Characteristics of the Airway Microbiome of Cystic Fibrosis Patients#
O. L. Voronina1,a*, N. N. Ryzhova1, M. S. Kunda1, E. V. Loseva1, E. I. Aksenova1, E. L. Amelina2, G. L. Shumkova2, O. I. Simonova3, and A. L. Gintsburg1
1Gamaleya National Research Center for Epidemiology and Microbiology, Ministry of Health of Russia, 123098 Moscow, Russia2Pulmonology Research Institute, Federal Medical-Biological Agency, 115682 Moscow, Russia
3National Medical Research Center for Children’s Health, Ministry of Health of Russia, 119296 Moscow, Russia
# This study is dedicated to the 80th anniversary of the Department of Biochemistry, Lomonosov Moscow State University (see vol. 84, no. 11, 2019).
* To whom correspondence should be addressed.
Received June 4, 2019; Revised July 29, 2019; Accepted September 10, 2019
Microbiota as an integral component of human body is actively investigated, including by massively parallel sequencing. However, microbiomes of lungs and sinuses have become the object of scientific attention only in the last decade. For patients with cystic fibrosis, monitoring the state of respiratory tract microorganisms is essential for maintaining lung function. Here, we studied the role of sinuses and polyps in the formation of respiratory tract microbiome. We identified Proteobacteria in the sinuses and samples from the lower respiratory tract (even in childhood). In some cases, they were accompanied by potentially dangerous basidiomycetes. The presence of polyps did not affect formation of the sinus microbiome. Proteobacteria are decisive in reducing the biodiversity of lung and sinus microbiomes, which correlated with the worsening of the lung function indicators. Soft mutations in the CFTR gene contribute to the formation of safer microbiome even in heterozygotes with class I mutations.
KEY WORDS: microbiome, cystic fibrosis, airway, chronic rhinosinusitis, ProteobacteriaDOI: 10.1134/S0006297920010010
Abbreviations: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane regulator; FDR, false discovery rate; FEV1, forced expiratory volume in one second; ITS, internal transcribed spacer; MLST, multilocus sequence typing; OTU, operational taxonomic unit; PCoA, principal coordinate analysis; PERMANOVA, permutational multivariate analysis of variance; SRA, sequence read archive; ST, sequence type related to MLST.
Microbiota is currently recognized as an essential component of human
body that affects gastrointestinal, immune, nervous, and other systems.
The term microbiome was introduced in the XXI century to denote
combined microbial genomes in an ecological niche or a biological
sample. However, the majority of studies defines microbiome as combined
microbiota-derived ribosomal RNA (rRNA) genes.
Woese et al. demonstrated that these genes are universal in living organisms and, therefore, can be used as molecular clocks in phylogenetics [1]. Stackebrandt and Woese discussed the role of rRNAs in the phylogeny and taxonomy of prokaryotes [2]. Accumulation of rRNA gene data has started in 1970s and was significantly promoted by the development of the DNA sequencing method by Sanger in 1977. The obtained data needed systemization, and in 1982, GenBank NCBI was formed to serve as a nucleic acid sequence database [3]. In 1990s, Woese et al. conducted two projects on the generation of rRNA sequence database [4]. Altogether, these achievements have helped to avoid the chaos by further automatization of nucleic acid sequencing and development of new generation sequencers, resulting in obtaining large-scale data arrays that have allowed to update existing databases with novel information. Woese aptly noted that in the XX century, biology has turned into an engineering discipline [1], and it has been thoroughly computerized. Newly developed and constantly improving software, as well as advancing hardware, allow to analyze sequencing data to be further compared with available sequencing databases.
These developments made it possible to start in 2008 a new project called The Human Microbiome Project (HMP) based on the existing informational environment and sequencing experience obtained during completing The Human Genome Project [5]. The 16S rDNA gene and, later, its fragment consisting of several variable regions (usually V1-V4 or shorter, depending on the possibilities of the used sequencing platform) was chosen for sequencing in the process of optimization of the ongoing studies. Examining microbiome in the context of 16S rDNA gene segments allows to get an insight into the bacteriome, i.e., phylogenetic bacterial diversity.
However, metagenomic studies aimed at the examination of microbial community metabolism are expensive and therefore available solely within the framework of some highly funded projects.
The Human Microbiome Project has targeted bacterial communities in the gut, skin, and urogenital tract, but not in the lungs (because of the dogma of lung sterility) [5, 6]. This misconception had been refuted only with the onset of the study started in 2014 and called The Integrative Human Microbiome Project (iHMP) [5]. The studies of microbiomes in the lungs and parts of the upper respiratory tract both in normal and pathological conditions have started only in 2010s using the methods of massively parallel sequencing.
Normally, the formation of lung microbiome is determined by two events: aspiration of microbes from the oral cavity and mucociliary clearance that removes most of the foreign matter from the lower respiratory tract. That provides the development of a balanced microbial community controlled by the host immune system that is able to eliminate pathogenic species via activity of phagolysosomes.
Monogenic cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane regulator (CFTR) gene resulting in altered chloride ion transport activity that leads to the multi-organ damage. The physicians’ focus in CF is the state of the lower respiratory tract because of the deteriorated respiratory function that affects the quality of life and life expectancy of CF patients.
It should be noted that the mutant chloride ion channel impairs the mucociliary clearance not only inside the lungs, bronchi, and trachea, but also in the nasal cavity, including sinuses. Denser secretions hinder the ciliated epithelium activity, which results in a higher microbial burden. These microorganisms are attacked but not eliminated by immune cells, as altered chloride ion channel activity impairs proper acidification of the phagolysosomal lumen. Moreover, neutrophils recruited by the macrophage-released cues also express the mutant CFTR. Necrosis of immune cells contributes to the dysregulation of inflammatory response upon chronic infection caused by the propagating microbes [7].
