Verification of the Stabilized Protein Design Based on the Prediction of Intrinsically Disordered Regions: Ribosomal Proteins L1
G. S. Nagibina1,a*, V. V. Marchenkov1, K. A. Glukhova1, T. N. Melnik1, and B. S. Melnik1
1Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia* To whom correspondence should be addressed.
Received August 9, 2019; Revised September 27, 2019; Accepted September 27, 2019
In our previous papers, we proposed the idea that programs predicting intrinsically disordered regions in amino acid sequences can be used for finding weakened sites in proteins. The regions predicted by such programs are suitable targets for the introduction of protein-stabilizing mutations. However, for each specific protein, it remains unclear what determines protein stabilization – the amino acid sequence (and accordingly, prediction of weakened sites) or the 3D structure. To answer this question, it is necessary to study two proteins with similar structures but different amino acid sequences and, consequently, different predictions of weakened regions. By introducing identical mutations into identical elements of the two proteins, we will be able to reveal whether predictions of the weakened sites or the 3D protein structure are the key factors in the protein stability increase. Here, we have chosen ribosomal proteins L1 from the halophilic archaeon Haloarcula marismortui (HmaL1) and extremophilic bacterium Aquifex aeolicus (AaeL1). These proteins are identical in their structure but different in amino acid sequences. A disulfide bond introduced into the region predicted as the structured one in AaeL1 did not lead to the increase in the protein melting temperature. At the same time, a disulfide bond introduced into the same region in HmaL1 that was predicted as a weakened one, resulted in the increase in the protein melting temperature by approximately 10°C.
KEY WORDS: ribosomal protein L1, globular protein stabilization, protein intrinsically disordered regions, disulfide bondsDOI: 10.1134/S0006297920010083
Abbreviations: AaeL1, ribosomal protein L1 from the extremophilic bacterium Aquifex aeolicus; HmaL1, ribosomal protein L1 from the halophilic archaeon Haloarcula marismortui.
Proteins lacking fixed tertiary structure (the so-called disordered
proteins) were discovered in the middle of 20th century [1]. Active studying of these proteins has started in
the end of 1990s and yielded numerous on this topic in recent years [2, 3]. The interest to
intrinsically disordered proteins has stimulated active development of
theoretical programs that use amino acid sequence to predict the class
of protein, whether it is fully structured or intrinsically disordered
[2, 3]. Such programs used
quite different theoretical approaches (e.g., neuronal networks in
DISOPRED [4] or calculation of occurrence of
individual amino acid residues in different proteins in the regions
undetectable by X-ray diffraction in DisEMBL [5]).
There are also programs that combine several different approaches, the
so-called metapredictors (PONDR-FIT) [6].
Despite different theoretical approaches, the predictions by different program are in a rather good agreement. Moreover, according to these programs, virtually any densely packed protein has small fragments of the polypeptide chain predicted as intrinsically disordered, although such fragments are densely packed, structured, and not considerably different from the other regions of the analyzed protein [7]. We suggested that that algorithms used in these programs should be able to predict not only intrinsically disordered regions (mobile and unstructured), but also regions unable to independently acquire rigid packaging. In our previous work, we tested the idea that these regions could be targets for the introduction of structure-stabilizing mutations, i.e., mutations leading to the formation of artificial disulfide bonds at the protein surface [8]. Our purpose was to stabilize the Gαo protein from Drosophila melanogaster which could not be crystallized for a long time. We proposed that the problem could be associated with the mobility and instability of some regions of its polypeptide chain. The amino acid sequence of the Gαo protein was analyzed with the PONDR-FIT and IsUnstruct programs [6, 9] and one of its regions was predicted as intrinsically disordered. For this region, we designed a disulfide bond that stabilized the protein Gαo structure (increased the melting temperature by 4 K) [8]. Therefore, we demonstrated that programs predicting intrinsically disordered regions of the polypeptide chain can be used for searching for weakened regions in proteins.
To test this idea in more detail, it was necessary to make sure that the main parameter affecting protein stabilization after introduction of the stabilizing mutation was predicted weakness of the polypeptide region, but not specific features of protein secondary or tertiary structure.
For this, we had to study two proteins with the identical structure but considerably different amino acid sequences.
We have chosen ribosomal proteins L1 from the halophilic archaeon Haloarcula marismortui (HmaL1) and extremophilic bacterium Aquifex aeolicus (AaeL1). The 3D structure of AaeL1 was determined in [10]; the structure of HmaL1 was modeled in [11]. Both proteins have the structure typical for ribosomal proteins L1 that includes two domains connected with a double-chain linker. The N- and C-ends of the protein are located in domain I responsible for the binding of 23S rRNA. Domain II is involved in the rRNA recognition [11]. AaeL1 and HmaL1 have different amino acid sequences (identity, 33%), but identical spatial structure. This highly conserved structure was the reason why we chose ribosomal proteins L1 for our studies. Unfortunately, sufficiently different amino acid sequences with significantly different predictions of intrinsically disordered regions were found only in proteins “adapted” to functioning under very different conditions. Since in this work we did not intend to compare L1 proteins from different organisms, we had to compare wild-type and mutant proteins in order to reveal how introduction of disulfide bridges into identical regions of protein structure with different predictions of intrinsically disordered regions would influence protein stability. Obviously, each pair of proteins was compared under the same conditions.
