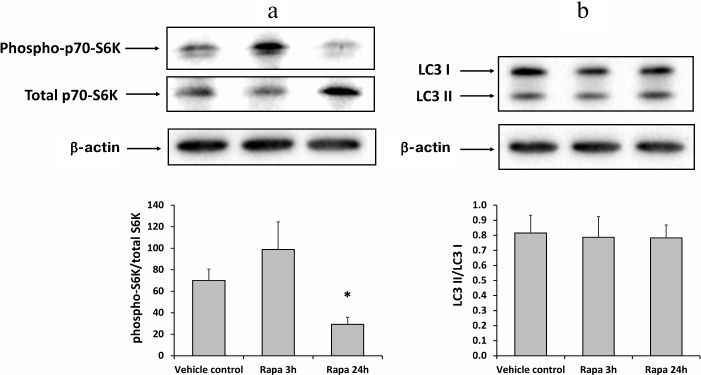
Inhibition of mTOR (mammalian target of rapamycin) and the absence of autophagy activation by rapamycin. To investigate the effect of rapamycin on the mTOR signaling, we analyzed the levels of phosphorylated and total p70-S6K protein which is known to be one of the main substrates of mTORC1 (mammalian target of rapamycin complex 1) [22]. Western blotting of kidney homogenates from rats injected with rapamycin 3 or 24 h before the tissue collection, revealed a decrease in the ratio phospho-p70-S6K to the total p70-S6K in the 24-h group (Fig. 4a). Therefore, rapamycin inhibited the activity of mTORC1 in the kidney tissue. However, the LC3 II/LC3 I ratio remained the same in all rat groups, indicating the absence of autophagy activation (Fig. 4b).
Fig. 4. Inhibition of mTORC1 signaling and autophagy activation in rat kidneys 3 or 24 h after administration of 1 mg/kg rapamycin (Rapa). a) Phosphorylated p70-S6K to total p70-S6K ratio; b) LC3 II/LC3 I ratio; * p < 0.05 compared to vehicle control (DMSO).
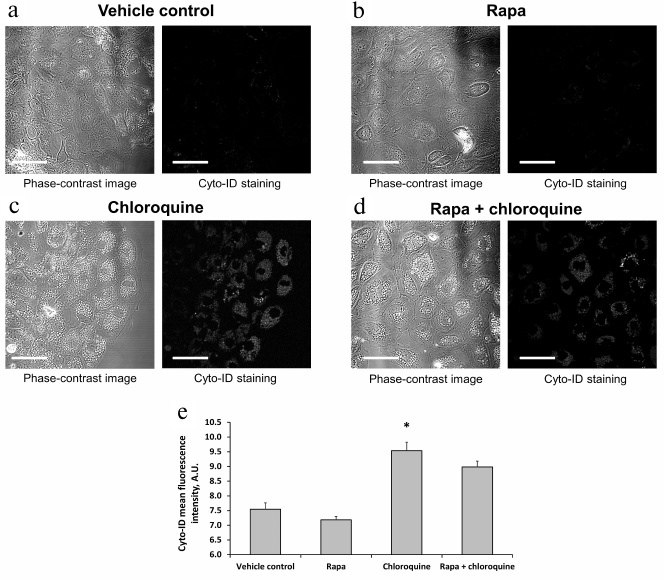
The lack of autophagy activation after rapamycin treatment was also confirmed by examination of primary RTCs cultures using Cyto-ID, which selectively accumulates in autophagosomes and autophagolysosomes [23]. The intensity of Cyto-ID fluorescence in RTCs did not increase after incubation with rapamycin for 3 h compared to the vehicle control or even exhibited a tendency to decrease (Fig. 5, a and e). To promote a possible effect of rapamycin on the autophagosome accumulation, we added chloroquine (autophagy late stage inhibitor) to the RTCs growth media for 3 h, which resulted in a significant increase in both fluorescence intensity of Cyto-ID and the number of Cyto-ID-positive structures in RTCs (Fig. 5, c and e). However, incubation with rapamycin and chloroquine simultaneously for 3 h did not lead to further increase in the mean fluorescence intensity of Cyto-ID, thus confirming the absence of autophagy activation by rapamycin (Fig. 5, d and e).