Changes in the microbiome of CF patients are detected in early age [8]. Periodic microbiome exanimations throughout the entire patient’s life period might help to unveil the causative trigger for the disease. The information accumulated so far has shaped our concept of the so-called healthy lower respiratory tract microbiome that includes microorganisms from the three major phyla: Actinobacteria, Bacteroidetes, and Firmicutes [9]. Despite the fact that Proteobacteria are found in a balanced microbiome in healthy people [10], their emergence in CF patients is a warning sign; therefore, the lower respiratory tract of CF patients has to be regularly monitored for the presence of these microorganisms. The data on patients with chronic lung infections are available both from the Russian Federation Cystic Fibrosis Patient Registry and the European Cystic Fibrosis Society Patient Registry [11, 12].
Comparing the composition of lung microbiomes derived from healthy volunteers and CF patients requires assessment of the bacterial burden, which was found to be low throughout the entire life period in healthy individuals and in early childhood in CF patients. The onset of changes in the microbiome of CF patients involves an increase in the bacterial burden and number of Proteobacteria and reduction of the microbial species diversity. These changes can be affected by many factors, the most important of which still remains unknown [13].
Proteobacteria that enter the human body from the environment are of particular significance in CF patients. The studies started in 1950s have proven that potentially pathogenic species Pseudomonas aeruginosa, Burkholderia spp., Achromobacter spp., and Stenotrophomonas maltophilia are pathogenic in CF patients [14]. In particular, it was found that infections caused by these bacteria impair lung function, reduce 5-year survival rate [15], and pose a threat of colonization in lung transplants [16].
Disease exacerbation, antibiotic therapy, mutant CFTR gene, and other factors affect the microbiota state, development of pathogenic microbes, and deterioration of lung functions [17-19].
However, even after successful antibiotic therapy, Proteobacteria with the genotype similar to the genotype of microorganisms previously colonizing lower respiratory tract may re-emerge in the lungs. The follow-up of Burkholderia-infected patients after lung transplantation demonstrated that depending on the patient’s state and Burkholderia virulence potential, bacteria of the same genotype colonized transplanted lungs either immediately or three, twelve, or more months after the surgical intervention and were detected in all the examined patients 2.5 years after the surgery [20].
Such evidence has prompted physicians to examine the upper respiratory tract, particularly, paranasal sinuses, as a potential reservoir of infection. In particular, we demonstrated in the pilot study that all analyzed CF patients were positive for pathogenic microbes with the same genotype that were detected both in the paranasal sinus lavage and sputum samples [21].
Since chronic rhinosinusitis and altered sinus mucociliary clearance have been observed in all CF patients, but only some of them displayed sinonasal polyposis and middle turbinate hypertrophy, we examined the role of morphological nasal structures in the shaping of microbiome composition of the upper respiratory tract and its impact on lung microbiome.
MATERIALS AND METHODS
Materials. In the study, we used 53 samples collected from 21 CF patients (15 adults, median age 27.3 years; 6 children, median age 8.7 years): 22 sputum samples, 14 maxillary sinus lavage samples, 2 nasopharyngeal swabs, 8 tracheal aspirates, and 7 polyp fragments obtained during polypectomy. Based on the upper respiratory tract state, all patients were diagnosed with (mostly severe) chronic rhinosinusitis with/without nasal polyps by an ENT (ear, nose and throat) doctor. All samples were collected by in-hospital medical specialists.
DNA from sputum samples was isolated with a Maxwell 16 Tissue DNA Purification Kit in accordance with the manufacturer’s recommendations and analyzed with a Maxwell MDX Instrument (Promega, USA).
Dominant microbial species in each biological sample was identified using 16S rDNA amplification and sequencing according to Voronina et al. [22]. Rapid test for detecting bacteria of the Order Burkholderiales in the respiratory tract of a CF patient was performed according to the genotyping protocol proposed by Voronina et al. [23]. Pseudomonas aeruginosa was identified by detecting trpE (the most variable target) in the MLST (multilocus sequence typing) scheme according to Curran et al. [24] with modifications.
Identification of the mecA gene within the staphylococcal cassette chromosome mec (SCCmec) responsible for methicillin resistance was performed using the primers Mec_10-11_For (5′-ATGTATGCTTTGGTCTTTCT-3′) and Mec_10-11_Rev (5′-TACACATATCGTGAGCAATGA-3′) designed by us. The amplification regime for the mecA gene fragment was as follows: 95°C – 10 min; 35 cycles of 95°C – 30 sec, 52°C – 1 min, 72°C – 1 min; 72°C – 5 min. Positive samples contains a 584-bp-long amplification product.
Mycosis-causing pathogens were identified by amplification and sequencing of the ITS1_5.8S_ITS2 region as suggested by Voronina et al. [25].
Microbiome composition in the upper respiratory tract was determined by massively parallel sequencing of the 16S rDNA gene amplicons with an MiSeq Illumina platform as proposed by Ryzhova et al. [19].
The data were analyzed with the Microbial Genomics module of the CLC Genomic Workbench v.11-12 software. The Greengenes v.13_8 database was used to identify OTUs (operational taxonomic units) with 97% similarity.
Sequencing data were deposited to the SRA (Sequence Read Archive) NCBI under the BioProject accession numbers PRJNA544655 and PRJNA544933 for the adult and children microbiome data, respectively.
Phylogenetic diversity index, Simpson diversity index, Shannon entropy, and Chao 1 bias-corrected estimator were used for assessing the alpha diversity (microbiota taxonomic diversity in samples). The beta-diversity (ratio between microbiota taxonomic diversities in various samples) was assessed by using the Jaccard index, Bray–Curtis index, Euclidean distance, and various UniFrac metrics (Unweighted UniFrac, Weighted UniFrac, Weighted unnormalized UniFrac, D_0 UniFrac, and D_0.5 UniFrac) accounting for the phylogenetic interspecific relationships [26].