Next, we analyzed domain II in these proteins, because predictions of intrinsically disordered regions in this domain were different for the two studied proteins. Thus, domain II was predicted to be intrinsically disordered in HmaL1, but partially structured in AaeL1. Therefore, designing mutations in domain II would have allowed us to compare the influence of disulfide bridge introduced into identical structural elements having different amino acid sequences and, consequently, different predisposition for intrinsic disorder in structurally identical proteins. Such studies would have helped to verify the assumption that theoretical approaches for the prediction of intrinsically disordered regions in proteins can be used to design mutations increasing protein stability.
Our assumptions were fully confirmed. The disulfide bond introduced into the region of HmaL1 amino acid sequence predicted to be intrinsically disordered led to the increase in the protein stability. The disulfide bond introduced into in the AaeL1 region with the same structure but predicted to be structured did not influence the protein stability.
MATERIALS AND METHODS
Cloning and expression of AaeL1 and HmaL1 proteins. The plasmid coding for AaeL1 (pET11a-PL-AaeL1) was expressed in Escherichia coli BL21(DE3) cells. The plasmid coding for the AaeL1 mutant was constructed based on the pET11a-PL-AaeL1 vector (Novagen, USA) by site-directed mutagenesis using a pair of corresponding primers (Eurogen, Russia; see table) and KOD Hot Start DNA Polymerase (Novagen) in accordance with the manufacturer’s instructions. The obtained construct was verified by sequencing (Eurogen). AaeL1 and AaeL1-D101C-K127C proteins were purified as described by Nikonova et al. [10]. The plasmid coding for the HmaL1-E82C-D114C mutant was constructed based on the pET11a-PL-HmaL1 plasmid (Novagen) carrying the gene for the wild-type protein by a standard PCR method using pET11a-PL vector as a template. To increase the efficiency of protein purification, the recombinant proteins were modified with the C-terminal His6-tag. For this, DNA fragment encoding the His6 sequence was cut out from the pET19mod plasmid (Novagen) using restriction endonucleases NdeI and XbaI (Thermo, USA) and subcloned into the pET11a-PL-HmaL1-E82C-D114C vector [12]. The obtained constructs were verified by sequencing. Escherichia coli Rosetta (DE3) cells (Novagen) were transformed with the HmaL1-encoding plasmids and inoculated into liquid LB medium (Amresco, USA). The cells were grown at 37°C with aeration (Eppendorf, Germany) to the optical density (OD600) of 0.8-1.0; protein expression was induced by adding IPTG to the final concentration of 1 mM and the cells was then grown at 37°C to OD600 of 1.8-2.0. Protein expression was analyzed by SDS-PAGE. The cells were precipitated by centrifugation (8000g, 15 min) (Hermle, Germany), resuspended in buffer (50 mM Tris-HCl, pH 7.5, 2 M LiCl, 100 mM imidazole, 50 mM MgCl2, 1 mM EDTA, 1 mM PMSF) (Sigma, Germany), disintegrated with a high-pressure homogenizer EmulsiFlex-C3 (Avestin, Canada), and centrifuged (Beckman, USA) for 40 min at 90,000g and 4°C. The His6-tagged recombinant proteins were purified by affinity chromatography on a HisTrap HP column (5 ml) (GE Healthcare, USA) equilibrated with 50 mM Tris-HCl (pH 7.5) containing 2 M NaCl, 20 mM imidazole, and 50 mM MgCl2. Elution was performed with a 20-200 mM gradient of imidazole concentration in the same buffer. Fractions containing HmaL1 were collected and concentrated for further purification. Gel-filtration was performed on a HiLoad 26/600 Superdex 75 prep grade column (GE Healthcare) equilibrated with 50 mM Tris-HCl, pH 7.5, 3 M NaCl, 50 mM MgCl2, 500 mM NH4Cl. The purity of the obtained protein preparations was assessed by electrophoresis according to Laemmli [13]. The purified protein was concentrated and stored at 4°C. HmaL1-E82C-D114C protein was purified similarly, except for addition of 5 mM β-mercaptoethanol to prevent formation of intermolecular disulfide bonds.
Primer sequences used in the work

Oxidation of free SH-groups. Free SH-groups in AaeL1-D101C-K127C protein underwent spontaneous oxidation in solution. For oxidizing free SH-groups in HmaL1-E82C-D114C, we used a buffer containing 200 mM Tris-HCl (pH 8.8), 2 M NaCl, 50 mM MgCl2, 500 mM NH4Cl, and 1 mM EDTA. The protein was oxidized in the presence of 10 mM oxidized glutathione and 2 mM reduced glutathione. Incubation was performed at room temperature for 24 h. After incubation, the protein was transferred to the buffer containing 50 mM Tris-HCl, pH 7.5, 3 M NaCl, 50 mM MgCl2, and 500 mM NH4Cl. Formation of disulfide bridges in the protein mutants was assayed by Laemmli’s electrophoresis with modifications; the presence of disulfide bonds in the mutant proteins was controlled by adding β-mercaptoethanol.