Fig. 5. Accumulation of autophagosomes in response to rapamycin and/or chloroquine treatment. a-d) Confocal images of RTCs incubated with 0.5 μM rapamycin, or 30 mM chloroquine, or both for 3 h. e) Mean fluorescence intensity of Cyto-ID in RTCs incubated with the indicated substances for 3 h; * p < 0.05 compared to vehicle control (DMSO). Scale bar, 50 μm.
DISCUSSION
The aim of this study was to determine whether the autophagy inducer rapamycin protects RTCs from AKI caused by renal I/R or cisplatin-induced nephrotoxicity. We found that rapamycin proved to be inefficient under all studied conditions.
Although the involvement of autophagy in AKI pathogenesis is commonly accepted, the effects of autophagy activators or inhibitors on AKI remain unexplored. However, recent data have confirmed the protective role of autophagy, since autophagy suppression was found to aggravate AKI. For instance, the autophagy inhibitor chloroquine increased the severity of AKI in animals [10, 14]. Surprisingly, lower doses of chloroquine alleviated AKI caused by kidney exposure to I/R [24]. Because the characteristics of the autophagic/lysosomal system were not explored in [24], it is impossible to elucidate the mechanisms responsible for the protective effect of chloroquine at low doses. Experiments with another autophagy inhibitor, 3-methyladenine, which prevents autophagophore formation by inhibiting phosphatidylinositol 3-kinase, also demonstrated deleterious consequences of autophagy inhibition [12, 25, 26].
There are only a few experimental papers that have directly studied the effect of rapamycin pretreatment on AKI. For instance, intravenous (i.v.) injection of rapamycin at a dose of 1 mg/kg 15 min before renal I/R improved renal function evaluated from the content of SCr and BUN 24 h after I/R [25]. Rapamycin pretreatment decreased the histological renal tissue injury scores associated with the apoptosis suppression [25]. Similar data was obtained by i.p. injection of 1 mg/kg rapamycin 2 h before the induction of kidney ischemia [26] and by injection of 10 mg/kg rapamycin 1 h after I/R injury [27]. Apart from the kidneys, the protective effects of rapamycin were observed in the liver exposed to I/R. Rapamycin administered at the doses of 1 and 5 mg/kg (i.p.) 1 h before the ischemia induction protected the liver from I/R injury. Rapamycin pretreatment significantly decreased the serum levels of ALT (alanine aminotransferase) and AST (aspartate aminotransferase) and preserved liver architecture [28].
To compare our results with the data of previous studies, we used rapamycin at the same dose (1 mg/kg body weight) 1 h before the induction of ischemia in the kidneys. However, rapamycin failed to reduce the pathologically elevated levels of BUN and SCr (Fig. 1, a and b). Furthermore, additional analysis of AKI biomarkers in the urine revealed no decrease in the NGAL level in the rapamycin-pretreated group (Fig. 1c). Our findings are in agreement with studies published several years prior to the articles highlighting the beneficial effects of rapamycin. Since rapamycin and its analogues were originally introduced as immunosuppressive agents, the first studies of rapamycin effects on the kidneys compared the effects of this drug to those of other known immunosuppressants. For instance, comparison of methylprednisolone with rapamycin (1 mg/kg) injected 6 and 1 h before ischemia did not reveal any differences between the groups with regard to the SCr levels and histological changes [29]. In another paper, the effects of rapamycin (3 mg/kg) administered for 2 days before the surgical procedure and then 24 h after the surgery or for 7 days were compared to the action of the immunosuppressant cyclosporine A. Neither of these agents affected the functioning of kidneys injured by I/R [30].