Similarity of microbiota in the samples was assessed using the Principal Coordinate Analysis (PCoA) based on the multidimensional data scaling [27]. Adult CF patients were stratified based on the following parameters: 1) age: 18-25 and 26-30 years; 2) FEV1 (forced expiratory volume in one second) range: 70-115, 40-69, or <40%; 3) type of sample: derived from the upper or lower respiratory tract; 4) clinical score of lung disease: mild, moderate, or severe; 5) prevalent microbial species in community (eight groups); 6) class of mutations in the CFTR gene according to the LOVD v.0.1 database compiled by the Research Centre for Medical Genetics [28].
Statistical analysis for the significance of differences between the groups was performed using the PERMANOVA (permutational multivariate analysis of variance) test [29]. Each pairwise comparison was analyzed by calculating the pseudo-f statistic criteria, significance p-value, and Bonferroni-adjusted p-values. Inter-group significance level was set at p < 0.05.
RESULTS
Comparison of the microbiome composition in the upper vs. lower respiratory tract in pediatric CF patients. Studies published over the last decade have proven that the composition of the upper respiratory tract microbiome in CF patients should be continuously monitored. It is unclear, however, at what age microbes harmful to the lung function colonize the upper respiratory tract, especially paranasal sinuses and middle and superior turbinates. It is difficult to collect such biological samples in children, because the collection procedure is painful. Hence, polyp fragments excised during polypectomy can be used as a source of biological samples.
Analysis of samples obtained from CF patients demonstrated that four out of five patients were positive for Staphylococcus aureus, which is an autochthonous microorganism of the nasal cavity. In addition, methicillin sensitivity of S. aureus was confirmed by the lack of mecA gene signal in pediatric samples, suggesting an additional evidencing in favor of the autochthonous origin of this bacterial species. Patient 3-CHP, whose polyp-derived sample was positive for S. aureus, demonstrated normal lung function in the follow-up observations, as well as the absence of Proteobacteria in the tracheal aspirate sample that was dominated by Streptococcus spp.
Polyp and tracheal aspirate samples in four patients were positive for P. aeruginosa. In all the four patients, the genotypes of bacteria from the lower and upper parts of the respiratory tract were the same. It should be noted that the tracheal aspirates of patient 2-CHP at the age from 9 to 12 years were positive solely for Achromobacter xylosoxidans ST251 (a member of Proteobacteria); after its eradication, the lower respiratory tract was inhabited by P. aeruginosa with the genotype identical to the genotype of the bacterium previously detected in the nasal polyps.
Polyp samples from the twin patients 4-CHP and 5-CHP contained the fungus Auriculariopsis ampla (Fungi; Dikarya; Basidiomycota; Agaricomycotina; Agaricomycetes; Agaricomycetidae; Agaricales; Schizophyllaceae) along with P. aeruginosa of the same genotype, whereas the tracheal aspirates from both patients contained the yeast-like fungus Candida albicans. The microbiome composition differed even in the twin patients: S. aureus dominated in the polyp microbiome in patient 4-CHP, while Haemophilus influenzae (earlier detected in the tracheal aspirate sample) prevailed in the polyp sample from patient 5-CHP.
Therefore, the obtained data confirm that as early as in the childhood age, the upper respiratory tract in CF patients may become colonized by bacterial species able to infect the lungs.
Comparison of bacterial microbiome alpha diversity in pediatric and adult CF patients. Proteobacteria of the order Burkholderiales are hazardous to CF patients because of the potential to generate epidemic strains that can be transmitted by the airborne route. In Russia, the most clinically significant for CF patients microorganisms are Burkholderia cenocepacia ST709 (sequence type) and Achromobacter ruhlandii ST36 [21]. No eradication of these bacteria from the airway tract was observed during infection transition to the chronic stage. Hence, when the patient 82-CF aged 4 years 9 months tested positive for B. cenocepacia ST709, this patient was subjected to intensive therapy and surgical irrigation of the sinuses. It was found that even after eradicating these bacteria from the lungs, the sinus lavage sample contained various Burkholderiales spp. and other Proteobacteria. Despite the fact that antibiotic therapy was able to get rid of Firmicutes, the alpha diversity index (max 12) in the tracheal aspirate and sinus samples from the patient 82-CF was relatively high (Fig. 1) and markedly exceeded the alpha diversity index (max 7) in adult CF patients, including patient 61-CF with favorable prognosis, preserved lung function, and mild chronic rhinosinusitis with clean sinuses.
Fig. 1. Microbiome alpha diversity in samples from CF patients. A) Pediatric patient 82-CF: a, tracheal aspirate; n, sinus lavage; B) adult patients: s, sputum; n, sinus lavage.
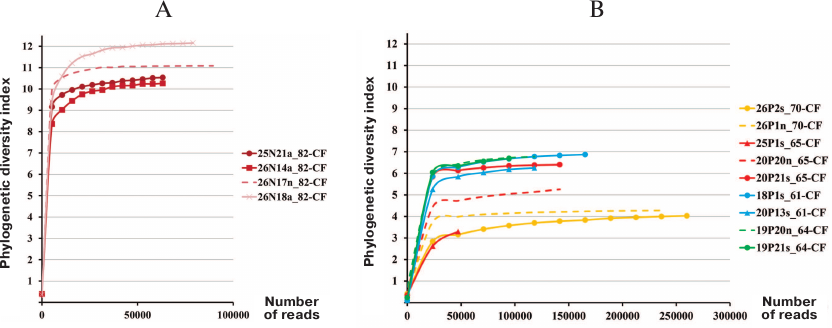
These data confirm that regular exacerbation of the disease and repeated antibiotic therapy reduce the microbiome diversity in CF patients, thereby lowering the ability of the “healthy” microbiota to withstand pathogenic microorganisms.