Concentration of AaeL1 protein was determined from the absorption at 280 nm using extinction coefficient A1cm1 mg/ml = 0.59 [10]. The extinction coefficient for HmaL1 (A1cm1 mg/ml = 0.176) was calculated with the Vector-NTI program.
Intrinsic protein fluorescence was measured with a Varian Cary 100 spectrofluorometer (Varian, Australia) in quartz cuvettes with the optical path length of 1 cm at the excitation wavelength of 280 nm. Fluorescence emission spectra were recorded at 290-500 nm. All measurements were performed in 20 mM sodium phosphate buffer (pH 7.5) in the presence of 4 M urea in the temperature range of 21-98°C. The working protein concentration was 0.2 mg/ml.
Differential scanning microcalorimetry. Experiments with the AaeL1 and AaeL1-D101C-K127C proteins were performed using a SCAL-1 differential scanning microcalorimeter (SCAL, Russia) with a 0.33-ml glass cell at a heating rate of 1°C/min (1.0 K/min) at a pressure of 2.5 atm [14]. The samples were transferred into the buffer solution (20 mM sodium phosphate buffer, pH 7.5, 4 M urea) by dialysis. Protein concentration in the experiments (1.0 mg/ml) was measured with a spectrophotometer (Ecrom, Russia). The data were recorded using the SCAL program. The temperature dependence of the molar heat capacity was calculated by the standard method [15] and analyzed with the Sigma Plot program (SigmaPlot, USA).
Circular dichroism (CD) spectra were recorded with a Chirascan spectropolarimeter (Applied Photophysics, England). Far-UV CD spectra were recorded using cuvettes with the optical path of 0.1 cm. The protein concentration for recording far-UV CD spectra was 0.2 mg/ml for AaeL1 and AaeL1-D101C-K127C and 0.4 mg/ml for HmaL1 and HmaL1-E82C-D114C. The spectra were recorded at 200-250 nm. The molar ellipticity was calculated using equation:
[θ] = [θ] mes·M/(L·C),
where C is the protein concentration (mg/ml); L is the optical path length (mm); [θ] mes is the measured ellipticity (degrees); and M is the mean molecular mass of the protein (Da). The temperature dependence of the ellipticity at 215 nm was measured at the heating rate of 1°C/min. AaeL1 and AaeL1-D101C-K127C were studied in 20 mM sodium phosphate buffer (pH 7.5) in the presence of 4 M urea. HmaL1 and HmaL1-E82C-D114C were studied in 50 mM Tris-HCl (pH 7.5), 3 M NaCl, 50 mM MgCl2, and 50 mM NH4Cl.
A fragment of 23S rRNA from H. marismortui (helices 76-78) with the following sequence was kindly provided by S. V. Tishchenko (Institute of Protein Research, Russian Academy of Sciences, Laboratory of Structural Studies of Translational Apparatus):
ggGUCGCCGAUGUGCAGCAUAGGUAGGAGACACUACACAGGUACCCGCGCUAGCGGGCCACCGAGUCAACAGUGAAAUACUACCCGUCGGUGACcc.
The 23S rRNA fragment was incubated at 70°C for 10 min and then cooled on ice. To obtain the RNA–protein complex, the protein was dialyzed against the buffer containing 50 mM Tris-HCl (pH 7.5), 3 M NaCl, 500 mM NH4Cl, and 10 mM MgCl2 and then added to the 23S rRNA fragment solution at the rRNA/protein molar ratio of 1 : 1 and incubated at room temperature for 30 min.
Formation of the 23S rRNA–protein complex was assessed in vitro by size-exclusion chromatography on a Varian ProStar HPLC system (Varian, Australia) equipped with a Superdex 200 (24 ml) column for gel-filtration (GE Healthcare, USA) equilibrated with 50 mM Tris-HCl (pH 7.5) containing 3 M NaCl, 500 mM NH4Cl, and 20 mM MgCl2. The obtained 23S rRNA/protein mixture (0.2 mg/ml, 200 μl) was applied onto the column and eluted at 0.4 ml/min. The calibration curve was prepared using BSA, ovalbumin, ribonuclease, and aprotinin (Bio-Rad, USA).
RESULTS
Comparison of spatial structures and amino acid sequences of AaeL1 and HmaL1. The spatial structure of ribosomal proteins L1 is highly conserved. It includes two domains (I and II) connected by a flexible linker that allows them to alter their mutual orientation. Two variants of the domain arrangement in a solution are known: “open” and “closed” [10]. The “open” conformation is characteristic for archaeal L1 proteins (including HmaL1). In this arrangement, the RNA-binding sites of the domains are exposed and accessible to RNA. In the closed conformation, the domains are brought together, and their RNA-binding sites are hidden, as in bacterial L1 proteins (including AaeL1) [10].