Finally, a number of studies have demonstrated that rapamycin can exacerbate the severity of I/R injury. Rapamycin administration at a dose of 3 mg/kg 1 day before and on the day of I/R procedure was associated with more pronounced impairment in the renal function 48 and 120 h after I/R as compared to the animals undergoing I/R alone [31]; however, rapamycin in a dose of 1.5 mg/kg caused neither renal dysfunction nor any improvements. Similar results were obtained by Lui et al. in mice treated with rapamycin by oral gavage (2 mg/kg daily), starting 1 day before the kidney I/R. One day after I/R, the SCr levels in the rapamycin-treated mice were higher, while kidney histology demonstrated significant tubular damage [32]. Similarly, Lieberthal et al. observed rapamycin to impair recovery from acute renal failure by inhibiting proliferation and inducing cell cycle arrest and apoptosis in tubular cells [33].
It is important to mention that rapamycin is a potent immunosuppressive agent that acts by inhibiting proliferation and clonal expansion of stimulated T-cells [34]. Therefore, its effect in vivo is a combination of autophagy activation and immune response suppression, the latter playing a significant role in ischemic and other forms of AKI [35]. In this regard, we evaluated the action of rapamycin not only in the ischemia model but also using OGD in primary cultures of renal tubular cells. Since this experimental system does not contain immune cells, its application excludes the influence of inflammatory and immune components on AKI.
To address mTOR signaling, we measured the levels of total and phosphorylated forms of the p70-S6 kinase in the rat kidneys after rapamycin administration at a standard dose of 1 mg/kg. The p70-S6 kinase is one of the major effectors of the mTOR pathway and it is phosphorylated by activated mTORC1 [22]. We also assayed kidney homogenates for the autophagy-associated signaling by measuring the LC3 II/LC3 I ratio and evaluated the intensity of Cyto-ID fluorescence in rapamycin-treated cells. Although we observed mTORC1 inhibition evidenced by a decrease in the level of phosphorylated p70-S6 kinase 24 h after rapamycin administration (Fig. 4), no autophagy activation was detected in vivo and in vitro (Figs. 4b and 5). We conclude that inhibition of mTOR did not result in further autophagy activation; rapamycin administration was unable to prevent or reduce cell death after OGD.
The observed decrease in the proliferation rate of RTCs incubated with rapamycin could be explained by the antiproliferative effects of rapamycin originally described in T-cells and tumor cells [36, 37]. Proliferation inhibition by rapamycin was also shown in the culture of proximal epithelial cells, where it caused a decrease in the number of dividing renal cells [33]. Kidney sections from rapamycin-treated mice showed a significantly lower number of PCNA (proliferating cell nuclear antigen)-positive cells after I/R injury [32].
We also analyzed the effect of rapamycin on the cisplatin-induced nephrotoxicity (another AKI model). Since the pathogenic mechanisms of cisplatin-induced injury of renal cells may differ from the mechanisms triggered by I/R, the action of rapamycin in this model may be very different. Activation of autophagy after cisplatin-induced AKI has been demonstrated both in vitro and in vivo in numerous studies [5, 6]. Moreover, inhibition of autophagy by chloroquine promoted AKI, whereas upregulation of autophagy by rapamycin rescued renal functions and histology, thus corroborating the protective role of autophagy in the cisplatin-induced AKI [38].
However, we did not observe any protective effect of rapamycin administration in vivo in rats with the cisplatin-induced nephrotoxicity or in vitro in RTCs subjected to different doses and protocols of rapamycin treatment (Figs. 1d-1f and 3). Our data is consistent with the study of Nakagawa et al. [39] who showed that plasma creatinine concentration and Kim-1 levels were higher in rats treated with a combination of everolimus (rapamycin analogue) and cisplatin. The authors concluded that mTOR inhibition by everolimus reduced renal function. They observed that cisplatin activated mTOR, while administration of everolimus inhibited this activation. No protective effect of everolimus on kidney function was found [39].