Comparison of the sinus and lower respiratory tract microbiomes in adult CF patients. Adult CF patients were divided into three groups according to the severity of lung disease: mild (Group 1), moderate (Group 2) and severe (Group 3). The only patient (61-CF) in Group 1 did not have Proteobacteria in the sputum sample; the sputum samples from all other patients were positive for species from the genera Burkholderia, Achromobacter, and Pseudomonas regardless of the severity score.
The sinus lavage sample from patient 61-CF was negative for bacterial and fungal species. Patient 74-CF contained trace amount of Burkholderia in the sinus lavage sample analyzed after polypectomy and sinus surgery (Fig. 2); S. aureus was prevalent in the sinus lavage sample, whereas B. cenocepacia ST709 comprised up to 90% total lung microbiome, which might be accounted for by an extremely impaired nasal airflow due to enlarged polyps.
Fig. 2. Prevalence of bacterial phyla (a) and Proteobacteria taxa (b) in the respiratory tract samples from adult CF patients: n, maxillary sinus lavage (nasopharyngeal swabs enriched in nasal sinus content in the case of patients 70-CF and 71-CF); s, sputum. Curly brackets denote samples from each patient.
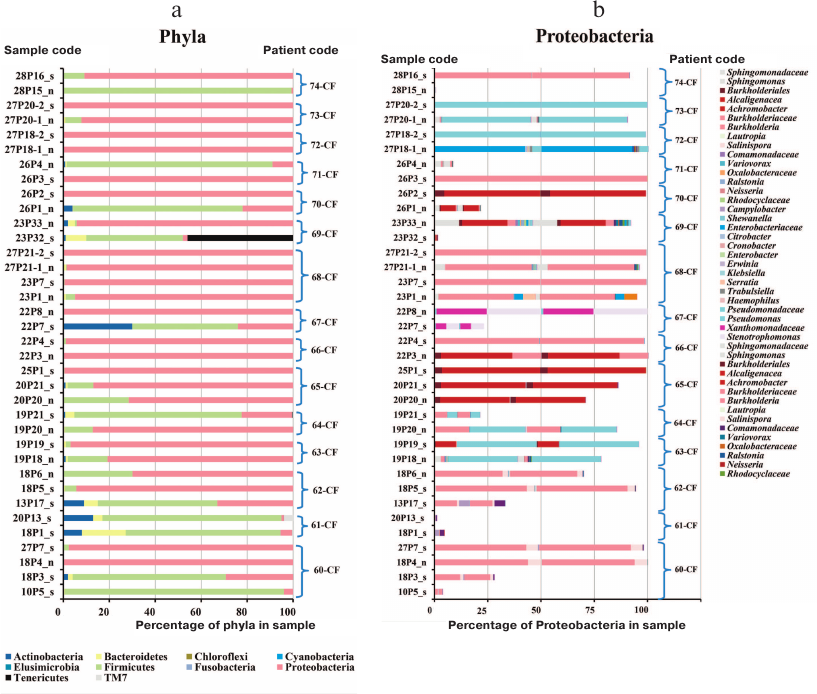
The prevalence of Firmicutes in the upper respiratory tract samples (“n”) from patients 70-CF and 71-CF could result from the difficulty in collecting biological material. Due to the disease severity, the sample were collected via sinus puncture allowing to obtain nasal washings that contained not only purulent nasal discharge, but also pharyngeal microbes. Therefore, these samples should be considered as nasopharyngeal washings enriched with the sinus discharge. Although “n” samples from these patients contained 80-90% Firmicutes in the microbiome (Fig. 2), the genotype of the identified Proteobacteria matched the genotype of bacteria earlier found in the sputum samples.
In other patients (e.g., patients 60-CF and 64-CF), Proteobacteria dominated in the sinus microbiome even during a period of favorable lung microbiome state.
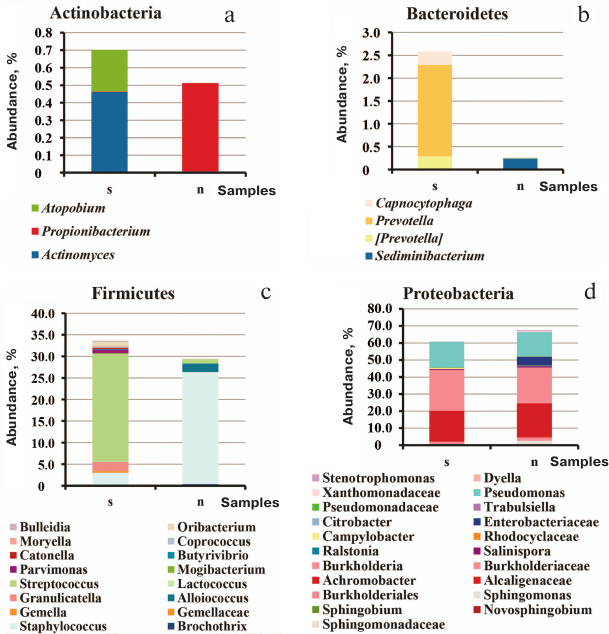
Comparison of all sputum and sinus lavage samples revealed a significant difference in Unweighted UniFrac (p = 0.00118) and D_0 UniFrac (p = 0.0001) (Fig. 3). The sinus and sputum microbiomes differed in the prevalence of Firmicutes, as well as minor Actinobacteria and Bacteroides (see Fig. 3 and table for the taxon composition and significance levels). In the sinus samples, the prevailing Firmicutes were Staphylococcus species (as expected), whereas in the sputum, they were Streptococcus species. Bacteroides Prevotella and Capnocytophaga detected in the sputum samples were virtually absent in the sinus microbiome. The phylum Actinobacteria was mainly represented by Actinomyces and Atopobium in the sputum samples and Propionibacterium in the sinus microbiome. The species composition of Proteobacteria prevalent in the sinus and lung microbiomes did not differ. The sinus microbiome also contained Enterobacteria. Therefore, we obtained another evidence indicating that paranasal sinuses serve as a reservoir for Proteobacteria able to perpetuate lung infection in CF patients.