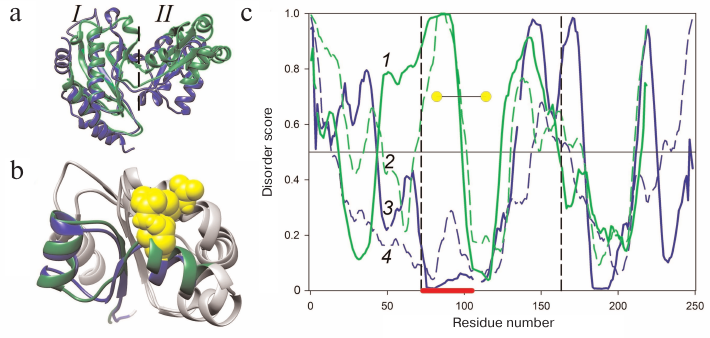
Figure 1a presents the superposition of the AaeL1 spatial structure (blue) and the model of HmaL1 spatial structure (green). Both proteins have the two-domain structure characteristic for L1 proteins. The N- and C-termini are in domain I which is connected through the double-chain linker with domain II. It can be seen that the major elements of domain I secondary structure in both proteins (β-sheet consisting of four antiparallel β-strands limited by two α-helices at one side) overlap well, with the exception of the C-terminal α-helix in domain I of AaeL1. Comparison of the structures of these two proteins reveals a characteristic difference between archaeal and bacterial L1 proteins, which is a different orientation of domain II relatively to domain I. However, the structures of domains II overlap (Fig. 1b). Both these domains have the Rossmann fold topology [16], with the β-sheet composed of four parallel β-strands surrounded by two α-helices at one side and four α-helices at the other side.
Fig. 1. Comparison of spatial structures and amino acid sequences of AaeL1 and HmaL1 proteins. a) Superposition of the spatial structure of AaeL1 (PDB:3QOY) (blue) and model of HmaL1 spatial structure (green). Dashed line separates domains I and II. b) Superposition of spatial structures of domains II from AaeL1 and HmaL1. The region of AaeL1 polypeptide chain predicted as structured by the PONDR-FIT and IsUnstruct programs is shown in blue. The region of HmaL1 polypeptide chain predicted as disordered is shown in green. Amino acid residues chosen by us for substitution with cysteine residues are shown in yellow. c) The probability of amino acid residues in HmaL1 (green lines 1 and 2) and AaeL1 (blue lines 3 and 4) to be disordered (to be in intrinsically disordered state) calculated with the PONDR-FIT (solid lines) and IsUnstruct (dashed lines) programs. The vertical lines indicate the regions of amino acid sequences forming domain II. Red line at the bottom of the graph indicates the region predicted as structured in AaeL1 and disordered in HmaL1. Yellow circles show positions for the introduction of disulfide bonds. (Colored version of Fig. 1 is available in electronic version of the article on the site http://sciencejournals.ru/journal/biokhsm/)
Based on the comparison of amino acid sequences of AaeL1 and HmaL1 using the BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi), the identity of these sequences is 33%. Figure 2 shows the alignment of AaeL1 and HmaL1 amino acid sequences (identical amino acids are shown in gray). AaeL1 and HmaL1 differ in their length: AaeL1 consists of 242 a.a.; HmaL1 consists of 212 a.a. HmaL1 contains ~25% negatively charged residues (aspartate and glutamate) located mainly on the surface of domain II – a feature that is lacking from L1 proteins of other organisms [11].
Fig. 2. Comparison of HmaL1 and AaeL1 amino acid sequences. Identical residues are shown in gray.

Search for disordered regions and design of disulfide bonds in AaeL1 and HmaL1. We used the PONDR-FIT and IsUnstruct programs to search for the disordered regions in the AaeL1 and HmaL1 protein sequences [6, 9]. Figure 1c shows the probability of different amino acid residues of AaeL1 and HmaL1 (blue and green, respectively) to be in the intrinsically disordered state. The predictions considerably differed for a fragment of domain II sequence indicated with red line at the bottom of Fig. 1c: in AaeL1 this region was predicted as stable, whereas in HmaL1 – as disordered. Therefore, we chose this region for verifying our hypothesis. If the results of calculations with the PONDR-FIT and IsUnstruct programs can be interpreted as predictions of stable and weakened parts of a protein polypeptide chain, then the introduction of disulfide bond into the predicted disordered region in HmaL1 will increase the stability and reduce the mobility of this protein, whereas introduction of disulfide bond into the same region in AaeL1 (predicted to be structured) will not affect protein stability or might even decrease it. If the spatial structure of proteins is more important for their stabilization, then the introduced mutation will have an identical effect in both proteins.
We also tested whether the amino acid substitutions at positions D101 and K127 in AaeL1 and E82 and D114 in HmaL1 affect the predictions of the PONDR-FIT and IsUnstruct programs. We found that insignificant changes in the amino acid sequences of proteins (substitution of 2 a.a.) only slightly altered the curves presented in Fig. 1c. At least, the influence of these substitution was much less than the difference between the predictions of PONDR-FIT and IsUnstruct programs.