Therefore, the studies on the effects of rapamycin and its analogues can be divided into two different categories. On the one hand, a great number of studies strongly prove that autophagy plays an important role in the AKI pathogenesis. Furthermore, activation of autophagy (e.g., by caloric restriction) was shown to protects kidneys from different forms of AKI [15, 40, 41]. On the other hand, the attempts to activate autophagy by pharmacological substances, such as rapamycin and its analogues, were frequently unable to demonstrate effective protection against AKI. Moreover, in some cases, rapamycin administration caused even more pronounced deterioration of kidney function. Therefore, it is important to take into consideration that rapamycin is not the ideal mimetic of caloric restriction, because the latter inhibits mTOR signaling through various signaling cascades (e.g., AMPK and Akt pathways) involving phosphorylation/dephosphorylation of their components [42]. Rapamycin acts as an allosteric inhibitor of mTOR. It forms a gain-of-function complex with the FK506-binding protein that interacts with and inhibits mTOR [43]. It is likely that caloric restriction has numerous cell targets, unlike rapamycin, which acts as the mTORC1 inhibitor only. Therefore, the conclusion about the equivalence between the rapamycin action and the impact of caloric restriction is too premature [44, 45].
Note that the majority of papers demonstrating the protective effect of rapamycin on AKI were published after 2010, when the idea of the beneficial effect of autophagy and its activators on the lifespan and healthy aging has become very popular. Communications reporting the negative effect or the absence of any effect of rapamycin on AKI had been publisher earlier, when rapamycin had been considered as an immunosuppressive agent.
We would like to highlight that researchers should be very cautious when applying pharmacological interventions imitating physiological activation of autophagy. It is unreasonable to believe that mTOR inhibitors act identically to naturally occurring physiological processes, such as caloric restriction. The effects of rapamycin on ischemic and cisplatin-induced AKI and its protective action in acute pathologies remain controversial, which might explain why the attempts to use rapamycin in clinical practice have been unsuccessful so far.
Funding. This work was supported by the Russian Science Foundation (project 18-15-00058).
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical norms. The rats were treated according to the protocols evaluated and approved by the Animal Ethics Committee of Belozersky Institute of Physico-Chemical Biology. All procedures were in accordance with the guidelines of the Federation of Laboratory Animal Science Associations (FELASA).
REFERENCES
1.Levine, B., and Kroemer, G. (2008) Autophagy in the
pathogenesis of disease, Cell, 132, 27-42, doi:
10.1016/j.cell.2007.12.018.
2.Mizushima, N., Levine, B., Cuervo, A. M., and
Klionsky, D. J. (2008) Autophagy fights disease through cellular
self-digestion, Nature, 451,1069-1075, doi:
10.1038/nature06639.
3.Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba,
E., Moullan, N., Nicolet-Dit-Felix, A. A., Williams, E. G., Jha, P., Lo
Sasso, G., Huzard, D., Aebischer, P., Sandi, C., Rinsch, C., and
Auwerx, J. (2016) Urolithin A induces mitophagy and prolongs lifespan
in C. elegans and increases muscle function in rodents, Nat.
Med., 22, 879-888, doi: 10.1038/nm.4132.
4.Andreux, P. A., Blanco-Bose, W., Ryu, D., Burdet,
F., Ibberson, M., Aebischer, P., Auwerx, J., Singh, A., and Rinsch, C.
(2019) The mitophagy activator urolithin A is safe and induces a
molecular signature of improved mitochondrial and cellular health in
humans, Nat. Metab., 1, 595-603, doi:
10.1038/s42255-019-0073-4.
5.Periyasamy-Thandavan, S., Jiang, M., Wei, Q.,
Smith, R., Yin, X.-M., and Dong, Z. (2008) Autophagy is cytoprotective
during cisplatin injury of renal proximal tubular cells, Kidney
Int., 74, 631-640, doi: 10.1038/ki.2008.214.
6.Takahashi, A., Kimura, T., Takabatake, Y., Namba,
T., Kaimori, J., Kitamura, H., Matsui, I., Niimura, F., Matsusaka, T.,
Fujita, N., Yoshimori, T., Isaka, Y., and Rakugi, H. (2012) Autophagy
guards against cisplatin-induced acute kidney injury, Am. J.
Pathol., 180, 517-525, doi:
10.1016/j.ajpath.2011.11.001.