Significance level for the differences in the taxon composition of
Actinobacteria, Bacteroides, and Firmicutes in the sputum and sinus
lavage samples
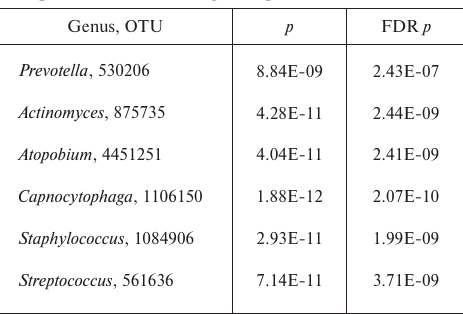
Note: FDR, false discovery rate.
Fig. 3. Species composition of the phyla Actinobacteria (a), Bacteroidetes (b), Firmicutes (c), and Proteobacteria (d) in the sputum and upper respiratory tract samples from adult CF patients: s, 21 sputum samples; n, 13 maxillary sinus lavage samples and 2 nasopharyngeal swabs enriched in the nasal sinus content.
Comparison of the sinus microbiome composition in adult CF patients with and without nasal polyps. Chronic rhinosinusitis was diagnosed by an ENT specialist in all adult CF patients enrolled in the study. Only patient 61-CF had a mild disease course; the rest 14 patients were characterized by severe rhinosinusitis. Eight of them fully lacked polyps, and six patients had grade 2 polyps on both sides. In patient 68-CF, the sinus microbiome composition was analyzed before and after polypectomy and sinus surgery, whereas in patient 72-CF, it was assessed in the sinus lavage two months after the third polypectomy and sinus surgery performed over the last 16 years.
As seen from the PCoA plots for the sinus microbiome data presented in Fig. 4a, no polyp-dependent clustering was observed. However, the microbiome composition differed significantly in groups I and II dominated by Burkholderia and Pseudomonas, respectively. Moreover, group I included both samples collected from patient 68-CF. In patient 72-CF, the sinus microbiome, even after the third surgery, contained only Proteobacteria (100%) with E. coli as a dominant species, whereas P. aeruginosa prevailed in the lung microbiome (<10%) (Fig. 2). Therefore, surgical intervention alone is insufficient for eradication of Proteobacteria and should be accompanied by prolonged antibiotic therapy via inhalation, which requires patient’s compliance with treatment.
Fig. 4. PCoA microbial diversity in samples collected from adult CF patients. a) Maxillary sinus lavage (n = 13) and nasopharyngeal samples (n = 2) from patents with (red circles) and without (blue circles) polyps. Subgroup I, samples containing Burkholderia; subgroup II, samples containing Pseudomonas. Asterisks denote nasopharyngeal swab samples. b) All samples obtained from adult patients. Subgroup I, samples containing Burkholderia; subgroup II, samples containing Achromobacter; subgroup III, samples containing Pseudomonas.
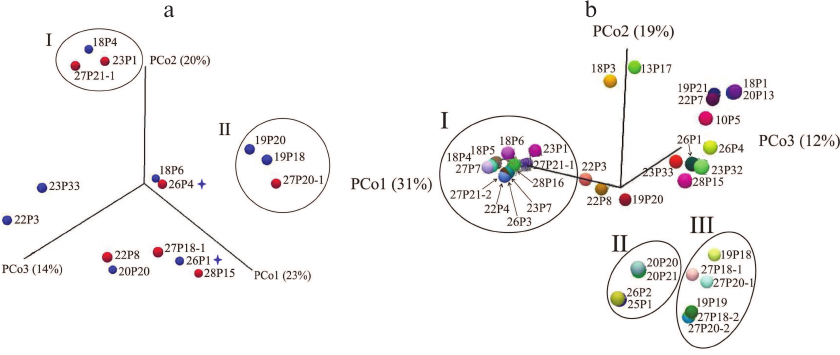
Comparison of the respiratory tract microbiome composition in groups of adult CF patients. Analysis of PCoA data for all respiratory tract samples (Fig. 4b) revealed three significantly different groups featured by different dominant microbial species. The most abundant Group I (Burkholderia) differed from Group II (Achromobacter) by eight diversity indices (Bray–Curtis p = 0.00016) and from Group III (Pseudomonas) – by all indices except Euclidean (Bray–Curtis p = 0.000116). The difference between Group II and Group III was confirmed by assessing eight diversity indices (Bray–Curtis p = 0.00433).
Of note is location of samples obtained from patient 61-CF with the healthiest microbiome composition (the most distant points on the PCo3 axis, 12%). An increased percentage of Firmicutes and decreased abundancy of Proteobacteria (down to 30%) in the sputum samples from patients 60-CF and 62-CF obtained during the favorable period resulted in the shift of the sample points upwards on the PCo2 axis (19%).
At the same time, sinus lavage samples with a complex combination of several Proteobacteria species (22P3, 22P8, 19P20) were clustered along the PCo1 axis 31%, whereas the sputum samples containing a combination of Proteobacteria species (19P21 and 22P7) were shifted along the PCo2 axis 19%.
The microbiome composition in the subgroups with different FEV1 values (70-115% vs. <40%) differed significantly (Bray–Curtis p = 0.00899).
Comparison of subgroups identified according to the Class of mutations revealed that they differed significantly between Class II/Class I and Class V/Class I (Bray–Curtis p = 0.02857), as well as between Class II/Class II and Class V/Class I (Euclidean p = 0.02573). These data emphasize the contribution of mild mutations (Class V), even in the heterozygous state in a combination with Class I mutations.