Figure 1b shows the superposition of the spatial structures of domains II from AaeL1 and HmaL1. The regions chosen by us for the introduction of disulfide bonds in AaeL1 and HmaL1 based on the predictions by the PONDR-FIT and IsUnstruct programs are shown in blue and green, respectively. Amino acid residues selected for the substitution with cysteine residues (D101 and K127 in AaeL1 and E82 and D114 in HmaL1) are shown in yellow. These residues were chosen based on two criteria: (i) the distance between their Cβ-atoms should be ~5 Å and (ii) the residues should be directed toward each other in space.
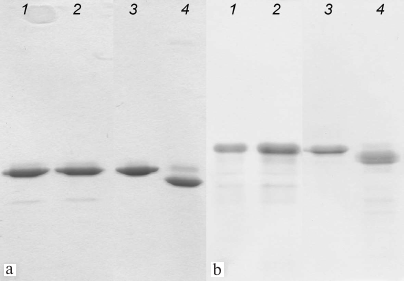
Formation of disulfide bonds in AaeL1-D101C-K127C and HmaL1-E82C-D114C proteins. Mutant forms of AaeL1 and HmaL1 proteins were purified as described in “Materials and Methods”. Formation of disulfide bonds in the mutants was verified by SDS-PAGE, which was possible due to the difference in the hydrodynamic radius of the denatured proteins with and without the disulfide bond. Figure 3 shows the results of SDS-PAGE for the AaeL1 and AaeL1-D101C-K127C proteins (Fig. 3a) and HmaL1 and HmaL1-E82C-D114C proteins (Fig. 3b). When the disulfide bonds were reduced with β-mercaptoethanol (Fig. 3, a and b, lanes 1 and 2), the wild-type and the mutant proteins demonstrated identical electrophoretic mobility. When the disulfide bonds remained intact (in the absence of β-mercaptoethanol), the mobility of the wild-type and mutant proteins was different (Fig. 3, a and b, lanes 3 and 4). The results presented in Fig. 3 indicated that the introduced disulfide bonds were projected correctly and formed spontaneously in AaeL1-D101C-K127C or upon oxidation in HmaL1-E82C-D114C (see “Materials and Methods”).
Fig. 3. SDS-PAGE of AaeL1 and AaeL1-D101C-K127C (a) and HmaL1 and HmaL1-E82C-D114C (b) under reducing conditions (lanes 1 and 2) and with formed disulfide bonds (lanes 3 and 4).
Stability of AaeL1 protein with the introduced disulfide bond. AaeL1 is characterized by a high thermostability typical for a protein from a thermophilic bacterium. To study its melting, we had to first slightly decrease its stability by adding a denaturing agent. Figure 4 shows the AaeL1 melting curve (studied by fluorescence) in the presence of different concentrations of urea. In the absence of the denaturing agent, the protein melting temperature was above 100°C. In the presence of 1 and 4 M urea, the melting temperature was ~90 and 75°C, respectively. Figure 5a shows that the CD spectra for the wild-type AaeL1 in the absence of denaturing agent (curve 1) and in the presence of 4 M urea (curve 3) coincide. Therefore, 4 M urea did not influence the protein native structure but destabilized it with the increase in temperature. Hence, further experiments with AaeL1 were performed in the presence of 4 M urea.
Fig. 4. Changes in the fluorescence intensity during thermal denaturation of the wild type AaeL1 under native conditions (1) and in the presence of 1 M (2) and 4 M urea (3). The measurements were performed in 20 mM sodium phosphate buffer, pH 7.5. (Colored version of Fig. 4 is available in electronic version of the article on the site http://sciencejournals.ru/journal/biokhsm/)
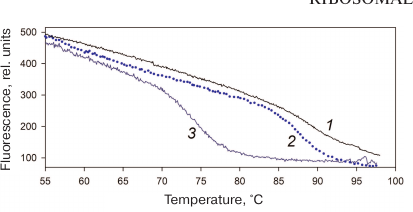
Fig. 5. Stability of the AaeL1 protein with the introduced disulfide bond. a) CD spectra of AaeL1 (curves 1 and 3) and AaeL1-D101C-K127C with the introduced disulfide bond (curves 2 and 4) in the presence of different concentrations of urea: 1, 2 (gray and light-blue colors, respectively), in the absence of denaturing agent; 3, 4 (black and dark-blue, respectively), in the presence of 4 M urea. The measurements were performed in 20 mM sodium phosphate buffer (pH 7.5). b) Temperature dependence of the molar ellipticity at 215 nm of AaeL1 (curve 3, black) and AaeL1-D101C-K127C (curve 4, dark-blue) in the presence of 4 M urea. c) Temperature dependence of the excessive heat capacity of AaeL1 (curve 3, black) and AaeL1-D101C-K127C with the disulfide bridge (curve 4, dark-blue) in the presence of 4 M urea. (Colored version of Fig. 5 is available in electronic version of the article on the site http://sciencejournals.ru/journal/biokhsm/)
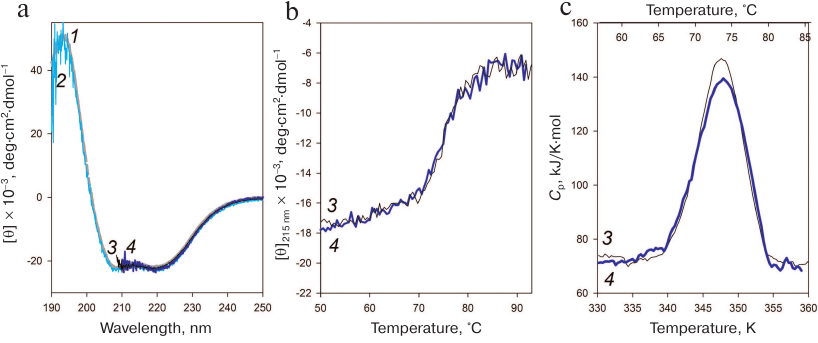
Next, we checked whether the disulfide bond disturbed the protein native arrangement. Figure 5a shows the CD spectra of the wild-type and mutant AaeL1 proteins in the absence of denaturing agent and in the presence of 4 M urea. Under native conditions (in the absence of urea), the spectra of the wild-type and mutant proteins coincided and were typical for α-helical proteins. The spectra of both proteins in the presence of 4 M urea coincided with each other (despite the noise) and with their CD spectra obtained under native conditions. Therefore, neither the presence of 4 M urea, nor the introduced disulfide bond affected the secondary structure of AaeL1.