7.Liu, S., Hartleben, B., Kretz, O., Wiech, T.,
Igarashi, P., Mizushima, N., Walz, G., and Huber, T. B. (2012)
Autophagy plays a critical role in kidney tubule maintenance, aging and
ischemia-reperfusion injury, Autophagy, 8, 826-837, doi:
10.4161/auto.19419.
8.Li, L., Wang, Z. V., Hill, J. A., and Lin, F.
(2014) New autophagy reporter mice reveal dynamics of proximal tubular
autophagy, J. Am. Soc. Nephrol., 25, 305-315, doi:
10.1681/ASN.2013040374.
9.Li, T., Liu, Y., Zhao, J., Miao, S., Xu, Y., and
Liu, K., Liu, M., Wang, G., and Xiao, X. (2017) Aggravation of acute
kidney injury by mPGES-2 down regulation is associated with autophagy
inhibition and enhanced apoptosis, Sci. Rep., 7, 10247,
doi: 10.1038/s41598-017-10271-8.
10.Mei, S., Livingston, M., Hao, J., Li, L., Mei,
C., and Dong, Z. (2016) Autophagy is activated to protect against
endotoxic acute kidney injury, Sci. Rep., 6, 22171, doi:
10.1038/srep22171.
11.Karagiannidis, I., Kataki, A., Glustianou, G.,
Memos, N., Papalois, A., and Alexakis, N., Zografos, G. C., and
Konstadoulakis, M. M. (2016) Extended cytoprotective effect of
autophagy in the late stages of sepsis and fluctuations in signal
transduction pathways in a rat experimental model of kidney injury,
Shock, 45,139-147, doi: 10.1097/SHK.0000000000000505.
12.Ko, G. J., Bae, S. Y., Hong, Y.-A., Pyo, H. J.,
and Kwon, Y. J. (2016) Radiocontrast-induced nephropathy is attenuated
by autophagy through regulation of apoptosis and inflammation, Hum.
Exp. Toxicol., 35, 724-736, doi:
10.1177/0960327115604198.
13.Kimura, T., Takabatake, Y., Takahashi, A.,
Kaimori, J., Matsui, I., Namba, T., Kitamura, H., Niimura, F.,
Matsusaka, T., Soga, T., Rakugi, H., and Isaka, Y. (2011) Autophagy
protects the proximal tubule from degeneration and acute ischemic
injury, J. Am. Soc. Nephrol., 22, 902-913, doi:
10.1681/ASN.2010070705.
14.Jiang, M., Wei, Q., Dong, G., Komatsu, M., Su,
Y., and Dong, Z. (2012) Autophagy in proximal tubules protects against
acute kidney injury, Kidney Int., 82, 1271-1283, doi:
10.1038/ki.2012.261.
15.Andrianova, N. V., Jankauskas, S. S., Zorova, L.
D., Pevzner, I. B., Popkov, V. A., Silachev, D. N., Plotnikov, E. Y.,
and Zorov, D. B. (2018) Mechanisms of age-dependent loss of dietary
restriction protective effects in acute kidney injury, Cells,
7, 178, doi: 10.3390/cells7100178.
16.Kuo, S.-Y., Castoreno, A. B., Aldrich, L. N.,
Lassen, K. G., Goel, G., Dancik, V., Kuballa, P., Latorre, I., Conway,
K. L., Sarkar, S., Maetzel, D., Jaenisch, R., Clemons, P. A.,
Schreiber, S. L., Shamji, A. F., and Xavier, R. J. (2015)
Small-molecule enhancers of autophagy modulate cellular disease
phenotypes suggested by human genetics, Proc. Natl. Acad. Sci.
USA, 112, 4281-4287, doi: 10.1073/pnas.1512289112.
17.Blagosklonny, M. V. (2017) From rapalogs to
anti-aging formula, Oncotarget, 8, 35492-35507, doi:
10.18632/oncotarget.18033.
18.Plotnikov, E. Y., Kazachenko, A. V., Vyssokikh,
M. Y., Vasileva, A. K., Tcvirkun, D. V., Isaev, N. K., Kirpatovsky, V.
I., and Zorov, D. B. (2007) The role of mitochondria in oxidative and
nitrosative stress during ischemia/reperfusion in the rat kidney,
Kidney Int., 72, 1493-502, doi:
10.1038/sj.ki.5002568.