DISCUSSION
Personalized medicine that takes into account the impact of genetic and environmental factors on human health [30], cannot ignore an essential influence of the microbiome-derived gene set. Pediatric and adult CF Centers have developed a systemic approach to the treatment of this disease that affects multiple organs and tissues. One of the therapeutic strategy components is microbiological diagnostics. Collection of biological samples by pulmonologists and ENT specialists and long-term follow-up with the consideration of antibiotic regime, physiotherapy, and nutrition, as well as patient migration across different geographic regions, help in the interpretation of changes in the patient’s respiratory tract microbiome in order to provide timely adjustments to the treatment and stabilization of the patient’s condition.
Studies of the upper respiratory tract microbiome have revealed another reservoir of the infection, as well as justified and facilitated active introduction of inhalation therapy in CF patients. Morphological changes in the nasal cavity (e.g., polyps) that can decrease the efficacy of inhalation therapy have recently come into attention of ENT specialists and microbiologists.
According to Pletcher et al. [18], sinonasal polyps are found in more than 40% children with CF. Polypectomy in childhood results in better sinonasal blood flow, which indirectly improves patient’s quality of life, but does not guarantee the absence of further polyp recurrence. For instance, patient 72-CF in our study underwent polypectomy three times.
Examining microbiome composition in polyp washings after surgical removal demonstrated that as early as at the age of seven years, sinuses of CF patients were infected with P. aeruginosa that was found to be prevalent in our small patient cohort. Detection of the basidiomycete Auriculariopsis ampla from the Schizophyllaceae family in the samples from the twin patients is a warning sign. Thus, Schizophyllum commune (another microorganism from this family) is known to infect humans since 1950, mainly affecting the respiratory system and causing bronchopulmonary disease in 63% cases and sinusitis in 31% cases [31].
It should be noted that the twin patients had individual changes in the respiratory tract microbiome. A 3-year follow-up of the lower respiratory tract microbiome allowed to identify such changes even before detecting P. aeruginosa, when the microbiome of one of twins was dominated by H. influenzae, now found in the sinus samples. However, in the latest tests, the tracheal aspirate samples from the twin patients, otherwise similar in the majority of microbial species, differed in the composition of prevalent Flavobacterium spp.: patient 4-CHP was positive for Capnocytophaga spp., while patient 5-CHP – for Chryseobacterium spp.
Polyps were observed in 40% adult CF patients. Comparison of the phylogenetic diversity of the sinonasal microbiome in both groups revealed no significant differences between them. Polypectomy and sinus surgery did not result in the short-term changes in the sinus microbiome composition in two post-surgery patients; therefore, long-lasting antibiotic therapy under compliance with physician’s recommendations might be more efficient in affecting the sinus microbiome. Currently, we can state that the altered mucociliary clearance is the determining factor in the development of sinonasal infections.
Non-CF patients with chronic rhinosinusitis were studied by Biswas et al., who demonstrated no difference in the phylogenetic diversity or the level of inflammatory markers between the patients with or without polyps [32].
Proteobacteria (the most dangerous pathogenic microbes) of the same genotype as was detected in both respiratory tract regions in 13 out of 15 adult CF patients. Moreover, in 10 patients, this species comprised up to 70-100% of sinus microbiome; in three of these patients, the prevalence of Proteobacteria was observed even during the period of relatively beneficial lung microbiome composition. In addition, we also found Burkholderia, Pseudomonas, and Achromobacter and their combination, as well as Stenotrophomonas and E. coli combined with Pseudomonas. Biswas et al. observed decreased sinus microbiome diversity in samples with Proteobacteria spp., such as Pseudomonas, Haemophilus, and Achromobacter [32].
Eight out of analyzed 14 adult patients displayed markedly overlapping patterns of microbial species in the sinus and lung microbiomes. However, six CF patients contained sinus microbial species that were absent in the lung microbiome, e.g., E. coli in patient 72-CF. According to some researchers (see Lucas et al. [33]), the presence of a pathogenic microbial species in the sinus does not predict its emergence later in the lungs [33], whereas Fothergill et al. [34] demonstrated that Pseudomonas bacteria in the sinus can acquire adaptations ensuring their efficient colonization of the lower respiratory tract.
Therefore, further lung colonization by sinonasal microbes is solely a matter of time, and properly chosen therapeutic strategy may serve as a restriction factor.
Currently, because of the emergence of targeted drugs allowing partial restoration of the chloride channel activity, the CF therapeutic strategy is determined by the class of mutation in the CFTR gene. Here, we demonstrated a correlation between the class of mutation and diversity of microbial communities in the respiratory tract. In particular, a mild mutation (Class V) even combined with the Class I mutation improved the microbiome composition. The most unusual microbiome composition was found in patient 67-CF carrying the [delta]F508/P205S (Class II/Class V) mutations: the lung microbiome contained Actinobacteria (Rothia, 29%), Firmicutes (Lactobacillus, 6%; Lactococcus, 2%; Streptococcus, 37%), and Proteobacteria (Pseudomonas, 1%; Xanthomonadaceae, 10%; Stenotrophomonas, 12%), whereas the sinus microbiome contained Proteobacteria (Pseudomonas, 2%; Xanthomonadaceae, 47%; Stenotrophomonas, 51%).
Hence, our data support the concept of microbial translocation occurring across the respiratory tract in CF patients, but disprove the role of sinonasal polyps in shaping the composition and diversity of microbial communities. Most likely, an individual state of mucosal layers and mucociliary clearance, as well as anatomical features of patient’s paranasal sinuses, are of importance. The lack of pathogenic microbes in one region of the respiratory tract, but their presence in another region is likely a temporary event related to the antibiotic resistance and the used treatment strategy.
Our study demonstrated a need for monitoring the microbiome composition of the sinuses and lungs and proved a higher value of molecular and genetic approaches vs. bacterial cultivation. The continued microbiome analysis, including assessment of microbiome minor components, will provide better understanding of the triggers in the infectious process development.