The melting of AaeL1 and AaeL1-D101C-K127C proteins was studied by CD spectroscopy. Figure 5b shows the temperature dependence curves of the molar ellipticity at 215 nm. The curves for AaeL1 and AaeL1-D101C-K127C with the introduced disulfide bond coincided. To determine more accurately the melting temperatures of AaeL1 and AaeL1-D101C-K127C, we studied the melting of these proteins using differential scanning microcalorimetry. Figure 5c shows the melting curves of AaeL1 and its mutant with the introduced disulfide bond in the presence of 4 M urea. The melting temperatures were the same for the wild-type and mutant proteins and equaled 347 K (74°C).
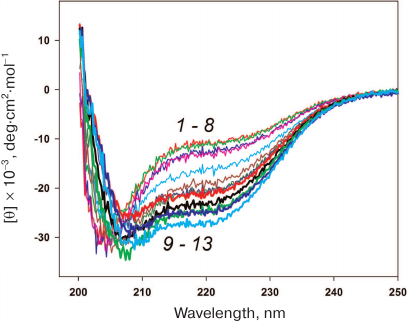
Stability of HmaL1 protein with the introduced disulfide bond. HmaL1 is a protein from an extremely halophilic organism living at high (near-saturation) concentrations of salt [17]. Therefore, we first selected conditions (NaCl concentration) ensuring the structured state of this protein. Figure 6 shows the CD spectra of HmaL1 in the presence of different NaCl concentrations. HmaL1 was disordered or structured poorly at 0-2 M NaCl (curves 1-8). Most of the HmaL1 secondary structure was observed in the presence of 3 M NaCl (curve 13, blue), which was the minimal NaCl concentration required for retaining the structured state of HmaL1.
Fig. 6. CD spectra of HmaL1 in the presence of different NaCl concentrations: from 0 M (1) to 3.5 M (13) with the step of 0.25 M NaCl. The measurements were performed in 50 mM Tris-HCl, pH 7.5. (Colored version of Fig. 6 is available in electronic version of the article on the site http://sciencejournals.ru/journal/biokhsm/)
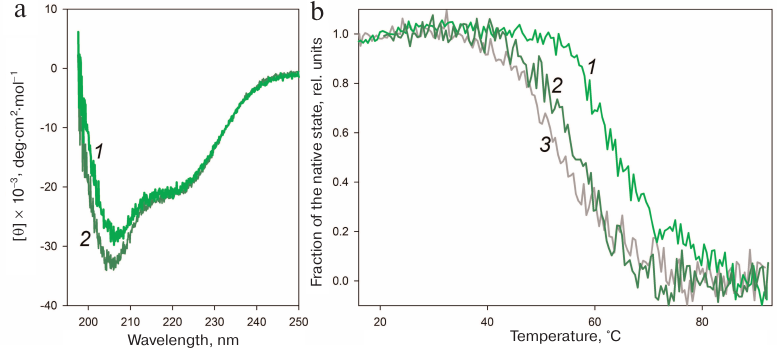
The disulfide bond in HmaL1 was designed based on the model of HmaL1 structure proposed by Gabdulkhakov et al. [11]. To ensure correct formation of the disulfide bonds in this protein, we performed oxidation as described in “Materials and Methods”. The effect of the introduced disulfide bond on the HmaL1 structure was studied by CD spectroscopy. Figure 7a shows CD spectra of HmaL1 and HmaL1-E82C-D114C with the disulfide bond. It can be seen that introduction of the disulfide bond into the disordered region increased the amount of secondary structure in the protein. The melting of Hmal1 was studied by CD spectroscopy. Figure 7b shows the temperature dependence curves of the protein portion with the native structure in HmaL1 and HmaL1-E82C-D114C with the introduced disulfide bond, as well as the dependence of the calculated molar ellipticity at 215 nm on temperature. The melting temperatures for Hmal1 and HmaL1-E82C-D114C were 55 and 65°C, respectively, which confirmed our hypothesis that the disulfide bond introduced into the region predicted as disordered (despite its structured state within the protein) leads to the increase in the protein stability.