19.Schneider, C. A., Rasband, W. S., and Eliceiri,
K. W. (2012) NIH image to imageJ: 25 years of image analysis, Nat.
Methods, 9, 671-675.
20.Giaever, I., and Keese, C. R. (1984) Monitoring
fibroblast behavior in tissue culture with an applied electric field,
Proc. Natl. Acad. Sci. USA, 81, 3761-3764.
21.Popkov, V. A., Andrianova, N. V., Manskikh, V.
N., Silachev, D. N., Pevzner, I. B., and Zorova, L. D., Sukhikh, G. T.,
Plotnikov, E. Y., and Zorov, D. B. (2018) Pregnancy protects the kidney
from acute ischemic injury, Sci. Rep., 8, 14534, doi:
10.1038/s41598-018-32801-8.
22.Tavares, M. R., Pavan, C. B., Amaral, C. L.,
Meneguello, L., Luchessi, A. D., and Simabuco, F. M. (2015) The S6K
protein family in health and disease, Life Sci., 131,
1-10, doi: 10.1016/j.lfs.2015.03.001.
23.Stankov, M., Panayotova-Dimitrova, D., Leverkus,
M., Klusmann, J.-H., and Behrens, G. (2014) Cytometric analysis of
autophagic activity with cyto-ID staining in primary cells,
Bio-Protocol, 4, doi: 10.21769/BioProtoc.1090.
24.Todorovic, Z., Medic, B., Basta-Jovanovic, G.,
Radojevic Skodric, S., Stojanovic, R., Rovcanin, B., and Prostran, M.
(2014) Acute pretreatment with chloroquine attenuates renal I/R injury
in rats, PLoS One, 9, 92673, doi:
10.1371/journal.pone.0092673.
25.Zhang, Y.-L., Zhang, J., Cui, L.-Y., and Yang, S.
(2015) Autophagy activation attenuates renal ischemia-reperfusion
injury in rats, Exp. Biol. Med., 240, 1590-1598, doi:
10.1177/1535370215581306.
26.Guan, X., Qian, Y., Shen, Y., Zhang, L., Du, Y.,
Dai, H., Qian, J., and Yan, Y. (2015) Autophagy protects renal tubular
cells against ischemia/reperfusion injury in a time-dependent manner,
Cell Physiol. Biochem., 36, 285-298, doi:
10.1159/000374071.
27.Ling, H., Chen, H., Wei, M., Meng, X., Yu, Y.,
and Xie, K. (2016) The effect of autophagy on inflammation cytokines in
renal ischemia/reperfusion injury, Inflammation, 39,
347-356, doi: 10.1007/s10753-015-0255-5.
28.Zhu, J., Lu, T., Yue, S., Shen, X., Gao, F.,
Busuttil, R. W., Kupiec-Weglinski, J. W., Xia, Q., and Zhai, Y. (2015)
Rapamycin protection of livers from ischemia and reperfusion injury is
dependent on both autophagy induction and mammalian target of rapamycin
complex 2-Akt activation, Transplantation, 99, 48-55,
doi: 10.1097/TP.0000000000000476.
29.Parra, C., Salas, P., and Dominguez, J. (2010)
Effects of immunosuppressive drugs on rat renal ischemia reperfusion
injury, Transplant. Proc., 42, 245-247, doi:
10.1016/j.transproceed.2009.11.018.
30.Pereira, B. J., Castro, I., Burdmann, E. A.,
Malheiros, D. M. A., and Yu, L. (2010) Effects of sirolimus alone or in
combination with cyclosporine A on renal ischemia/reperfusion injury,
Braz. J. Med. Biol. Res., 43, 737-744.
31.Goncalves, G. M., Cenedeze, M. A., Feitoza, C.
Q., de Paula, C. B., Marques, G. D., Pinheiro, H. S., de Paula Antunes
Teixeira, V., Antonia dos Reis, M., Pacheco-Silva, A., and Camara, N.