Funding. The study was conducted within the framework of the 2018 State Assignment no. 056-00108-18-00 and 2019-2020 Planning Period and the 2019 State Assignment no. 056-00078-19-00 and 2020-2021 Planning Period for the Gamaleya National Research Center for Epidemiology and Microbiology, Ministry of Health of the Russian Federation.
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical standards. An informed consent was obtained from adult and CF patients over 15 years old; parental or guardian consent was obtained for pediatric patients under 15 years old. All procedures used to examine biological samples from the patients with CF and congenital lung malformation were approved by the Biomedical Ethics Committee at the Gamaleya National Research Center for Epidemiology and Microbiology, Ministry of Health of the Russian Federation (Protocol no. 1, 17.05.2012).
REFERENCES
1.Woese, C. R. (2004) A new biology for a new
century, Microbiol. Mol. Biol. Rev., 68, 173-186, doi:
10.1128/MMBR.68.2.173-186.2004.
2.Stackebrandt, E., and Woese, C. R. (1984) The
phylogeny of prokaryotes, Microbiol. Sci., 1,
117-122.
3.Land, M., Hauser, L., Jun, S. R., Nookaew, I.,
Leuze, M. R., Ahn, T. H., Karpinets, T., Lund, O., Kora, G., Wassenaar,
T., Poudel, S., and Ussery, D. W. (2015) Insights from 20 years of
bacterial genome sequencing, Funct. Integr. Genomics, 15,
141-161, doi: 10.1007/s10142-015-0433-4.
4.Olsen, G. J., Larsen, N., and Woese, C. R. (1991)
The ribosomal RNA database project, Nucleic Acids Res.,
19 (Suppl.), 2017-2021, doi: 10.1093/nar/19.suppl.2017.
5.NIH Human Microbiome Project
(https://hmpdacc.org/).
6.Proctor, L. M. (2011) The Human Microbiome Project
in 2011 and beyond, Cell Host Microbe, 10, 287-291, doi:
10.1016/j.chom.2011.10.001.
7.Nichols, D. P., and Chmiel, J. F. (2015)
Inflammation and its genesis in cystic fibrosis, Pediatr.
Pulmonol., 50 (Suppl. 40), S39-S56, doi:
10.1002/ppul.23242.
8.Salsgiver, E. L., Fink, A. K., Knapp, E. A.,
LiPuma, J. J., Olivier, K. N., Marshall, B. C., and Saiman, L. (2016)
Changing epidemiology of the respiratory bacteriology of patients with
cystic fibrosis, Chest, 149, 390-400, doi:
10.1378/chest.15-0676.
9.Dickson, R. P., Erb-Downward, J. R., Martinez, F.
J., and Huffnagle, G. B. (2016) The microbiome and the respiratory
tract, Annu. Rev. Physiol., 78, 481-504, doi:
10.1146/annurev-physiol-021115-105238.
10.Zakharkina, T., Heinzel, E., Koczulla, R. A.,
Greulich, T., Rentz, K., Pauling, J. K., Baumbach, J., Herrmann, M.,
Grunewald, C., Dienemann, H., von Muller, L., and Bals, R. (2013)
Analysis of the airway microbiota of healthy individuals and patients
with chronic obstructive pulmonary disease by T-RFLP and clone
sequencing, PLoS One, 8, e68302, doi:
10.1371/journal.pone.0068302.
11.Voronkova, A. Yu., Amelina, E. L., Kashirskaya,
N. Yu., Kondrat’eva, E. I., Krasovskiy, S. A., Starinova, N. I.,
and Kapranov, N. I. (2019) in The Russian Federation Cystic Fibrosis
Patient Registry 2017 (Voronkova, A. Yu., ed.) [in Russian],
Medpraktika-M, Moscow, p. 68.
12.European Cystic Fibrosis Society Patient
Registry. Annual data report (year 2016), v. 1.2018
(www.ecfs.eu/sites/default/files/general-content-images/working-groups/ecfs-patient-registry/ECFSPR_Report2016_06062018.pdf).
13.Einarsson, G. G., Zhao, J., LiPuma, J. J.,
Downey, D. G., Tunney, M. M., and Elborn, J. S. (2019) Community
analysis and co-occurrence patterns in airway microbial communities
during health and disease, ERJ Open Res., 5, 00128-2017,
doi: 10.1183/23120541.00128-2017.
14.Caverly, L. J., and LiPuma, J. J. (2018) Cystic
fibrosis respiratory microbiota: unraveling complexity to inform
clinical practice, Expert Rev. Respir. Med., 12, 857-865,
doi: 10.1080/17476348.2018.1513331.
15.Isles, A., Maclusky, I., Corey, M., Gold, R.,
Prober, C., Fleming, P., and Levison, H. (1984) Pseudomonas
cepacia infection in cystic fibrosis: an emerging problem, J.
Pediatr., 104, 206-210, doi:
10.1016/s0022-3476(84)80993-2.
16.Lobo, L. J., Tulu, Z., Aris, R. M., and Noone, P.
G. (2015) Pan-resistant Achromobacter xylosoxidans and
Stenotrophomonas maltophilia infection in cystic fibrosis does
not reduce survival after lung transplantation, Transplantation,
99, 2196-2202, doi: 10.1097/TP.0000000000000709.
17.Voronina, O., Ryzhova, N., Kunda, M., Sharapova,
N., Aksenova, E., Amelina, E., Shumkova, G., Simonova, O., Egorov, M.,
Kondratyeva, E., Chuchalin, A., and Gintsburg, A. (2018) Changes in
airways bacterial community with cystic fibrosis patients’ age
and lung function decline, 41st Eur. Cystic Fibrosis Conf., J.
Cystic Fibrosis, 17 (Suppl. 3), S78, doi:
10.1016/S1569-1993(18)30366-7.
18.Pletcher, S. D., Goldberg, A. N., and Cope, E. K.
(2019) Loss of microbial niche specificity between the upper and lower
airways in patients with cystic fibrosis, Laryngoscope,
129, 544-550, doi: 10.1002/lary.27454.