Fig. 7. Stability of the HmaL1 protein with the introduced disulfide bond. a) CD spectra of HmaL1 (curve 2, dark green) and HmaL1-E82C-D114C (curve 1, green). The measurements were performed in 50 mM Tris-HCl (pH 7.5) containing 3 M NaCl and 50 mM MgCl2. b) Temperature dependence of the protein portion with the native structure in HmaL1 (curve 2, dark green) and in HmaL1-E82C-D114C with the formed (curve 1, green) and reduced (curve 3, gray) disulfide bond. The native state fraction was calculated from the molar ellipticity at 215 nm. (Colored version of Fig. 7 is available in electronic version of the article on the site http://sciencejournals.ru/journal/biokhsm/)
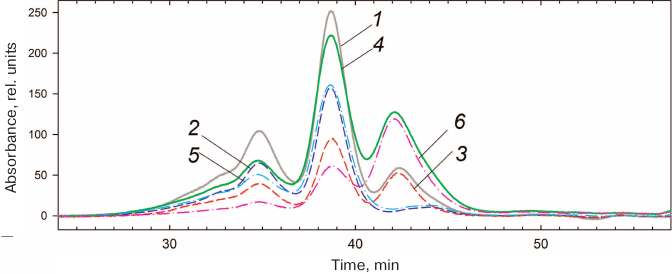
HmaL1 is poorly structured and contains no tryptophan residues in its sequence. It also requires the presence of high concentrations of salt, as it has a tendency for aggregation. Because of this, it was impossible to study HmaL1 melting based on its intrinsic fluorescence or using differential scanning microcalorimetry. We decided to verify whether the introduced disulfide bond influenced the biological function of HmaL1. Protein L1 is a highly conserved ribosomal protein that binds 23S rRNA. The binding occurs via domain I, while domain II is involved in the RNA recognition. We studied the HmaL1/rRNA complex by gel-filtration chromatography. HmaL1 binding with rRNA was performed as described in “Materials and Methods”. Figure 8 shows the elution profiles of free H77-H78 23S rRNA and its complexes with HmaL1 and HmaL1-E82C-D114C, as well as contribution of the proteins and 23S rRNA to these complexes. The heights of the peaks in the elution profiles (Fig. 8) indicate that 2/3 of the HmaL1 binds RNA, whereas 1/3 of the protein remains free. In the case of HmaL1-E82C-D114C with the introduced disulfide bond, 1/3 of the protein formed complexes with rRNA, whereas 2/3 of the protein remained in the free state. Therefore, introduction of the disulfide bond in domain II did not abolish the biological function of the protein, although decreases it. This might be associated with the decrease in the domain II structure mobility (because of the disulfide bond) that presumably affected the recognition of the target rRNA (which might require domain II mobility).
Fig. 8. Elution profiles of 23S rRNA complexes with HmaL1 (1) and HmaL1-E82C-D114C (4). Contributions of HmaL1 (3) and HmaL1-E82C-D114C (6) to the complex; curves 2 and 5 indicate contribution of 23S rRNA to the complexes with HmaL1 and HmaL1-E82C-D114C with the introduced disulfide bond, respectively. (Colored version of Fig. 8 is available in electronic version of the article on the site http://sciencejournals.ru/journal/biokhsm/)
DISCUSSION
The purpose of this study was to confirm our hypothesis that introduction of disulfide bonds into the protein regions predicted as intrinsically disordered by the PONDR-FIT and IsUnstruct programs leads to the increase in the protein stability and reduction of structural mobility, whereas introduction of the disulfide bond into a similar region predicted as structured should not influence the protein structure. Using AaeL1 and HmaL1 proteins, we identified the regions with identical spatial structure that were predicted as structured in AaeL1 and disordered in HmaL1. We designed and introduced disulfide bonds into identical positions in both proteins and studied their influence on the stability of the protein structure. As a result, the following conclusions were made: 1) the disulfide bond introduced into the region predicted as structured in AaeL1did not influence the protein stability; 2) the disulfide bond introduced into the region predicted as intrinsically disordered in HmaL1 led to the increase in the protein melting temperature; 3) HmaL1 mutant with the increased stability retained its functional activity. Therefore, we confirmed our suggestion that predictions made by the PONDR-FIT and IsUnstruct programs correlated with the weakness or stability of regions in the L1 polypeptide chain and not with specific features of the protein spatial structure.
The results of our studies from this and previous publications [8] allowed us to propose a hypothesis that protein stability depends not so much on the structure of the protein native state (rigidly packed), but rather on the characteristics of its unfolded and intermediate states (mobile states with unformed contacts). This idea seems simple from the viewpoint of a theorist, but it is very difficult to verify experimentally, especially from a viewpoint of an experimenter facing the task to increase the stability of a particular protein. Our studies allow researchers to use programs predicting intrinsically disordered regions as a tool for finding out weakened regions in proteins, which, in their turn, can be the reason for the existence of more mobile unstable intermediate states and, therefore, can influence the transition between the native and other states during protein melting or unfolding with denaturing agents.