O. (2007) The role of immunosuppressive drugs in aggravating renal
ischemia and reperfusion injury, Transplant. Proc., 39,
417-420, doi: 10.1016/j.transproceed.2007.01.027.
32.Lui, S. L., Chan, K. W., Tsang, R., Yung, S.,
Lai, K. N., and Chan, T. M. (2006) Effect of rapamycin on renal
ischemia-reperfusion injury in mice, Transpl. Int., 19,
834-839, doi: 10.1111/j.1432-2277.2006.00361.x.
33.Lieberthal, W., Fuhro, R., Andry, C. C., Rennke,
H., Abernathy, V. E., Koh, J. S., Valeri, R., and Levine, J. S. (2001)
Rapamycin impairs recovery from acute renal failure: role of cell-cycle
arrest and apoptosis of tubular cells, Am. J. Physiol. Renal
Physiol., 281, 693-706, doi:
10.1152/ajprenal.2001.281.4.F693.
34.Martel, R. R., Klicius, J., and Galet, S. (1977)
Inhibition of the immune response by rapamycin, a new antifungal
antibiotic, Can. J. Physiol. Pharmacol., 55, 48-51.
35.Kim, B. S., Lim, S. W., Li, C., Kim, J. S., Sun,
B. K., Ahn, K. O., Han, S. W., Kim, J., and Yang, C. W. (2005)
Ischemia-reperfusion injury activates innate immunity in rat kidneys,
Transplantation, 79, 1370-1377.
36.Dumont, F. J., and Su, Q. (1996) Mechanism of
action of the immunosuppressant rapamycin, Life Sci., 58,
373-395.
37.Law, B. K. (2005) Rapamycin: an anti-cancer
immunosuppressant? Crit. Rev. Oncol. Hematol., 56, 47-60,
doi: 10.1016/j.critrevonc.2004.09.009.
38.Kaushal, G. P. (2012) Autophagy protects proximal
tubular cells from injury and apoptosis, Kidney Int., 82,
1250-1253, doi: 10.1038/ki.2012.337.
39.Nakagawa, S., Nishihara, K., Inui, K., and
Masuda, S. (2012) Involvement of autophagy in the pharmacological
effects of the mTOR inhibitor everolimus in acute kidney injury,
Eur. J. Pharmacol., 696, 43-54, doi:
10.1016/j.ejphar.2012.09.010.
40.Mitchell, J. R., Verweij, M., Brand, K., van de
Ven, M., Goemaere, N., van den Engel, S., Chu, T., Forrer, F., Muller,
C., de Jong, M., van IJcken, W., IJzermans, J. N., Hoeijmakers, J. H.,
and de Bruin, R. W. (2010) Short-term dietary restriction and fasting
precondition against ischemia reperfusion injury in mice, Aging
Cell, 9, 40-53, doi: 10.1111/j.1474-9726.2009.00532.x.
41.Ning, Y.-C., Cai, G.-Y., Zhuo, L., Gao, J.-J.,
Dong, D., Cui, S.-Y., Shi, S. Z., Feng, Z., Zhang, L., Sun, X. F., and
Chen, X. M. (2013) Beneficial effects of short-term calorie restriction
against cisplatin-induced acute renal injury in aged rats, Nephron.
Exp. Nephrol., 124, 19-27, doi:
10.1159/000357380.
42.Tokunaga, C., Yoshino, K., and Yoneezawa, K.
(2004) mTOR integrates amino acid- and energy-sensing pathways,
Biochem. Biophys. Res. Commun., 313, 443-446.
43.Li, J., Kim, S. G., and Blenis, J. (2014)
Rapamycin: one drug, many effects, Cell Metab., 19,
373-379, doi: 10.1016/j.cmet.2014.01.001.
44.Lee, S.-H., and Min, K.-J. (2013) Caloric
restriction and its mimetics, BMB Rep., 46, 181-187.
45.Lamming, D. W. (2016) Inhibition of the
mechanistic target of rapamycin (mTOR) – rapamycin and beyond,
Cold Spring Harb. Perspect. Med., 6, a025924, doi:
10.1101/cshperspect.a025924.