19.Ryzhova, N. N., Voronina, O. L., Loseva, E. V.,
Aksenova, E. I., Kunda, M. S., Sharapova, N. E., Sherman, V. D., and
Gintsburg, A. L. (2019) Respiratory tract microbiome in children with
cystic fibrosis, Sib. Med. Obozrenie, 2, 19-28,
doi: 10.20333/2500136-2019-2-19-28.
20.Voronina, O. L., Ryzhova, N. N., Kunda, M. S.,
Aksenova, E. I., Sharapova, N. E., Amelina, E. L., Lazareva, A. V.,
Chernevich, V. P., Simonova, O. I., Zhukhovitskiy, V. G., Zhilina, S.
V., Semykin, S. Yu., Polikarpova, S. V., Asherova, I. K., Orlov, A. V.,
and Kondratenko, O. V. (2019) Major trends in altered diversity of
Burkholderia spp. infecting cystic fibrosis patients in the
Russian Federation, Sib. Med. Obozrenie, 2, 80-88, doi:
10.20333/2500136-2019-2-80-88.
21.Voronina, O. L., Kunda, M. S., Ryzhova, N. N.,
Aksenova, E. I., Sharapova, N. E., Semenov, A. N., Amelina, E. L.,
Chuchalin, A. G., and Gintsburg, A. L. (2018) On Burkholderiales order
microorganisms and cystic fibrosis in Russia, BMC Genomics,
19 (Suppl. 3), 74, doi: 10.1186/s12864-018-4472-9.
22.Voronina, O. L., Kunda, M. S., Ryzhova, N. N.,
Aksenova, E. I., Semenov, A. N., Lasareva, A. V., Amelina, E. L.,
Chuchalin, A. G., Lunin, V. G., and Gintsburg, A. L. (2015) The
variability of the order Burkholderiales representatives in the
healthcare units, BioMed Res. Int., 2015, 680210,
doi: 10.1155/2015/68021.
23.Voronina, O. L., Kunda, M. S., Aksenova, E. I.,
Orlova, A. A., Chernukha, M. Yu., Lunin, V. G., Amelina, E. L., and
Chuchalin, A. G., and Gintsburg, A. L. (2013) Express test for
detecting microbes infecting respiratory tract in patients with cystic
fibrosis, Klin. Lab. Diag., 11, 53-57.
24.Curran, B., Jonas, D., Grundmann, H., Pitt, T.,
and Dowson, C. G. (2004) Development of a multilocus sequence
typing scheme for the opportunistic pathogen Pseudomonas
aeruginosa, J. Clin. Microbiol., 42, 5644-5649, doi:
10.1128/JCM.42.12.5644-5649.2004.
25.Voronina, O. L., Ryzhova, N. N., Kunda, M. S.,
Aksenova, E. I., Ovchinnikov, R. S., Fedosova, N. F., Amelina, E. L.,
Lunin, V. G., Chuchalin, A. G., and Gintsburg, A. L. (2015) Developing
approaches to identify lung mycosis pathogens directly in respiratory
tract clinical samples from patients with cystic fibrosis, Lab.
Sluzhba, 4, 11-17.
26.Chen, J., Bittinger, K., Charlson, E. S.,
Hoffmann, C., Lewis, J., Wu, G. D., Collman, R. G., Bushman, F.
D., and Li, H. (2012) Associating microbiome composition with
environmental covariates using generalized UniFrac distances,
Bioinformatics, 28, 2106-2113, doi:
10.1093/bioinformatics/bts342.
27.A consensus report on clinical effects of genetic
variants in the Federal State Budgetary Scientific Institution
“Research Centre for Medical Genetics” [in Russian], Leiden
Open Variation Database, v. 3.0 (http://seqdb.med-gen.ru/).
28.Clustering and Classification Methods for
Biologists, Manchester Metropolitan University
(http://www.angelfire.com/planet/biostats/upload.htm).
29.Anderson, M. J. (2001) A new method for
non-parametric multivariate analysis of variance, Austral.
Ecol., 26, 32-46, doi:
10.1111/j.1442-9993.2001.01070.pp.x.
30.Ginsburg, G. S., and Willard, H. F. (2009)
Genomic and personalized medicine: foundations and applications,
Transl. Res., 154, 277-287, doi:
10.1016/j.trsl.2009.09.005.
31.Chowdhary, A., Randhawa, H. S., Gaur, S. N.,
Agarwal, K., Kathuria, S., Roy, P., Klaassen, C. H., and Meis, J. F.
(2013) Schizophyllum commune as an emerging fungal pathogen: a
review and report of two cases, Mycoses, 56, 1-10, doi:
10.1111/j.1439-0507.2012.02190.x.
32.Biswas, K., Cavubati, R., Gunaratna, S., Hoggard,
M., Waldvogel-Thurlow, S., Hong, J., Chang, K., Wagner Mackenzie, B.,
Taylor, M. W., and Douglas, R. G. (2019) Comparison of subtyping
approaches and the underlying drivers of microbial signatures for
chronic rhinosinusitis, mSphere, 4, e00679-18, doi:
10.1128/mSphere.00679-18.
33.Lucas, S. K., Yang, R., Dunitz, J. M., Boyer, H.
C., and Hunter, R. C. (2018) 16S rRNA gene sequencing reveals
site-specific signatures of the upper and lower airways of cystic
fibrosis patients, J. Cyst. Fibros., 17, 204-212, doi:
10.1016/j.jcf.2017.08.007.
34.Fothergill, J. L., Neill, D. R., Loman, N.,
Winstanley, C., and Kadioglu, A. (2014) Pseudomonas aeruginosa
adaptation in the nasopharyngeal reservoir leads to migration and
persistence in the lungs, Nat. Commun., 5, 4780, doi:
10.1038/ncomms5780.