Funding. The study was supported by the Russian Foundation for Basic Research (project 18-34-00243 mol_a).
Acknowledgements. The authors are grateful to S. V. Tishchenko for kindly providing rRNA preparations.
Conflict of interest. The authors declare no conflict of interest in financial or any other area.
Ethical norm compliance. This article does not contain description of studies with the participation of people or animals performed by any of the authors.
REFERENCES
1.Jirgensons, S. C., Hnilica, B., and Capetillo, L.
S. (1966) Viscosity and conformation of calf thymus histones, Die
Makromol. Chem., 97, 216-224, doi:
10.1002/macp.1966.020970120.
2.DeForte, S., and Uversky, V. (2016) Order,
disorder, and everything in between, Molecules, 21, 1090,
doi: 10.3390/molecules21081090.
3.He, B., Wang, K., Liu, Y., Xue, B., Uversky, V. N.,
and Dunker, A. K. (2009) Predicting intrinsic disorder in proteins: an
overview, Cell Res., 19, 929-949, doi:
10.1038/cr.2009.87.
4.Jones, D. T., and Cozzetto, D. (2015) DISOPRED3:
precise disordered region predictions with annotated protein-binding
activity, Bioinformatics, 31, 857-863, doi:
10.1093/bioinformatics/btu744.
5.Linding, R., Jensen, L. J., Diella, F., Bork, P.,
Gibson, T. J., and Russell, R. B. (2003) Protein disorder prediction:
implications for structural proteomics, Structure, 11,
1453-1459, doi: 10.1016/j.str.2003.10.002.
6.Xue, B., Dunbrack, R. L., Williams, R. W., Dunker,
A. K., and Uversky, V. N. (2010) PONDR-FIT: a meta-predictor of
intrinsically disordered amino acids, Biochim. Biophys. Acta
Protein. Proteom., 1804, 996-1010, doi:
10.1016/j.bbapap.2010.01.011.
7.Li, J., Feng, Y., Wang, X., Li, J., Liu, W., Rong,
L., and Bao, J. (2015) An overview of predictors for intrinsically
disordered proteins over 2010-2014, Int. J. Mol. Sci.,
16, 23446-23462, doi: 10.3390/ijms161023446.
8.Nagibina, G. S., Tin, U. F., Glukhov, A. S.,
Melnik, T. N., and Melnik, B. S. (2016) Intrinsic disorder-based design
of stabilizing disulfide bridge in Gαo protein, Protein Pept.
Lett., 23, 176-184, doi:
10.2174/092986652302160105130540.
9.Lobanov, M. Y., and Galzitskaya, O. V. (2011) The
Ising model for prediction of disordered residues from protein sequence
alone, Phys. Biol., 8, 35004, doi:
10.1088/1478-3975/8/3/035004.
10.Nikonova, E. Y., Tishchenko, S. V., Gabdulkhakov,
A. G., Shklyaeva, A. A., Garber, M. B., Nikonov, S. V., and Nevskaya,
N. A. (2011) Crystal structure of ribosomal protein L1 from the
bacterium Aquifex aeolicus, Crystallogr. Rep., 56,
648-652, doi: 10.1134/S1063774511040158.
11.Gabdulkhakov, A., Tishchenko, S., Mikhaylina, A.,
Garber, M., Nevskaya, N., and Nikonov, S. (2017) Crystal structure of
the 23S rRNA fragment specific to r-protein L1 and designed model of
the ribosomal L1 stalk from Haloarcula marismortui,
Crystals, 7, 37, doi: 10.3390/cryst7020037.
12.Muller, M. P., Peters, H., Blumer, J.,
Blankenfeld, W., Goody, R., and Itzen, A. (2010) The Legionella
effector protein DrrA AMPylates the membrane traffic regulator Rab1b,
Science, 329, 946-949.
13.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685, doi: 10.1038/227680a0.
14.Senin, A. A., Potekhin, S. A., Tiktopulo, E. I.,
and Filimonov, V. V. (2000) Differential scanning microcalorimeter
SCAL-1, J. Therm. Anal. Calorim., 62, 153-160, doi:
10.1023/A:1010171013669.
15.Privalov, P. L., and Potekhin, S. A. (1986)
Scanning microcalorimetry in studying temperature-induced changes in
proteins, Methods Enzymol., 131, 4-51, doi:
10.1016/0076-6879(86)31033-4.
16.Rao, S. T., and Rossmann, M. G. (1972) Comparison
of super-secondary structures in proteins, J. Mol. Biol.,
4, 241-250, doi: 10.1016/0022-2836(73)90388-4.
17.Camacho Cordova, D. I., Camacho Ruiz, R. M.,
Mateos Diaz, J. C., Cordova Lopez, J. A., and Rodriguez Gonzalez, J. A.
(2014) Haloarcula marismortui, eighty-four years after its
discovery in the Dead Sea, Int. J. Eng. Res. Technol., 3,
1257-1267.